Document
advertisement

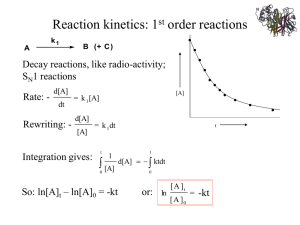
Enzyme kinetics Why study the rate of enzyme catalyzed reactions? • Study of reaction rates is an important tool to investigate the chemical mechanism of catalysis • Kinetic studies provide information on substrate and product affinity to the enzyme • Knowledge of the dynamic properties of enzyme catalysis is a prerequisite for the design of inhibitors (drugs) directed against a certain enzyme Chemical reaction kinetics Reaction order of a chemical process corresponds to the molecularity of the reaction, which is the number of molecules that must collide simultaneously to generate a product: aA + bB + cC product(s) The velocity for such a process is given by: v = k [A]a • [B]b • [C]c Where v is the velocity of the reaction and k the rate constant of the reaction; the reaction order is the sum of the exponentials in the rate equation. First and Second-order reactions For a first-order process: A P v = The reaction velocity, v, is given by: dA dP = = k [A] dt dt The reaction velocity for such a first-order reaction is proportional to the concentration of A. Such a reaction is also called a unimolecular reaction. A second-order reaction can involve the reaction of two identical or different substrate molecules: A + A product(s) or A + B or v =- product(s) The velocity of the reaction is then: dA = k [A]2 v = dP = dt dt dA = - dB = k [A] [B] dt dt The dimension of the rate constant The dimension of k depends on the reaction order. This is due to the fact that the velocity of a reaction is measured in mol product formed per time unit (e.g. sec). Therefore the unit of the rate constant of a firstorder reaction is sec-1 and that of a second-order reaction is M-1 sec-1. Determination of k for a first-order reaction For a first-order process: A P As before, the reaction velocity, v, is given by: v = dA = k [A] dt ln [A] ln [A]t=0 slope = -k dA = dln [A] = - k dt [A] Integration with [A]t=0 to [A]t yields: [A]t ∫ dln [A] [A]t=0 t t ∫ = - k dt ln [A]t = ln [A]t=0 - kt t=0 [A]t = [A]t=0 e-kt Example: radioactive decay Relationship of half-life and rate constant The half-life (t1/2) is a constant (dimension: time). Radioactive decay is usually expressed in half-life rather than the first-order rate constant: [A]t=0 The half-life is defined as the time required for [A]t=0 to decrease to 2 [A]t=0 = [A]t=0 e-kt1/2 2 t 1/2 = ln2 k Rate constant for a second-order reaction Consider the following reaction: A + A v=[A]t ∫- [A]t=0 dA = k [A]2 dt 1 [A]t t dA [A]2 = k product(s) ∫ dt t=0 slope = k 1 [A]t=0 1 = 1 + kt [A]t [A]t=0 The half-life for such a reaction is: t1/2 = 1 • [A]t=0 1 k t This type of plot can be used to distinguish between a unimolecular and bimolcular reaction involving A Note: half-life depends on [A]t=0 The enzyme-substrate complex Adrian Brown Brown and Henri investigated the substrate-dependence of an enzymecatalyzed reaction and found that the reaction reached a maximum velocity at high substrate concentrations (Vmax) Brown and Henri’s conclusion: „The enzyme works by forming a complex (like a lock and a key) with the susbtrate and acting on it for a finite period of time.“ binding E + S enzyme + substrate reaction E-S dissociation E-P enzyme-substrate enzyme-product complex complex E + P enzyme + product Schematic representation of an enzyme-catalyzed reaction Kinetics of enzyme reactions Recall: Brown observed that the rate at which sucrose is degraded by invertase shows saturation behavior, that is at high sucrose concentrations the rate becomes independent of the sucrose concentration („zero-order“ reaction with respect to sucrose). It was concluded that an enzyme-catalyzed reaction proceeds in two steps: [E] + [S] k1 k-1 [ES] k2 [P] + [E] When [S] >> [E] then all of the enzyme is in the complex [ES] and the formation of product is given by: V = d[P] = k [ES] 2 dt The concentration of [ES] is a complex function depending on the individual rate constants k1, k-1 and k2: d[ES] dt = k1 [E] [S] - k-1 [ES] - k2 [ES] Two approaches 1. Michaelis-Menten kinetic (1913) („rapid equilibrium“ assumption) 2. Briggs-Haldane kinetic (1925) („steady-state“ assumption) Title page of Michaelis & Menten’s original paper in “Biochemische Zeitschrift” in 1913 Leonor Michaelis (1875-1940) Maud L. Menten (1879-1960) The Michaelis-Menten approach k1 [E] + [S] [ES] k-1 k2 [P] + [E] Assumption: k-1 >> k2 i.e. the equilibrium of [E], [S] and [ES] is not affected by k2: KS = dissociation constant k-1 [E] [S] KS = k1 = [ES] [ES] = „Michaelis-Menten“ complex Since we assume equilibrium it follows: [E] [S] k1 = [ES] k-1 solving for [E] = In addition we know that: k-1 k1 [ES] [S] (1) [E]total = [E] + [ES] (2) This relationship is called the „enzyme conservation equation“ The Michaelis-Menten approach [E] = k-1 k1 [ES] [S] (1) [E]total = [E] + [ES] (2) Solving equation (2) for [E] and substituting [E] in equation (1): [E]total = [ES] (1 + k-1 k1 [S] ) (3) We also know that the velocity of the reaction equals: v = k2 [ES] (4) Solving equation (3) and (4) for [ES] and then substituting [ES] in equation (3) with [ES] = v / k2 then yields: v= k2 (1 + [E]total k-1 k1 [S] = k2 [E]total [S] k-1 [S] + k1 ) We define k-1/ k1 as KM, the Michaelis-Menten constant and the maximal velocity as vmax = k2 [E]total This simplifies the above equation to: v = vmax [S] [S] + KM if [S] >> KM then v = vmax vmax if [S] = KM then v = 2 Therefore KM can be viewed as the substrate concentration with halfmaximal velocity (dimension M, typically mM to nM) Michaelis-Menten plot v Linear plot of substrate concentration versus velocity yields a hyperbolic relationship: vmax vmax 1st order zero order 2 KM [S] The Briggs-Haldane approach [E] + [S] k1 k-1 Assumption: k-1 ~ k2 i.e. during substrate turnover the concentration of [ES] is constant („steady-state“ assumption). The assumption is less restrictive than the „rapidequilibrium“ assumption by Michaelis-Menten. d[ES] =0 dt [ES] k2 [P] + [E] The Briggs-Haldane approach d[ES] = 0 = k1 [E] [S] - k-1 [ES] - k2 [ES] dt k1 [E] [S] = k-1 [ES] + k2 [ES] We also know that: [E]total = [E] + [ES], solving this equation For [E] and substituting in the „steady-state“ equation yields: k1 ([E]total - [ES]) [S] = k-1 [ES] + k2 [ES] k1 [E]total [S] - k1 [ES] [S] = k-1 [ES] + k2 [ES] k1 [E]total [S] = (k-1 + k2) [ES] + k1 [ES] [S] : k1 [E]total [S] = (k-1 + k2) [ES] + [ES] [S] k1 Solving this equation for [ES] yields: [ES] = [ES] = [E]total [S] (k-1 + k2) k1 [E]total [S] KM + [S] k2 [E]total [S] v = KM + [S] Same equation! KM = (k-1 + k2) + [S] and with v = k2 [ES] and with vmax = k2 [E]total v = vmax [S] KM + [S] k1 Michaelis-Menten vs. Briggs-Haldane Although both approaches yield the same basic equation for the velocity of an enzyme catalysed reaction, the meaning of the Michaelis-Menten parameter, KM, differs: In the „rapid-equilibrium“approach by Michaelis-Menten KM is equivalent to the true dissociation constant Ks In the „steady-state“ approach by Briggs-Haldane the rate of the „chemical step“, k2, is part of the KM and hence it is not equivalent to the dissociation constant. v = KM = KM = vmax [S] KM + [S] k-1 k1 (k-1 + k2) k1 Analysis of kinetic data - the Lineweaver-Burk plot We have seen that at high substrate concentration, the initial velocity of the reaction approaches the maximal velocity vmax asymptotically. In practice this asymptotic value is difficult to determine in a direct (hyperbolic) plot of velocity vs. substrate concentration. Therefore Hans Lineweaver and Dean Burk have linearized the Michaelis-Menten equation to: 1 = v ( ) [S]1 + v1 KM vmax max In this double-reciprocal plot 1/v is plotted vs 1/ [S]. The y-axis intercept yields 1/vmax whereas the x-axis intercept yields 1/KM. The slope of the straight line is equivalent to KM/vmax. Catalytic efficiency of enzymes For an enzyme that obeys the Michaelis-Menten kinetics: vmax = k2 [E]total k2 is also called kcat or turnover number because it reflects directly the commitment to catalysis and therefore we can also write: vmax = kcat [E]total or kcat = vmax [E]total When [S] << KM then [E] ~ [E]total v = k2 [E]total [S] KM + [S] kcat v = KM [E] [S] kcat/KM is a 2nd order rate constant (M-1 sec-1) and as such reflects the efficiency of „ E to react with S“ These consideration also allow us to determine how fast an enzyme catalyzed reaction can proceed: Maximal velocity of an enzyme-catalyzed reaction k1 k2 kcat k2 = = k-1 + k2 KM KM In the case of efficient catalysis k2 >> k-1 This means that the Michaelis-Menten complex decays rapidly to the product(s) and the back-reaction to free enzyme and substrate are much slower („enzyme is committed to catalysis“). In such a case the equation above can be rewritten as: kcat = k1 KM Since k1 is the rate at which the Michaelis-Menten complex forms, the limiting value for this rate constant is the rate of encounter of enzyme and substrate, i.e. the rate limited by diffusion. This rate is of the order 108-109 M-1 sec-1. Hence enzymes that operate in this range have achieved maximal velocity (catalase: 4 x 108 M-1 sec-1) Determination of rates • From steady-state measurements two enzyme parameters are obtained: 1) The Michaelis-Menten Parameter (KM) which may be equivalent to the enzyme-substrate dissociation constant 2) kcat (turnover number) which may be a microscopic rate constant or a combination of several • To observe individual rates the approach to steady-state needs to be observed („pre-steady-state kinetics“); the rates are typically on the order of 1- 10-7 sec! The Continuous Flow Method • Hartridge & Roughton (1923) • Reactants are compressed at a constant rate generating a constant flow • At a constant flow rate the age of the solution is linearly proportional to the distance down the flow tube from „Structure and mechanism in protein science“, Alan Fersht The Stopped Flow Method • Roughton (1934); improved by Chance (1940) • Amenable for reactions that undergo spectral changes (UV-Vis absorbance, fluorescence, CD) from „Structure and mechanism in protein science“, Alan Fersht The Rapid Quench Flow Technique • Require quenching of the reaction and concomittant analysis of the sample collected by an appropriate analytical method (HPLC, GC-MS, etc.) from „Structure and mechanism in protein science“, Alan Fersht