Presentazione di PowerPoint
advertisement

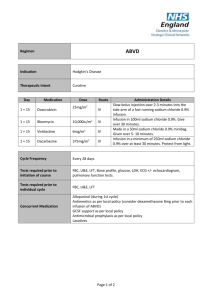
The Ideal DMPK Profile versus Lead Optimization Clinical DMPK Profile High oral bioavailability: Half-life between 12 and 24 hr: Multiple elimination pathways: No reactive metabolites: No human-specific metabolites: No inhibition of CYP450 enzymes: No induction of CYP enzymes: Rationale Low inter-subject variability/ cost of goods QD dosing/ acceptable accumulation Drug-drug interactions (DDI) less likely Avoid safety issues/ idiosyncratic AEs Simplifies safety program & risk assessment Drug unlikely to cause DDIs Avoid autoinduction or DDIs Drug Safety DMPK Profile Good PK with developable form: Acceptable exposure multiples: Stable and predictable exposure: Clean in AMES test: Crystalline form - reduced bioavailability? Human risk assessment Reliably target appropriate exposure Avoid mutagens Discovery DMPK In Vitro Assays Assay CYP 450 Inhibition: CYP 450 Profiling: Rat liver slice - induction: hPXR : Hepatocyte clearance: Inter-species hepatocyte met ID : Plasma stability: Plasma protein binding: Mock hERG drug assay: Caco-2: Rationale Avoid drug-drug interactions (DDI)? Avoid Polymorphism? Avoid DDI? Predict in vivo rat enzyme induction. Predict CYP 3A4 induction in humans. Predict human metabolic rate. Look for human-specific metabolites. Ex vivo degradation? Normalize exposure based on ‘free’ drug Confirm actual concentration. Predict human absorption potential. Discovery DMPK In Vivo Studies Study Full” PK (IV & PO): Single-rising dose: 14-Day Rat multiple dose: Rationale Predict human PK Adequate exposure for safety testing Steady-state; tumorigenic induction ‘Hot’ PK (3H-SCH) : Absorption and circulating metabolites Metabolite Pathway Elucidation: Identify metabolites in ‘safety’ species Mass Balance of 3H-SCH: Avoid unusual retention of metabolites Melanin Binding (LE rats): Assess phototoxicity potential PK/PD: Developable form acceptability: Estimate efficacious drug exposure Adequate exposure for safety testing Pharmacokinetics and Pharmacodynamics Pharmacokinetics (PK) Pharmacodynamics (PD) Concentration vs Time Effect vs Concentration PK/PD Effect vs Time Assumption: The magnitude of the desired effect (or side effect) is a function of the drug concentration at the site of action PK-PD relationship Drug excretion (Metabolism-Elimination) Drug at Absorption Site Drug in Tissues (Distribution) Pharmacokinetics PK PD Drug at Drug at Effect Effect SiteSite Response!! Pharmacodynamics What your body does to the drug What the drug does to your body Different potency in different species The PK-PD relationship The test compound has a full effect at the dose of 10 mg/kg po Pharmacological effect 16 cutoff 800 14 700 12 600 10 500 8 400 6 300 4 200 2 100 0 0 0 30 60 120 180 240 360 Plasma level ng/ml Veh 10 mpk, po Time (min) At 10 mg/kg po the temporal profile of the plasma level and the pharmacological activity profile are similar Safety and preclinical toxicology Safety Effect on the principal physiological system (cardiovascular, renal, nervous, etc). Safety pharmacology Precinical toxicology To 1. 2. 3. 4. 5. establish : safe dose MTD (maximum tolerated dose) terapeutic window target organs Reversibility of toxicity Toxic effects could mechanism-based or compound-based. Preclinical toxicology •Acute toxicity profile •Chronic toxicity profile • 14 days toxicity test in one rodent and one non-rodent species before use in man. • 3 months study read out at 28 days • longer studies (12 & 24 month) • Mutagenicity tests in vitro and in vivo What a therapeutic window is? Therapeutic effect Tolerability Toxicity Sedation Ansiolitic effect Respiratory depression P2Y12 antagonists Therapeutic index evaluation (van Giezen and Humphries Semin Thrombosis Hemost 2005) It is possible to obtain a clear separation between antithrombotic and bleeding effects Reasons for Failure in Development Toxicity (22%) Lack of Efficacy (31%) Market Reasons (6%) Poor Biopharmaceutical (PK) Properties (41%) Moving from Animals to Man 1. 2. 3. Humans and animals have different biochemistry, physiology and anatomy Predictions of a drug’s PK profile in humans using animal PK data must account for these differences Allometric scaling is used to predict differences based only on size. • • • • The relationship of some PK parameters across species can be correlated with body weight. One can determine an empirical relationship log PK parameters and log Body Weight These parameters can be used to extrapolate PK parameters in humans when parameters have been determined in lower species (mouse, rat, dog, monkey, etc.) The relationship is not always predictive, but it can often give a good estimate. Summary The information collected in ADME and DMPK study are necessary to establish the dose that will be used in human Questions?? R&D process for a new drug CANDIDATE POC Exploratory development Developpability Fase 0 or Preclinical development Safety Fase I (A and B) Full development Therapeutic efficacy Fase II Study in the patient D R U G Pre-marketing Fase III Study in the patient Registration Fase IV Post marketing Surveillance Formulation study • Pharmaceutical form: tablet , capsule, cream, injection, etc •To achieve the best effect is necessary to identify not only the best form but also the most suitable formulation. Example of composition of a tablet: •Active principle, filler, binder, lubricant, disintegrant, surfactant. Objectives of Clinical Trials • Phase I: First in man safety e tolerability • Phase II: First in patient IIa safety and tolerability IIb dose, dosage form • Phase III: Value (is better than existing treatments) • Post marketing surveillance or Phase IV : Monitor the drug in the real clinical setting Clinical trials Uncontrolled Controlled Randomized Open or blind Sequential or cross-over