- No category
Nitride Semiconductors Handbook: Properties, Physics, Growth
advertisement
Hadis Morkoç
Handbook of Nitride
Semiconductors and Devices
Related Titles
Piprek, J. (ed.)
Nitride Semiconductor Devices: Principles and Simulation
2007
ISBN: 978-3-527-40667-8
Adachi, S.
Properties of Group-IV, III-V and II-VI Semiconductors
2005
ISBN: 978-0-470-09032-9
Ruterana, P., Albrecht, M., Neugebauer, J. (eds.)
Nitride Semiconductors
Handbook on Materials and Devices
2003
ISBN: 978-3-527-40387-5
Ng, K. K.
Complete Guide to Semiconductor Devices
2002
ISBN: 978-0-471-20240-0
Hadis Morkoç
Handbook of Nitride Semiconductors
and Devices
Vol. 1: Materials Properties, Physics and Growth
The Author
Prof. Dr. Hadis Morkoç
Virginia Commonwealth University
Dept. of Chemical Enigineering
Richmond, VA
USA
Cover
SPIESZDESIGN GbR,
Neu-Ulm, Germany
All books published by Wiley-VCH are carefully
produced. Nevertheless, authors, editors, and
publisher do not warrant the information contained
in these books, including this book, to be free of
errors. Readers are advised to keep in mind that
statements, data, illustrations, procedural details or
other items may inadvertently be inaccurate.
Library of Congress Card No.: applied for
British Library Cataloguing-in-Publication Data
A catalogue record for this book is available from the
British Library.
Bibliographic information published by
the Deutsche Nationalbibliothek
Die Deutsche Nationalbibliothek lists this
publication in the Deutsche Nationalbibliografie;
detailed bibliographic data are available in the
Internet at <http://dnb.d-nb.de>.
# 2008 WILEY-VCH Verlag GmbH & Co. KGaA,
Weinheim
All rights reserved (including those of translation into
other languages). No part of this book may be
reproduced in any form – by photoprinting,
microfilm, or any other means – nor transmitted or
translated into a machine language without written
permission from the publishers. Registered names,
trademarks, etc. used in this book, even when not
specifically marked as such, are not to be considered
unprotected by law.
Typesetting Thomson Digital, Noida, India
Printing betz-druck GmbH, Darmstadt
Binding Litges & Dopf GmbH, Heppenheim
Printed in the Federal Republic of Germany
Printed on acid-free paper
ISBN: 978-3-527-40837-5
V
Contents
Preface XIII
Color Tables XXI
1
1.1
1.2
1.2.1
1.2.2
1.2.3
1.3
1.3.1
1.3.2
1.3.3
1.3.4
1.4
1.4.1
1.4.2
1.4.3
1.4.4
1.4.5
1.5
1.5.1
1.5.2
1.5.3
1.5.4
1.5.5
General Properties of Nitrides 1
Introduction 1
Crystal Structure of Nitrides 1
Gallium Nitride 30
Chemical Properties of GaN 35
Mechanical Properties of GaN 35
Thermal Properties of GaN 47
Aluminum Nitride 62
Mechanical Properties of AlN 62
Thermal and Chemical Properties of AlN
Electrical Properties of AlN 69
Brief Optical Properties of AlN 71
Indium Nitride 75
Crystal Structure of InN 77
Mechanical Properties of InN 77
Thermal Properties of InN 79
Brief Electrical Properties of InN 81
Brief Optical Properties of InN 84
Ternary and Quaternary Alloys 89
AlGaN Alloy 90
InGaN Alloy 92
InAlN Alloy 97
InAlGaN Quaternary Alloy 99
Dilute GaAs(N) 105
References 110
Handbook of Nitride Semiconductors and Devices. Vol. 1. Hadis Morkoç
Copyright # 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 978-3-527-40837-5
66
VI
Contents
2
2.1
2.1.1
2.1.2
2.1.3
2.1.4
2.1.5
2.2
2.3
2.4
2.5
2.6
2.7
2.8
2.8.1
2.8.2
2.9
2.9.1
2.9.2
2.10
2.10.1
2.10.2
2.10.3
2.10.4
2.10.5
2.10.6
2.10.7
2.10.8
2.11
2.11.1
2.11.2
2.11.3
2.12
2.12.1
2.12.2
2.12.3
2.12.3.1
2.12.3.2
2.12.3.3
2.12.4
2.12.4.1
2.12.4.2
2.12.5
Electronic Band Structure and Polarization Effects 131
Introduction 131
Band Structure Calculations 132
Plane Wave Expansion Method 134
Orthogonalized Plane Wave (OPW) Method 134
Pseudopotential Method 135
Augmented Plane Wave (APW) Method 135
Other Methods and a Review Pertinent to GaN 136
General Strain Considerations 154
Effect of Strain on the Band Structure of GaN 159
kp Theory and the Quasi-Cubic Model 160
Quasi-Cubic Approximation 167
Temperature Dependence of Wurtzite GaN
Bandgap 169
Sphalerite (Zinc blende) GaN 172
AlN 176
Wurtzite AlN 177
Zinc Blende AlN 183
InN 184
Wurtzitic InN 185
Zinc Blende InN 200
Band Parameters for Dilute Nitrides 202
GaAsN 205
InAsN 208
InPN 209
InSbN 209
GaPN 210
GaInAsN 210
GaInPN 212
GaAsSbN 212
Confined States 212
Conduction Band 216
Valence Band 224
Exciton Binding Energy in Quantum Wells 227
Polarization Effects 230
Piezoelectric Polarization 236
Spontaneous Polarization 241
Nonlinearity of Polarization 245
Origin of the Nonlinearity 250
Nonlinearities in Spontaneous Polarization 253
Nonlinearities in Piezoelectric Polarization 256
Polarization in Heterostructures 264
Ga-Polarity Single AlGaN/GaN Interface 272
Ga-Polarity Single AlxIn1xN/GaN Interface 276
Polarization in Quantum Wells 278
Contents
2.12.5.1
2.12.5.2
2.12.6
2.12.7
Nonlinear Polarization in Quantum Wells 280
InGaN/GaN Quantum Wells 286
Effect of Dislocations on Piezoelectric Polarization
Thermal Mismatch Induced Strain 290
References 299
3
Growth and Growth Methods for Nitride Semiconductors 323
Introduction 323
Substrates for Nitride Epitaxy 324
Conventional Substrates 326
Compliant Substrates 327
van der Waals Substrates 328
A Primer on Conventional Substrates and their Preparation
for Growth 329
GaAs 329
A Primer on GaAs 330
Surface Preparation of GaAs for Epitaxy 331
Si 332
A Primer on Si 332
Surface Preparation of Si for Epitaxy 333
SiC 334
A Primer on SiC 334
Surface Preparation of SiC for Epitaxy 338
Sapphire 342
A Primer on Sapphire 343
Surface Preparation of Sapphire for Epitaxy 346
ZnO 350
A Primer on ZnO 351
Substrate Preparation for Epitaxy 353
LiGaO2 and LiAlO2 355
LiGaO2 Substrates 355
LiAlO2 Substrates 358
AlN and GaN 359
Seedless Growth of GaN 362
Seedless Growth of GaN by High Nitrogen Pressure Solution
Growth (HNPSG) for Substrates 362
Seeded Growth of GaN by HNPSG Method for Substrates 363
Pertinent Surfaces of GaN 365
GaN Surface Preparation for Epitaxy 369
Other Substrates 371
GaN Epitaxial Relationship to Substrates 372
Epitaxial Relationship of GaN and AlN with Sapphire 372
Epitaxial Relationship of GaN and AlN with SiC 381
Epitaxial Relationship of GaN and AlN with Si 381
Epitaxial Relationship of GaN with ZnO 381
3.1
3.1.1
3.1.2
3.1.3
3.2
3.2.1
3.2.1.1
3.2.1.2
3.2.2
3.2.2.1
3.2.2.2
3.2.3
3.2.3.1
3.2.3.2
3.2.4
3.2.4.1
3.2.4.2
3.2.5
3.2.5.1
3.2.5.2
3.2.6
3.2.6.1
3.2.6.2
3.2.7
3.2.7.1
3.2.7.1.1
3.2.7.1.2
3.2.7.2
3.2.7.3
3.2.8
3.3
3.3.1
3.3.2
3.3.3
3.3.4
289
VII
VIII
Contents
3.3.5
3.4
3.4.1
3.4.1.1
3.4.1.2
3.4.1.3
3.4.1.3.1
3.4.1.3.2
3.4.1.3.3
3.4.1.3.4
3.4.1.3.5
3.4.1.3.6
3.4.2
3.4.2.1
3.4.2.2
3.4.2.3
3.4.2.4
3.4.2.5
3.4.2.6
3.4.2.7
3.4.2.8
3.4.2.9
3.4.2.9.1
3.4.2.9.2
3.4.2.9.3
3.4.2.9.4
3.5
3.5.1
3.5.1.1
3.5.1.2
3.5.2
3.5.3
3.5.3.1
3.5.3.2
3.5.3.2.1
3.5.3.2.2
3.5.3.2.3
3.5.3.2.4
3.5.3.2.5
3.5.3.3
3.5.3.4
Epitaxial Relationship of GaN with LiGaO2 and LiAlO2 and
Perovskites 382
Nitride Growth Techniques 384
Vapor Phase Epitaxy 385
Hydride Vapor Phase Epitaxy 385
Organometalic Vapor Phase Epitaxy 393
Modeling of OMVPE Growth 398
Thermal Decomposition of GaN as it Relates to Growth 398
Ga and N Precursor Adsorption and Decomposition 400
Ga and N2 Desorption from the Surface 401
Ga and N Surface Diffusion 403
Kinetic Model: Balance Between Adsorption and Desorption 405
Comparison of Model with Growth Conditions for Surface
Morphology 406
Molecular Beam Epitaxy 409
Adsorption 411
Desorption 412
Surface Diffusion 413
Incorporation 415
Decomposition 416
Reflection High-Energy Electron Diffraction 417
Plasma-Assisted MBE (PAMBE) or RF MBE, Primarily N
Source 428
Reactive Ion MBE 435
Principles of RMBE and PAMBE Growth 436
Growth by RMBE 437
Growth by PAMBE 446
Which Species of N is Desirable? 451
The Effect of III/V Ratio and Substrate Temperature on Surface
Morphology 455
The Art and Technology of Growth of Nitrides 462
Sources 467
HVPE Buffer Layers and Laser Liftoff 468
Benchmark HVPE Layers/Templates 472
Growth on GaAs Substrates 477
Growth on SiC: Nucleation Layers and GaN 479
Stacking and Interfacial Relationship 480
Nucleation Layers on SiC 482
High-Temperature AlN Nucleation Layers on SiC 486
Low-Temperature GaN Nucleation Layers on SiC 488
High-Temperature GaN Nucleation Layers on SiC 489
Alloy and Multiple Layer Nucleation Layers on SiC 491
Nucleation Layers on SiC by MBE 492
Substrate Misorientation and Domain Boundaries 495
Polarity of GaN on SiC 498
Contents
3.5.3.4.1
3.5.3.5
3.5.3.6
3.5.4
3.5.5
3.5.5.1
3.5.5.1.1
3.5.5.1.2
3.5.5.1.3
3.5.5.2
3.5.5.2.1
3.5.5.2.2
3.5.5.2.3
3.5.5.2.4
3.5.5.2.5
3.5.5.2.6
3.5.5.3
3.5.5.3.1
3.5.5.4
3.5.5.5
3.5.6
3.5.6.1
3.5.7
3.5.8
3.5.9
3.5.10
3.5.11
3.5.11.1
3.5.11.2
3.5.11.3
3.5.12
3.5.13
3.5.14
3.5.14.1
3.5.15
3.5.15.1
3.5.15.1.1
3.5.15.1.2
3.5.15.2
3.5.15.2.1
3.5.15.2.2
3.5.15.2.3
3.5.15.3
GaN Growth on SiC 499
Growth on Porous SiC 503
Zinc Blende Phase Growth 507
Growth on Si 507
Growth on Sapphire 512
OMVPE Low-Temperature Nucleation Buffer Layers 513
The Effect of V/III Ratio on Nucleation Buffer Layer 523
Effect of Epitaxial Growth Temperature 525
Effect of Process Pressure 525
Epitaxial Lateral Overgrowth 528
Selective Epitaxial Growth and Lateral Epitaxial Overgrowth
with HVPE 537
Lateral Epitaxial Overgrowth on Si 539
Pendeo-Epitaxy 540
Pendeo-Epitaxy on SiC Substrates 542
Pendeo-Epitaxy on Silicon Substrates 544
Point Defect Distribution in ELO Grown GaN 558
Nanoheteroepitaxy and Nano-ELO 564
SiN and TiN Nanonets 569
Selective Growth Using W Masks 583
Low-Temperature Buffer Interlayer 584
Polarity and Surface Structure of GaN Layers, Particularly on
Sapphire 586
MBE Buffer Layers 597
Growth on ZnO Substrates 598
Growth on LiGaO2 and LiAlO2 Substrates 603
Growth on GaN Templates 605
Growth on Spinel (MgAl2O4) 611
Growth on Non c-Plane Substrates 611
The a-Plane GaN Growth 613
Epitaxial Lateral Overgrowth of a-plane GaN 616
00) m-Plane GaN Growth 623
The (11
Growth of p-Type GaN 627
Growth of InN 629
Growth of AlN 638
Surface Reconstruction of AlN 642
Growth of Ternary and Quaternary Alloys 652
Growth of AlGaN 653
Growth of p-Type AlGaN 666
Ordering in AlGaN 668
Growth of InGaN 671
Doping of InGaN 678
Phase Separation in InGaN 679
Surface Reconstruction of InGaN 689
Growth of AlInN 695
IX
X
Contents
3.5.15.3.1
3.5.15.4
3.5.16
3.5.16.1
3.5.16.2
3.5.16.3
3.5.16.4
3.5.16.4.1
3.5.16.4.2
3.5.16.5
3.5.16.5.1
3.5.16.5.2
3.6
Miscibility Gap in InAlN 697
InGaAlN Quaternary Alloy 699
Growth of Quantum Dots 706
Quantum Dots by MBE 711
Quantum Dots by OMVPE 719
Quantum Dots by Other Techniques 723
Preparation and Properties of Nanostructures 725
Approaches for Synthesis 726
Vapor Phase Growth 726
Nanowires and Longitudinal Heterostructures 737
Coaxial Heterostructures 755
Nanotubes 756
Concluding Remarks 759
References 760
4
Extended and Point Defects, Doping, and Magnetism 817
Introduction 817
A Primer on Extended Defects 818
Dislocations 819
Misfit Dislocations 822
Threading Dislocations 822
Edge Dislocations 824
Screw Dislocations 828
Mixed Dislocations 836
Nanopipes or Hollow Pipes 840
Planar Defects: Domain Boundaries 844
Stacking Faults 851
Grain Boundaries 862
Electronic Structure of Extended Defects 863
Open Core Versus Close Core in Screw Dislocations 865
Edge and Mixed Dislocations 866
Simple Stacking Faults: Electrical Nature 884
TEM Analysis of High Nitrogen Pressure (HNP) Solution
Growth (HNPSG) and HVPE-Grown GaN 886
Extended Defect Characterization 887
Pyramidal Defects 894
V-Shaped Defects (Pits) in InGaN Multiple Quantum
Wells (MQWs) 905
Structural Defect Analysis by Chemical Etch Delineation 905
Structural Defect Observations with Surface Probes 910
Point Defects and Autodoping 917
Theoretical Studies of Point Defects in GaN 919
Hydrogen and Impurity Trapping at Extended Defects 924
Vacancies, Antisites, Interstitials, and Complexes 928
Vacancies 929
4.1
4.1.1
4.1.1.1
4.1.1.2
4.1.1.2.1
4.1.1.2.2
4.1.1.2.3
4.1.2
4.1.3
4.1.4
4.1.5
4.1.6
4.1.6.1
4.1.6.2
4.1.6.3
4.2
4.2.1
4.2.2
4.2.3
4.2.4
4.2.5
4.3
4.3.1
4.3.1.1
4.3.1.2
4.3.1.2.1
Contents
4.3.1.2.2
4.3.2
4.3.2.1
4.3.2.2
4.3.2.3
4.3.2.4
4.4
4.4.1
4.4.2
4.4.3
4.4.4
4.5
4.6
4.6.1
4.6.2
4.7
4.8
4.9
4.9.1.1
4.9.1.2
4.9.1.3
4.9.2
4.9.2.1
4.9.2.2
4.9.2.3
4.9.2.4
4.9.3
4.9.3.1
4.9.3.1.1
4.9.3.1.2
4.9.3.1.3
4.9.3.2
4.9.3.3
4.9.3.4
4.9.3.5
4.9.3.6
4.9.3.7
4.9.3.8
4.9.4
4.9.4.1
4.9.4.2
4.9.5
4.9.6
Interstitials and Antisite Defects 934
Complexes 935
Shallow Donor – Gallium Vacancy Complexes 936
Shallow Acceptor – Nitrogen Vacancy Complexes 936
Hydrogen-Related Complexes 937
Other Complexes 938
Defect Analysis by Deep-Level Transient Spectroscopy 938
Basics of DLTS 939
Applications of DLTS to GaN 948
Dispersion in DLTS of GaN 970
Applications of DLTS to AlGaN, In-Doped AlGaN, and InAlN 977
Minority Carrier Lifetime 977
Positron Annihilation 982
Vacancy Defects and Doping in Epitaxial GaN 983
Growth Kinetics and Thermal Behavior of Vacancy Defects in GaN 993
Fourier Transform Infrared (FTIR), Electron Paramagnetic Resonance,
and Optical Detection of Magnetic Resonance 998
Role of Hydrogen 1004
Intentional Doping 1006
Shallow Donors 1007
Substitutional Acceptors 1007
Isoelectronic Impurities 1009
n-Type Doping with Silicon, Germanium, Selenium, and
Oxygen 1010
Si Doping 1010
Ge Doping 1011
Se Doping 1012
p-Type Doping 1013
p-Type Doping and Codoping with Donors and Acceptors 1014
Magnesium Doping 1014
Codoping for Improving p-Type Conductivity 1019
Use of Superlattices for Improving p-Type Conductivity 1032
Role of Hydrogen and Defects in Mg-Doped GaN 1034
Beryllium Doping 1038
Mercury Doping 1040
Carbon Doping 1040
Zinc Doping 1042
Calcium Doping 1042
Cadmium Doping 1043
Other Acceptors in GaN 1043
Doping with Isoelectronic Impurities 1044
Arsenic Doping 1044
Phosphorus Doping 1045
Doping with Rare Earths 1045
Doping with Transition Metals and Rare Earths 1046
XI
XII
Contents
4.9.6.1
4.9.6.2
4.9.6.3
4.9.6.4
4.9.6.5
4.9.6.5.1
4.9.6.5.2
4.9.6.5.3
4.9.6.6
4.9.6.6.1
4.9.6.7
4.9.6.7.1
4.9.6.7.2
4.9.7
4.9.7.1
4.9.7.2
4.9.7.3
4.9.8
4.9.9
4.9.10
4.9.11
4.9.12
4.10
4.11
Manganese Doping for Electronic Properties 1046
Other TM Doping for Electronic Properties 1060
General Remarks About Dilute Magnetic Semiconductors 1063
General Remarks About Spintronics 1075
Theoretical Aspects of Dilute Magnetic Semiconductor 1082
Carrier – Single Magnetic Ion Interaction 1084
Interaction Between Magnetic Ions 1086
Zener, Mean Field, RKKY, and Ab Initio Treatments 1089
A Primer to Magnetotransport Measurements 1103
Faraday Rotation, Kerr Effect, and Magnetic Circular
Dichroism (MCD) 1104
II–VI and GaAs-Based Dilute Magnetic Semiconductors 1123
II–VI-Based Dilute Magnetic Semiconductors 1124
III–V-Based DMS: (GaMn)As 1133
Experimental Results of TM-Doped GaN 1141
Magnetotransport Properties TM-Doped GaN 1141
Magnetic Properties of Mn-Doped GaN 1143
Magneto-Optical Measurements in TM-Doped GaN 1146
Magnetic, Structural, Optical, and Electrical Properties of
Cr-Doped GaN 1156
Other TM and Rare Earth Doped GaN:(Co, Fe, V, Gd,
and so on) 1163
TM-Doped Nanostructures 1166
Applications of Ferromagnetism and Representative Devices 1167
Summarizing Comments on Ferromagnetism 1186
Ion Implantation and Diffusion for Doping 1188
Summary 1190
References 1191
Index
1231
Appendix
1257
XIII
Preface
This three-volume handbook represents the only comprehensive treatise on semiconductor and device fundamentals and technology under the overall umbrella of
wide bandgap nitride semiconductors with comparison to GaAs when applicable.
As it stands, the book is a reference book, a handbook, and a graduate text book all in
one and would be beneficial to second-year graduate students majoring in semiconductor materials and devices, graduate research assistants conducting research
in wide bandgap semiconductors, researchers and technologists, faculty members,
program monitors, and managers. The philosophy of this endeavor is to present the
material as much clearly and comprehensively as possible, so that there is very little
need to refer to other sources to get a grasp of the subjects covered. Extreme effort
has been expended to ensure that concepts and problems are treated starting with
their fundamental basis so that the reader is not left hanging in thin air. Even though
the treatise deals with GaN and related materials, the concepts and methods
discussed are applicable to any semiconductor.
The philosophy behind Nitride Semiconductors and Devices was to provide an
adequate treatment of nitride semiconductors and devices as of 1997 to be quickly
followed by a more complete treatment. As such, Nitride Semiconductors and Devices
did not provide much of the background material for the reader and left many issues
unanswered in part because they were not yet clear to the research community at that
time. Since then, tremendous progress both in the science and engineering of
nitrides and devices based on them has been made. While LEDs and lasers were
progressing well even during the period when Nitride Semiconductors and Devices was
written, tremendous progress has been made in FETs and detectors in addition to
LEDs and lasers since then. LEDs went from display devices to illuminants for
lighting of all kinds. Lasers are being implemented in the third generation of DVDs.
The power amplifiers are producing several hundred watts of RF power per chip and
the detectors and detector arrays operative in the solar-blind region of the spectrum
have shown detectivities rivaling photomultiplier tubes. The bandgap of InN has
been clarified which now stands near 0.7 eV. Nanostructures, which did not exist
Handbook of Nitride Semiconductors and Devices. Vol. 1. Hadis Morkoç
Copyright # 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 978-3-527-40837-5
XIV
Preface
during the period covered by Nitride Semiconductors and Devices, have since become
available. The technological breakthroughs such as epitaxial lateral overgrowth, laser
liftoff, and freestanding GaN were either not fully developed or did not exist, neither
did the highly improved quantum structures and devices based on them. In the
interim period since then, the surfaces of nitrides and substrate materials, point
defects and doping, magnetic ion doping, processing, current conduction mechanisms, and optical processes in bulk and quantum structures have been more clearly
understood and many misconceptions (particularly, those dealing with polarization)
identified, removed and/or elucidated. The handbook takes advantage of the fundamental and technological developments for a thorough treatment of all aspects of
nitride semiconductors. In addition, the fundamentals of materials physics and
device physics that are provided are applicable to other semiconductors, particularly,
wurtzitic direct bandgap semiconductors.
The handbook presents a thorough treatment of the science, fundamentals, and
technology of nitride semiconductors and devices in such a width and depth that the
reader would seldom need to engage in time-consuming exploration of the literature
to fill in gaps. Last but not the least, the handbook contains seamless treatments of
fundamentals needed or relied on throughout the entire book. The following is a
succinct odyssey through the content of the three-volume handbook.
Volume 1, Chapter 1 discusses the properties of nitride-based semiconductors
with plenty of tables for reference. Volume 1, Chapter 2 treats the band structure of
III–V nitrides, theories applied to determining the band structure, features of each
theory with a succinct discussion of each, band structure of dilute III–V semiconductors doped with N, strain and stress, deformation potentials, and in-depth
discussion of piezo and spontaneous polarization with illustrative and instructive
artwork. Volume 1, Chapter 3 encompasses substrates that have been and are used
for growth of nitride semiconductors, mainly, structural and mechanical (thermal)
properties of those substrates, surface structure of planes used for growth, and
substrate preparation for growth. Orientation and properties of GaN grown on those
substrates are discussed along with commonly used surface orientations of GaN.
The discussion is laced with highly illustrative and illuminating images showing
orientations of GaN resulting through growth on c-plane, a-plane, m-plane, and
r-plane substrates whichever applicable and the properties of resulting layers provided. The treatment segues into the discussion of various growth methods used for
nitrides taking into account the fundamentals of growth including the applicable
surface-oriented processes, kinetics, and so on, involved. A good deal of growth
details for both OMVPE and MBE, particularly, the latter including the fundamentals of in situ process monitoring instrumentation such as RHEED, and dynamics of
growth processes occurring at the surface of the growing layer are given. Of
paramount interest is the epitaxial lateral overgrowth (ELO) for defect reduction.
In addition to standard single multistep ELO, highly attractive nanonetwork meshes
used for ELO are also discussed. Specifics in terms of growth of binary, ternary, and
quaternaries of nitride semiconductors are discussed. Finally, the methods used to
grow nanoscale structures are treated in sufficient detail.
Preface
Volume 1, Chapter 4 focuses on defects, both extended and point, doping for
conductivity modulation and also for rendering the semiconductor potentially
ferromagnetic segueing into electrical, optical, and magnetic properties resulting
in films, with sufficient background physics provided to grasp the material. A clear
discussion of extended defects, including line defects, are discussed with a plethora
of illustrative schematics and TEM images for an easy comprehension by anyone
with solid-state physics background. An in-depth and comprehensive treatment of
the electrical nature of extended defects is provided for a full understanding of the
scope and effect of extended defects in nitride semiconductors, the basics of which
can be applicable to other hexagonal materials. The point defects such as vacancies,
antisites, and complexes are then discussed along with a discussion of the effect
of H. This gives way to the methods used to analyze point defects such as deep level
transient spectroscopy, carrier lifetime as pertained to defects, positron annihilation,
Fourier transform IR, electron paramagnetic resonance, and optical detection of
magnetic resonance and their application to nitride semiconductors. This is followed by an extensive discussion of n-type and p-type doping in GaN and related
materials and developments chronicled when applicable. An in-depth treatment of
triumphs and challenges along with codoping and other methods employed for
achieving enhanced doping and the applicable theory has been provided. In addition, localization effects caused by heavy p-type doping are discussed. This gives way
to doping of, mainly, GaN with transition elements with a good deal of optical
properties encompassing internal transition energies related to ion and perturbations caused by crystal field in wurtzitic symmetry. To get the reader conditioned for
ferromagnetism, a sufficient discussion of magnetism, ferromagnetism, and measurement techniques (magnetic, magneto transport, magneto optics with underlying theory) applied to discern such properties are given. This is followed by an indepth and often critical discussion of magnetic ion and rare earth-doped GaN, as
well as of spintronics, often accompanied by examples for materials properties and
devices from well-established ferromagnetic semiconductors such as Mn-doped
GaN and Cr-doped ZnTe.
Volume 2, Chapter 1 treats metal semiconductor structures and fabrication
methods used for nitride-based devices. Following a comprehensive discussion of
current conduction mechanisms in metal semiconductor contacts, which are applicable to any metal semiconductor system, specific applications to metal-GaN contacts
are treated along with the theoretical analysis. This gives way to a discussion of
ohmic contacts, their technology, and their characterization. In particular, an ample
discussion of the determination of ohmic contact resistivity is provided. Then
etching methods, both dry (plasma) and wet, photochemical, process damage, and
implant isolation are discussed. Volume 2, Chapter 2 deals with determination of
impurity and carrier concentrations and mobility mainly by temperature-dependent
electrical measurements, such as Hall measurements. Charge balance equations,
capacitance voltage measurements, and their intricacies are treated and used for
nitride semiconductors, as well as a good deal of discussion of often brushed off
degeneracy factors.
XV
XVI
Preface
Volume 2, Chapter 3 is perhaps one of the most comprehensive discussions of
carrier transport in semiconductors with applications to GaN. After a discussion of
scattering processes in physical and associated mathematical terms, the methods
discussed are applied to GaN and other related binaries and ternaries with useful
ranges of doping levels, compositions, and lattice temperatures. Comparisons with
other semiconductors are also provided when applicable. This discussion segues
into the discussion of carrier transport at high electric fields applicable to field-effect
transistors, avalanche and pin (biased) photodiodes. This is followed by the measurement of mobility and associated details, which are often neglected in text and
reference books. The discussion then flows into magnetotransport beyond that
present in Hall measurements. Low, medium, and high magnetic field cases, albeit
only normal to the surface of the epitaxial layers, determination of which is affected
by the value of the mobility and various cases are treated. The treatise also includes
cases where the relaxation time, if applicable, is energy-dependent and somewhat
energy-independent. The discussion of the magnetotransport paves the way for a
fundamental and reasonably extensive discussion of the Hall factor for each of the
scattering mechanisms that often are not treated properly or are treated only in a
cursory manner in many texts leading to confusion. After providing the necessary
fundamental knowledge, the transport properties of GaN are discussed. This gives
way to the discussion of various scattering mechanisms in two-dimensional systems
that are relied on in high-performance FETs. For determining the mobility of each
layer (in the case of multiple layer conduction), quantitative mobility spectrum
analysis including both fundamentals and experimental data obtained in nitride
semiconductors is discussed. The quantum Hall effect and fractional quantum Hall
effect in general and as germane to GaN are discussed along with parameters such
as the effective mass determined from such measurements.
Volume 2, Chapter 4 is devoted to p–n junctions, beginning with the discussion of
band lineups, particularly, in the binary pairs from the point of view of theoretically
and experimentally measured values. Current conduction mechanisms, such as
diffusion, generation-recombination, surface recombination, Poole–Frenkel, and
hopping conductivity are discussed with sufficient detail. Avalanche multiplication,
pertinent to the high-field region of FETs, and avalanche photodiodes, are discussedfollowed by discussions of the various homojunction and heterojunction diodes
based on nitrides.
Volume 2, Chapter 5 is perhaps the most comprehensive discussion of optical
processes that can occur in a direct bandgap semiconductor and, in particular, in
nitride-based semiconductors and heterostructures inclusive of 3, 2-, and 0-dimensional structures as well as optical nonlinearities. Following a treatment of photoluminescence basics, the discussion is opened up to the treatment of excitons,
exciton polaritons, selection rules, and magneto-optical measurements followed
by extrinsic transitions because of dopants/impurities and/or defects with energies
ranging from the yellow and to the blue wavelength of the visible spectrum. Optical
transitions in rare earth-doped GaN, optical properties of alloys, and quantum wells
are then discussed with a good deal of depth, including localization effects and their
possible sources particularly media containing InN. The discussion then leads to the
Preface
treatment of optical properties of quantum dots, intersubband transitions in GaNbased heterostructures, and, finally, the nonlinear optical properties in terms of
second and third harmonic generation with illuminating graphics.
Volume 3, Chapter 1 is devoted, in part, to the fundamentals of light emitting
diodes, the perception of vision and color by human eye, methodologies used in
conjunction with the chromaticity diagram and associated international standards in
terms of color temperatures and color rendering index. Specific performances of
various types of LEDs including UV varieties, current spreading or the lack of related
specifics, analysis of the origin of transitions, and any effect of localization are
discussed. A good deal of white light and lighting-related standards along with
approaches employed by LED manufacturers to achieve white light for lighting
applications is provided. The pertinent photon conversion schemes with sufficient
specificity are also provided. Finally, the organic LEDs, as potential competitors for
some applications of GaN-based LEDs are discussed in terms of fundamental
processes that are in play and various approaches that are being explored for
increased efficiency and operational lifetime.
Volume 3, Chapter 2 focuses on lasers along with sufficient theory behind laser
operation given. Following the primer to lasers along with an ample treatment based
on Einsteins A and B coefficients and lasing condition, an analytical treatment of
waveguiding followed by specifics for the GaN system and numerical simulations
for determining the field distribution, loss, and gain cavity modes pertaining to
semiconductor lasers are given. An ample fundamental treatment of spontaneous
emission, stimulated emission, and absorptions and their interrelations in terms of
Einsteins coefficients and occupation probabilities are given. This treatment segues
into the extension of the gain discussion to a more realistic semiconductor with a
complex valence band such as that of GaN. The results from numerical simulations
of gain in GaN quantum wells are discussed, as well as various pathways for lasing
such as electron-hole plasma and exciton-based pathways. Localization, which is very
pervasive in semiconductors that are yet to be fully perfected, is discussed in the light
of laser operation. Turning to experimental measurements, the method for gain
measurement, use of various laser properties such as the delay on the onset of lasing
with respect to the electrical pulse, dependence of laser threshold on cavity length to
extract important parameters such as efficiency are discussed. The aforementioned
discussions culminate in the treatment of performance of GaN-based lasers in the
violet down to the UV region of the optical spectrum and applications of GaN-based
lasers to DVDs along with a discussion of pertinent issues related to the density of
storage.
Volume 3, Chapter 3 treats field effect transistor fundamentals that are applicable
to any semiconductor materials with points specific to GaN. The discussion primarily focuses on 2DEG channels formed at heterointerfaces and their use for FETs,
including polarization effects. A succinct analytical model is provided for calculating
the carrier densities at the interfaces for various scenarios and current voltage
characteristics of FETs with several examples. Experimental performance of GaNbased FETs and amplifiers is then discussed followed by an in-depth analysis of
anomalies in the current voltage characteristics owing to bulk and barrier states,
XVII
XVIII
Preface
including experimental methods and probes used for cataloging these anomalies.
This is followed by the employment of field spreading gate plates and associated
performance improvements. This segues into the discussion of noise both at the
low-frequency end and high-frequency end with sufficient physics and practical
approaches employed. The combined treatment of various low-frequency noise
contributions as well as those at high frequencies along with their physical origin
makes this treatment unique and provides an opportunity for those who are not
specialists in noise to actually grasp the fundamentals and implications of low- and
high-frequency noise. Discussion of high-power FETs would not be complete without a good discussion of heat dissipation and its physical pathways, which is made
available. Unique to GaN is the awareness of the shortfall in the measured electron
velocity as compared to the Monte Carlo simulation. Hot phonon effects responsible
for this shortfall are uniquely discussed with sufficient theory and experimental
data. A section devoted to reliability with specifics to GaN based high power HFETs
is also provided. Finally, although GaN-based bipolar transistors are not all that
attractive at this time, for completeness and the benefit of graduate students and
others who are interested in such devices, the theory, mainly analytical, of the
operation of heterojunction bipolar transistors is discussed along with available
GaN based HBT data.
Volume 3, Chapter 4 discusses optical detectors with special orientation toward
UV and solar-blind detectors. Following a discussion of the fundamentals of photoconductive and photovoltaic detectors in terms of their photo response properties, a
detailed discussion of the current voltage characteristic of the same, including all the
possible current conduction mechanisms, is provided. Because noise and detectors
are synonymous with each other, sources of the noise are discussed, followed by a
discussion of quantum efficiency in photoconductors and p–n junction detectors.
This is then followed by the discussion of vital characteristics such as responsivity
and detectivity with an all too important treatment of the cases where the detectivity
is limited by thermal noise, shot current noise, generation-recombination current
noise, and background radiation limited noise (this is practically nonexistent in the
solar-blind region except the man-made noise sources). A unique treatment of
particulars associated with the detection in the UV and solar-blind region and
requirements that must be satisfied by UV and solar-blind detectors, particularly,
for the latter, is then provided. This leads the discussion to various UV detectors
based on the GaN system, including the Si- and SiC-based ones for comparison.
Among the nitride-based photodetectors, photoconductive variety as well as the
metal-semiconductor, Schottky barrier, and homo- and heterojunction photodetectors are discussed along with their noise performance. Nearly solar-blind and truly
solar-blind detectors including their design and performance are then discussed,
which paves the way for the discussion of avalanche photodiodes based on GaN.
Finally, the UV imagers using photodetectors arrays are treated.
It is fair to state that I owe so much to so many, including my family members,
friends, coworkers, colleagues, and those who contributed to the field of semiconductors in general and nitride semiconductors in particular, in my efforts to bring
this manuscript to the service of readers. To this end, I thank my wife, Amy, and son,
Preface
Erol, for at least their understanding why I was not really there for them fully during
the preparation of this manuscript, which took longer than most could ever realize.
Also, without the support of VCU, with our Dean R. J. Mattauch, Assistant Dean
Susan Younce, Department Chair A. Iyer, and my coworkers and students, it would
not have been possible to pursue this endeavor. Special recognitions also go to Dr N.
Izyumskaya for reading the entire manuscript for consistency in terms of figures,
references, and so on, which had to have taken perseverance beyond that many could
muster; Dr Ü. Özgür for being the bouncing board and proofing many parts of the
book, particularly chapters dealing with optical processes, lasers and magnetism; my
colleague P. Jena for reading and contributing to the band structure section; my
coworker Professor M. Reshchikov for his contributions to the point defects and
doping sections; Professor A. Baski for her expert assistance in obtaining microprobe images; Dr D. Huang for his many contributions to the quantum dots section;
Dr Y-T Moon for his assistance in current crowding; C. Liu for her assistance with
ferromagnetism; Prof. A. Teke for reading the chapter on detectors; Dr. R. Shimada
for her contributions to the surface emitting laser section; Dr. J.-S. Lee for his help in
updating the LED chapter; Dr Q. Wang for her help in generating the accurate ball
and stick diagrams in Volume 1, Chapter 1; Dr V. Litvinov for calculating the energy
levels in quantum wells; students Y. Fu, Q. Fan, X. Ni, and S. Chevtchenko for their
contributions to various sections of the book with proofing equations, redoing
calculations, and so on; and to J. Leach who took it upon himself to be the local
expert in the latest in semiconductor and organic LEDs and helped with the chapter
on LEDs and read the chapter on transport as well as proofread some of the other
chapters and create the figures; Ms G. Esposito for reading a large portion of the text
for English. Undergraduate students K. Ngandu, D. Lewis, B. D. Edmonds, and M.
Mikkelson helped in reading various parts of the manuscript as well as helping with
the artwork. Unbeknown to them, many graduate students who took classes
from me helped in many immeasurable ways. Special thanks go to Professors
R. M. Feenstra, A. Matulionis, A. Blumenau, P. Ruterana, G. P. Dimitrakopulos,
P. Handel, K. T. Tsen, T. Yao, P. I. Cohen, S. Porowski, B. Monemar, B. Gil, P. Le
Febvre, S. Chichibu, F. Tuomisto, C. Van de Walle, M. Schubert, F. Schubert,
H. Temkin, S. Nikishin, L. Chernyak, J. Edgar, T. Myers, K. S. A. Butcher,
O. Ambacher, V. Fiorentini, A. di Carlo, F. Bernardini, V. Fiorentini,
M. Stutzmann, F. Pollak, C. Nguyen, S. Bedair, N. El-Masry, S. Fritsch, M. Grundman,
J. Neugebauer, M. S. Shur, J. Bowers, J. C. Campbell, M. Razhegi, A. Nurmikko,
M. A. Khan, J. Speck, S. Denbaars, R. J. Trew, A. Christon, G. Bilbro, H. Ohno,
A. Hoffmann, B. Meyer, B. Wessels, N. Grandjean, and D. L. Rode; and Drs
Z. Liliental-Weber, P. Klein, S. Binari, D. Koleske, J. Freitas, D, Johnstone, D. C.
Look, Z.-Q. Fang, M. MacCartney, I. Grzegory, M. Reine, C. W. Litton, P. J. Schreiber,
W. Walukiewicz, M. Manfra, O. Mitrofanov, J. Jasinski, V. Litvinov, Jan-Martin
Wagner, K. Ando, H. Saito, C. Bundesmann, D. Florescu, H. O. Everitt, H. M. Ng,
I. Vurgaftman, J. R. Meyer, J. D. Albrecht, C. A. Tran, S.-H. Wei, G. Dalpian,
N. Onojima, A. Wickenden, B. Daudin, R. Korotkov, P. Parikh, D. Green, A. Hansen,
P. Gibart, F. Omnes, M. G. Graford, M. Krames, R. Butte, and M. G. Ganchenkova
for either reading sections of the book, providing unpublished data, or providing
XIX
XX
Preface
suggestions. Many more deserve a great deal of gratitude for willingly spending
considerable time and effort to provide me with digital copies of figures and highquality images, but the available space does not allow for individual recognition. They
are acknowledged in conjunction with the figures. In a broader sense, I benefited
greatly from the counsel and support of Professor T. A. Tombrello of Caltech.
I also would like to use this opportunity to recognize a few of the unsung heroes,
namely, Dr Paul Maruska and Professor Marc Ilegems who truly started the epitaxy
of nitrides with the hydride VPE technique independently, and Dr S. Yoshida and
Professor T. Matsuoka for their pioneering work in AlGaN and InGaN, respectively.
Richmond, VA January 2008
Hadis Morkoç
XXI
Color Tables
Figure 1.4 A stick-and-ball stacking model of
crystals with (a, both top and bottom) 2H wurtzitic and (b, both top and bottom) 3C zinc blende
polytypes. The bonds in an A-plane (1 1 2 0) are
indicated with heavier lines to accentuate the
stacking sequence. The figures on top depict the
three-dimensional view. The figures at the
bottom indicate the projections on the (0 0 0 1)
and (1 1 1) planes for wurtzitic and cubic phases,
respectively. Note the rotation in the zinc blende
case along the h1 1 1i direction.
(This figure also appears on page 6.)
Handbook of Nitride Semiconductors and Devices. Vol. 1. Hadis Morkoc
Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 978-3-527-40837-5
XXII
Color Tables
Figure 1.9 The bandgaps of nitrides, substrates commonly used
for nitrides, and other conventional semiconductors versus
their lattice constants.
(This figure also appears on page 12.)
Figure 1.23 An artists view of the scanning thermal microscope.
Patterned after D.I. Florescu and F.H. Pollak.
(This figure also appears on page 57.)
Color Tables
50
40
q D = 800 K
–1
–1
Molar specific heat, Cp (cal mol K )
q D = 500 K
30
20
Cp data, GaN
q D = 500 K
q D = 600 K
10
q D = 700 K
q D = 800 K
0
0
200
400
600
800
1000
Temperature (K)
Figure 1.26 Molar specific heat at constant
pressure, Cp (cal mol1 K1), of GaN versus
temperature. Open circles represent the
experimental data. The solid lines are calculation
based on the Debye model for Debye
temperatures, yD, of 500, 600, 700, and 800 K.
Unfortunately, it is difficult to discern a Debye
temperature that is effective over a wide
temperature range because a large concentration
of defects and impurities is present in GaN.
However, a value of 600 K estimated by Slack is
used commonly. The data are taken from
Refs [215,216], as compiled in Ref. [88].
(This figure also appears on page 61.)
XXIII
Color Tables
50
40
Specific heat, Cp (J mol–1 K–1)
XXIV
30
20
Specific heat
AlN (J mol–1 K–1)
800 K
850 K
900 K
950 K
1000 K
1050 K
1100 K
10
0
0
200
400
600
800
1000
Temperature (K)
Figure 1.29 Molar specific heat at constant
pressure, Cp (J mol1 K1, 1 cal ¼ 4.186 J), of AlN
versus temperature. Open circles represent the
experimental data. The solid lines are calculation
based on the Debye model for Debye
temperatures, yD, in the range of 800–1100 K
with 50 K increments. The data can be fit with
Debye expression for yD ¼ 1000 K, which
compares with 950 K reported by Slack et al. The
data are taken from Ref. [88].
(This figure also appears on page 68.)
Color Tables
Figure 1.41 Bandgap versus composition for quaternary
AlxInyGa1xyN (assumed InN bandgap ¼ 0.8 eV).
(This figure also appears on page 101.)
Figure 1.42 Bandgap versus composition for quaternary
AlxInyGa1xyN (assumed InN bandgap ¼ 1.9 eV).
(This figure also appears on page 101.)
XXV
Color Tables
0.02
Spontaneous polarization (C m–2)
XXVI
+0.333
InxAl1–xN
InxGa1–xN
0.00
+0.193
–0.02
InxGa1–xN
–0.04
GaN
+0.095
–0.06
InxAl1–xN
InN
AIxGa1–xN
+0.037
–0.08
Random alloy
CH-like
CP-like
AIN
AIN
–0.10
0
0.2
0.4
0.6
1
Molar fraction, x
Figure 2.37 Spontaneous polarization versus
the molar fraction in all three ternary nitride
alloys. Circles, squares, and triangles represent
random alloy, CH-/LZ-, and CP-like structures,
respectively. The dashed/dotted lines (blue) with
solid triangles are for the CP-like alloys, the
dashed lines (green) with solid squares are for
CH-like alloys, and solid lines (black) with filled
circles are for random alloys. The black dashed
lines represent the data calculated using
Vegards law. Numbers indicated in the figure are
for CP and CH-/LZ-like ordered alloy bowing
parameters in terms of C m2. Courtesy of F.
Bernaridini and V. Fiorentini.
(This figure also appears on page 249.)
Color Tables
25
AlN
Si
6HSiC
4HSiC
3CSiC
LiGaO2
Al2O3
MgO
GaAs
ZnO
MgAl2O4
LiAlO2
ScMgAlO4
NdGaO3
Curvature (1 m–1)
20
15
10
5
0.0
–5
0.0
20
40
60
Thickness (μm)
Figure 2.59 A compilation of the variation of thermal curvature, a
measure of strain, in epitaxial GaN layers grown on different
substrates with respect to layer thickness [492].
(This figure also appears on page 294.)
80
100
XXVII
XXVIII
Color Tables
Figure 2.59 (Continued )
Color Tables
5108
Stress on various substrates (Pa)
0
–5108
–1109
AlN
Si
6HSiC
4HSiC
3CSiC
LiGaO2
Al2O3
MgO
GaAs
ZnO
MgAl2O4
LiAlO2
ScMgAlO4
NdGaO3
–1.5109
–2109
–2.5109
–3109
0100
210–5
410–5
610–5
810–5
Thickness (m)
Figure 2.60 A compilation of residual thermal stresses in
epitaxial GaN layer on different substrates with respect to layer
thickness [492].
(This figure also appears on page 296.)
110–4
XXIX
XXX
Color Tables
Stress versus thickness of GaN/potential substrates
5.00E+08
0.00E+00
0
0.00002
0.00004
0.00006
0.00008
0.0001
AlN
Si
MgO
–5.00E+08
3C-SiC
6H-SiC
4H-SiC
Stress (Pa)
–1.00E+09
ZnO
Al2O3
LiGaO2
–1.50E+09
MgAl2O4
GaAs
NdGaO3**
ScAlMgO**
–2.00E+09
LiAlO2
LSAT
–2.50E+09
–3.00E+09
Thickness (m)
Figure 2.60 (Continued )
Color Tables
Figure 3.1 The perspective view of the GaAs crystal (a) along
[1 0 0] (1 · 1 · 1 unit), (b) [1 1 0] (2 · 2 · 2 units), and (c) [1 1 1]
(2 · 2 · 2 units) directions [13].
(This figure also appears on page 330.)
Figure 3.2 The perspective view along (a) the [0 0 1], (b) [0 1 1],
and (c) [1 1 1] directions of a Si cell.
(This figure also appears on page 333.)
Figure 3.3 Tetragonal bonding of a carbon atom with its four
nearest silicon neighbors. The bond lengths depicted with a and
C–Si (the nearest neighbor distance) are approximately 3.08 and
1.89 Å, respectively. The right side is the three-dimensional
structure of 2H-SiC structure.
(This figure also appears on page 334.)
XXXI
XXXII
Color Tables
Figure 3.9 (a) Top view of the oxide structure on
SiC ð0 0 0 1Þ. The Si2O3 silicate adlayer
consisting of a honeycomb structure with
SiOSi bonds. At the center of the hexagons,
one carbon atom of the topmost substrate
bilayer is visible [the dark shaded area indicates
the (1 1; 1) unit cell and light shaded the
pffiffiffi pffiffiffi
ð 3 3ÞR30 -unit cell]; (b) side view of the
oxide structure on the SiC (0 0 0 1) in ð0 1 1 0Þ SiC
projection. Linear SiOSi bonds connect the
silicate layer and the underlying SiC substrate.
Courtesy of N. Onojima (patterned after
Ref. [40]).
(This figure also appears on page 342.)
Figure 3.10 (a) Top view (projection on the Si-plane of the basal
plane of SiC) and (b) side view of SiC after an in situ Ga exposure
indicating of the lack of silicate adlayer. Courtesy of N. Onojima.
(This figure also appears on page 342.)
Color Tables
Figure 3.12 The unit cell of sapphire: (a) rhombohedral unit cell;
(b) hexagonal unit cell. Smaller spheres are for O and large ones
are for Al [13].
(This figure also appears on page 344.)
Figure 3.13 Perspective views in (2 · 2 · 1) unit cells: (a) along the
[0 0 0 1] direction in a rhombohedral unit cell; (b) along the
(0 0 0 1) direction in hexagonal unit cell [13].
(This figure also appears on page 344.)
XXXIII
XXXIV
Color Tables
Side view of
sapphire r-plane
{1 0 1 2}
Oxygen
(a)
Al
Al2O3 [1 0 1 1]
Color Tables
a
2
m
C
b
m
LiGaO2
Lower O
Li
Upper O
Ga
Figure 3.24 Example of the exact fit of GaN atoms over the LiGaO2
lattice if there is no distortion. Courtesy H. Paul Maruska.
(This figure also appears on page 356.)
3
Figure 3.20 (a) Sapphire r-plane stacking
sequence showing O atoms in larger clear circles
and Al atoms in smaller, filled circles. The salient
feature is that each Al layer has an O layer above
and below it. (b) The atomic arrangement on
three layers (the uppermost one is O,
immediately below is Al and third layer down is
another O layer) on the r-plane of sapphire. The
lines are there just guides to eye and do not
represent bonds.
(This figure also appears on page 352.)
XXXV
Color Tables
LGO: orthorhombic
a = 5.402 Å
b = 6.372 Å
c = 5.007 Å
c
b
a
(a)
LGO: orthorhombic
a = 5.402 Å
b = 6.372 Å
c = 5.007 Å
c
b
a
(b)
Projection on c-plane
∆b = –0.19 %
aG
aN
=3
.18
9Å
∆a = +1.1%
bLGO = 6.372 Å
XXXVI
∆a = +1.9%
∆b = –1.1%
(c)
aLGO = 5.402 Å
Figure 3.25 Structure of (a) orthorhombic LiGaO2 (LGO), (b)
GaN, and (c) a detailed view of the relative orientation of GaN with
respect to LGO. Courtesy of H. Paul Maruska.
(This figure also appears on page 357.)
Color Tables
Figure 3.43 A schematic representation of a vertical OMVPE
system employed at Virginia Commonwealth University along
with a picture of the deposition chamber (a); a photograph of the
reactor chamber of the same (b).
(This figure also appears on page 394.)
XXXVII
XXXVIII
Color Tables
Figure 3.82 Cartoons illustrating the laser liftoff
process sequence of 20 GaN membranes.
(a) Laser lift-off of the GaN film coated with
silicone elastomer and affixed onto a support
template; (b) removal of sapphire following laser
scanning; (c) deposition of an approximately
3 mm thick thermoplastic adhesive layer at
120 C; (d) peeling-off of the silicone elastomer.
In the last step the GaN film is fully removed by
dissolving the thermoplastic adhesive in
acetone. Courtesy of M. Stutzmann, Ref. [382].
(This figure also appears on page 471.)
Figure 3.128 SEM and CL wavelength image of a cross section of
HVPE ELO sample. Courtesy of J. Christen and A.G. Hoffmann,
Ref. [695].
(This figure also appears on page 560.)
Color Tables
Figure 3.129 SEM (a) and CL wavelength (b)
images of two different regions, coherently
grown above the openings and overgrown above
the SiO2 stripes. The growth in the windows
(between the SiO2 stripes) and the wing
(coalesced regions over the SiO2 stripes) regions
indicated in the schematic drawing (c) are clearly
visible in the CL wavelength image. Courtesy of
J. Christen and A.G. Hoffmann, Ref. [697].
(This figure also appears on page 561.)
XXXIX
XL
Color Tables
Figure 3.130 A m-Raman scans along c-axis of overgrowth GaN (in
blue) and coherently grown GaN (in red): (a) free carrier density
and (b) biaxial compressive stress. Courtesy of J. Christen and
A. G. Hoffmann, Ref. [697].
(This figure also appears on page 562.)
Color Tables
Figure 3.160 Schematic representation of m-, a-, and c-planes
of GaN.
(This figure also appears on page 613.)
XLI
XLII
Color Tables
Figure 3.165 Cartoon of epitaxial lateral
overgrowth (ELO) on a-plane GaN with growth
along the [0 0 0 1] direction, representing the Gapolar growth front and along the ½0 0 0 1
direction, representing the N-polar growth front:
(a) top view and (b) side view. The growth along
the Ga-polar front is about a factor of g ¼ 3 times
faster. Courtesy of VCU students Vishal Kasliwal
and Xianfeng Ni. (c) Left 30 mm · 30 mm AFM
image for sample B. Right 4 mm · 4 mm AFM
image near the window and N-polar wing
boundary of sample B, showing different surface
pit densities for the window and the wing.
Courtesy of VCU students Vishal Kasliwal and
Xianfeng Ni.
(This figure also appears on page 618.)
Color Tables
Figure 3.168 (a) AFM and (b) NSOM scans from a 40 mm · 40 mm
area of a-GaN ELO sample B [875].
(This figure also appears on page 620.)
XLIII
XLIV
Color Tables
Figure 3.249 (a) I–V characteristics of a core
multishell (CMS) nanowire device, the top view of
which is shown in the inset in the form of a field
emission scanning electron microscopy image.
Scale bar is 2 mm. (b) Optical microscopy images
collected from around theopaque p-contact ofcore
multishell nanowire LEDs with increasing In
concentration in the shell quantum well and in
forward bias, showing purple, blue, cyan, green,
and near yellow emission, respectively. (c)
Normalizedelectroluminescencespectraobtained
from five representative multicolor CMS nanowire
LEDs. Courtesy of C.M. Lieber.
(This figure also appears on page 747.)
Color Tables
Figure 3.249 (Continued )
XLV
Color Tables
(c)
1.0
0.8
Normalized intensity (au)
XLVI
0.6
0.4
0.2
0.0
300
400
500
Wavelength (nm)
Figure 3.249 (Continued )
600
700
Color Tables
Figure 4.8 Side view (projection onto the
ð1 1 2 0Þ plane) of a relaxed and neutral screw
dislocation: (a) full-core screw dislocation; (b)
Ga-filled screw dislocation. Core of a full-core
screw dislocation (discussed in greater detail in
Figure 4.10 and the associated text) showing the
double helix of Ga bonds. The supercell is
repeated twice in the [0 0 0 1] for clarity. Note that
the bonds at the core are heavily distorted.
Courtesy of Blumenau et al. (patterned after
Ref. [29]).
(This figure also appears on page 829.)
XLVII
XLVIII
Color Tables
Figure 4.36 Potential profile across the
dislocation in n-type GaN deduced from the
holographic phase map compared with
theoretical profile (a). Courtesy of D. Cherns.
False colored map showing phase shifts
produced by edge dislocations viewed end-on in
nominally undoped GaN. Contour lines
emphasize dipole-like phase shifts near
dislocation cores (b). Line profile through
indicated dislocation in (b) allows quantification
of nominally undoped GaN of electric fields,
yielding an estimated bound surface charge of
4 · 1011 e cm2 on either side of the defect (c).
Courtesy of M. McCartney.
(This figure also appears on page 874.)
Color Tables
2
1.5
Phase (rad)
1
0.5
0
–0.5
–1
–1.5
–2
–150
–100
(c)
–50
0
Distance (nm)
50
100
Figure 4.36 (Continued )
Figure 4.42 Contour plots of dislocation-induced electronic gap
states for three edge dislocation configurations, namely, (a) fourcore, (b) full-core, and (c) open-core structures. The plots are
obtained by calculating atomic geometries with DFT theory used
as input to image simulations. Large (small) balls correspond to
Ga (N) atoms [125].
(This figure also appears on page 883.)
150
XLIX
Color Tables
Mean implantation depth (µm)
0
0.09
0.26
0.50
0.79
1.13
HVPE GaN
0.50
10–14 µm
Mg-doped reference
0.49
S parameter
L
1 µm
36–39 µm
5 µm
49–68 µm
VGa
0.48
0.47
0.46
Defect free
0.45
0
5
10
15
20
Positron energy (keV)
Figure 4.97 Ga vacancies and 1, 5, 10–14, 36–39, 49–68 mm thick
HVPE GaN layers indicating increased S parameter, thus
increased Ga vacancy concentration toward the GaN/Al2O3
interface in each of the films. A Mg-doped p-type sample with very
low, if any, Ga vacancy is shown as the reference. Courtesy of
K. Saarinen.
(This figure also appears on page 986.)
25
Color Tables
Figure 4.118 Hole concentration versus ND
where the acceptor–donor–acceptor complex
model of Ref. [495] is shown with thin and bold
lines for NA ¼ 1 · 1019 cm3 (i) and
1 · 1020 cm3 (ii), respectively. The optimum
hole concentration where NA ¼ 2ND, as expected
from the complex formation, is shown with
diamonds. For comparative purpose, the simple
compensation model which assumes a single
donor and (unpaired) acceptor is depicted with
thin and thick lines for NA ¼ 1 · 1019 cm3 (iii)
and 1 · 1020 (iv), respectively. The random pair
model is also plotted with thin and thick lines for
NA ¼ 1 · 1019 cm3 (v) and 1 · 1020 cm3 (vi),
respectively. Discussions with Dr R. Korotkov are
acknowledged.
(This figure also appears on page 1024.)
LI
Color Tables
Figure 4.126 Proposed bond center and antibonding site incorporation
of H in GaN and its passivation of Mg during growth (the Mg atom is
directly below the H atom). In part courtesy of C. Van de Walle.
(This figure also appears on page 1037.)
GaN: Mn
Mnd projected PDOS (/eV cell)
LII
t+
e-
t+
e+
t-
GaP: Mn
e-
t+
e+
t+
ee+
t-
t+
tt+
GaSb: Mn
e-
e+
t–3
t-
t+
t-
GaAs: Mn
–4
t-
–2
t-
t+
–1
–F
Energy (eV)
1
2
3
Figure 4.150 In Mn d projected partial density of states for a single Mn in GaN,
GaP, GaAs, and GaSb, where the symmetry (t2 and e) as well as the spin (þ and )
have been indicated. The shaded region represents the t2þ states (after Ref. [699]).
(This figure also appears on page 1097.)
Color Tables
3d ion
d n-1
t-(d )
e-(d )
t+(d)
Mn on Ga site
t-CFR
Anion
dangling
bonds
V Ga3-
CFR
e-
t+CFR
t+(p)
e+(d)
VBM
CFR
e+
DBH
t-
t-(p)
DBH
t+
Figure 4.151 A schematic energy-level diagram for the levels
(central panel) generated from the interaction between the crystal
field and exchange-split levels on the 3d transition metal ion (left
panel) with the anion dangling bond levels (right panel), when the
TM d levels are energetically shallower than the dangling bond
levels (after Ref. [700]).
(This figure also appears on page 1098.)
LIII
LIV
Color Tables
Figure 4.155 Interband transitions in GaAs
selected because of its well-known band
structure and also its well-established and wellcharacterized properties in terms of magnetic
ion doped diluted magnetic semiconductors:
(arrows indicate emission but the concept is just
as applicable to transitions from the valence
band subband to the conduction band as in
absorption). (a) Schematic band structure of
GaAs near the G point, the center of the Brillouin
zone. As for the terms, Eg is the bandgap and DSO
the spin–orbit splitting; CB, conduction band;
HH, valence heavy hole; LH, light hole; SO,
spin–orbit split-off subbands; G6,7,8 are the
corresponding symmetries at the k ¼ 0 point
representing conduction, HH, LH, spin–orbit
(SO) bands, or, more precisely, the irreducible
representations of the tetrahedron group Td (see,
e.g., Ref. [721]). The terms s1/2 and p3/2 and p1/2
represent the conduction band (s-like) and
valence band (p-like) type of orbitals. (b)
Selection rules for interband transitions between
the Jz projection of the angular momentum along
z-direction, sublevels for circularly polarized light
sþ(right-hand circular polarization or positive
helicity that results from transitions between the
Jz ¼ 1/2 conduction band states and Jz ¼ 3/2
heavy-hole states, and Jz ¼ þ1/2 conduction
band and Jz ¼ 1/2 light-hole states), and s
(left-hand circular polarization or negative
helicity, which results from transitions between
the Jz ¼ þ1/2 conduction band states and
Jz ¼ þ3/2 heavy-hole states, and Jz ¼ 1/2
conduction band and Jz ¼ þ1/2 light-hole
states). The numbers by each transition indicate
the relative transition intensities, with respect to
the light-hole subband to the conduction band
(absorption or excitation of carriers to higher
bands), or the conduction band to the light-hole
subband transition (emission), which apply to
both excitation and radiative recombination
(depicted by the arrows). The circular
polarization (s polarization) for light energies
that would not excite the spin split-off band is
ideally 50%, which becomes 0 if the spin–orbit
split-off band is also excited. For completeness,
the transitions between the Jz ¼ 1/2
conduction band states and Jz ¼ 1/2 light-hole
states, and Jz ¼ þ1/2 conduction band states
and Jz ¼ þ1/2 light-hole states, which are linearly
polarized (p polarization), are also shown as
depicted by two-way arrows in the figure. The
"
transition probability or the emission intensity
normalized to the Jz ¼ 1/2 conduction state to
the Jz ¼1/2 state transitions (indicated with 1)
are also indicated in numbers for GaAs. The
circular polarization resulting from the
conduction band to the heavy-hole states are
three times more intense than the circular
polarization resulting from the conduction band
states to the light-hole valence band states. The
linearly polarized transitions are twice as intense
as the circular polarization involving light-hole
states. (c) Removal of the valence band heavyand light-hole degeneracy by, for example, strain
inducing either by lattice mismatch or by
confinement in a quantum well, which increases
the electron polarization to nearly 100%. Note
that heavy- and light-hole states are no longer
degenerate. Both the tensile (left) and
compressive (right) in-plane biaxial strain cases
are shown. The respective ratios of various
transitions (oscillator strengths) have been
assumed to be the same as in the relaxed case.
Note that spin is indifferent to strain, which
means that spin-up and spin-down states are
moved in the same direction by strain, but not to
magnetic field, as spin-up and spin-down states
in a given band are split and moved in opposite
directions as shown in (d). In part courtesy of W.
Chen, Linko1ping University. (d) Removal of the
valence band heavy- and light-hole degeneracy as
well as splitting the spin-up and spin-down states
by application of magnetic field. The total
splitting is enhanced due to sp-d interaction in
DMS materials in the form of xN0a<Sz> for the
conduction band states, xN0b<Sz> for the HH
and LH valence band states, and (1/3)xN0a<Sz>
for the spin–orbit split-off band. Here, N0, x, a, b,
<Sz> represent the number of cations per unit
volume, mole fraction of magnetic ions, the
product of Bohr magneton and the g factor for
the respective bands, and average spin for each
magnetic ion site, respectively. Note that
magnetic field/magnetization causes Zeeman
splitting, and direction of splitting either up or
down in energy is spin dependent. If the
semiconductor is ferromagnetic as is the case of
GaMnAs, one can either couple polarized light to
the symmetry/splitting allowed bands or cause
polarized light emission by tuning the
wavelength.
(This figure also appears on page 1112.)
Color Tables
LV
LVI
Color Tables
Figure 4.155 (Continued )
Color Tables
(a) x = 0.053
B ⊥ plane
2K
55 K
25 K
100 K
125 K
300 K
0.00
0.5
Rsheet(kΩ)
R Hall (kΩ )
0.03
-5
25 K
2K
0.3
-5
-0.03
100 K
125 K
55 K
0.4
300 K
0
5
B (T)
0
B (T )
5
0.08
1/χ Hall(au)
(RHall R
)
sheet s
(b)
0.06
0.04
0.02
0.00
0
100
200
300
T (K)
120
(c)
T c (K)
80
40
0
0.00
0.04
x
Figure 4.170 (a, top) Temperature dependence
of the Hall resistance RHall for a 200 nm thick
Ga0.947Mn0.053As sample for which direct
magnetization measurements have been
performed but not shown. The inset shows the
temperature dependence of the sheet resistance
Rsheet. (b, center) Temperature dependence of
the saturation magnetization [RHall/Rsheet]S
obtained using Arrott plots (solid circles) and
inverse susceptibility 1/wHall (open circles), both
0.08
deduced from the transport data shown in (a).
Solid lines depict [RHall/Rsheet]S and (c, bottom)
1/wHall (bottom, c) calculated using the mean
field Brillouin theory with S ¼ 5/2 for the Mn spin
and the Curie–Weiss law, respectively. The
dependence of magnetic transition temperature
TC on Mn composition as determined from the
transport data. Courtesy of Ohno and
Matsukura [777].
(This figure also appears on page 1140.)
LVII
LVIII
Color Tables
+1/2
CB
Jz = −1/2
σ+
σ+
σ−
π
σ−
π
σ+
σ−
+3/2 HH
−3/2
−1/2
+1/2 LH
+1/2 CR
−1/2
VB;HH
Figure 4.172 The GCB
, GVB;LH
7 conduction band and G9
7
VB;CR
(spin–orbit split-off band), and G7
(crystal field split off band)
valance bands in wurtzitic GaN at the G point along with
polarization (sþ right-hand and s left-hand circular
polarizations) of various transitions between the conduction and
valence band states in the presence of a magnetic field.
(This figure also appears on page 1147.)
0.15
Cr: 1%
5K
0.05
Cr: 3%
0.05
Magnetization (emu g–1)
Magnetization (emu g–1)
0.1
0.2 μ B /Cr
Cr: 5%
Cr: 0.5%
0
–0.05
–0.1
–0.15
–10 000
Cr : 5%
Cr :1%
0
H c =100 Oe
–0.05
–1000
0
1000
Magnetic Field (Oe)
–5000
0
5000
10 000
Magnetic Field (Oe)
Figure 4.177 Magnetization curves for Cr-doped
GaN in atomic concentrations of 0.5, 1, 3, and
5% up to a magnetic field normal to the surface
of 10 000 Oe (1 T). Note that the film containing
5% Cr does not show any saturation
magnetization in the range measured and
appears to be paramagnetic. The blow-up
version near the origin indicates hysteresis for
1% Cr sample and a coercive field of 100 Oe.
Courtesy of F. Hasegawa.
(This figure also appears on page 1157.)
Color Tables
0.04
Magnetization (emu g–1)
H = 200(Oe)
Ferromagnetic
Cr: 1%
0.02
Paramagnetic + ferromagnetic
Cr: 3 %
Paramagnetic
Cr : 5%
0
0
50
100
150
200
250
Temperature (K)
300
Figure 4.178 Temperature dependence of magnetization for 1, 3,
and 5% Cr-containing GaN. As indicated, the film with 1%
Cr is consistent with ferromagnetic behavior. The films with 3
and 5% Cr exhibit a combination of ferromagnetic and
paramagnetic behavior, and paramagnetic behavior, respectively.
Courtesy of F. Hasegawa.
(This figure also appears on page 1158.)
350
LIX
LX
Color Tables
VG < 0
R
(kΩ)
Hall
0.04
1
1.5 K
5K
10 K
0
20 K
0.02
RHall (kΩ)
–1
–0.5
0.0
B(T)
VG > 0
0.5
0.00
22.5 K
VG =
0V
–0.02
+125 V
–125 V
0V
–0.04
–1.0
–0.5
0.0
0.5
1.0
B (mT)
Figure 4.186 Hall resistance RHall of an insulated
gate (In,Mn)As field effect transistor at 22.5 K as
a function of the magnetic field for three different
gate voltages. RHall is proportional to the
magnetization of the (In,Mn)As channel. Upper
right inset shows the temperature dependence of
RHall. Left inset shows schematically the gate
voltage control of the hole concentration and the
corresponding change of the magnetic phase.
Courtesy of Ohno et al. [855].
(This figure also appears on page 1172.)
Color Tables
Figure 4.187 Injection of spin-polarized holes
into a light-emitting p–n diode using a
ferromagnetic semiconductor (Ga,Mn)As. (a)
Sample structure. Spin-polarized holes hþ travel
through the nonmagnetic GaAs and recombine
with spin-unpolarized electrons in the (In,Ga)As
quantum well. I represents the current, and sþ
represents circularly polarized light emitted from
the edge of the quantum well. (b) Dependence of
the polarization DP of the emitted light on the
magnetic field B at temperatures of 6, 31, 52 K,
the latter above the Curie value. The solid and
hollow symbols represent the degree of
polarization when the magnetic field is swept in
the positive and negative directions, respectively.
The magnetic field was applied parallel to the
surface along the easy axis of magnetization of
the (Ga,Mn)As. The temperature dependence of
the residual magnetization M in (Ga,Mn)As,
where the degree of polarization of the zero
magnetic field seen in the emitted light exhibits
the same temperature dependence as the
magnetization (not shown). Dependence on
temperature for B ¼ 0 of the change in the
relative remanent polarization, DP, (hollow
circles) and magnetic moment measured by a
SQUID magnetometer (solid circles). Courtesy
of Ohno and coworkers [770].
(This figure also appears on page 1174.)
LXI
Color Tables
∆P
1.00
T=6K
Magnetization
6
16 K
0.75
4
0.50
31 K
2
0.25
52 K
0.00
0
20
40
60
Temperature (K)
(c)
Figure 4.187 (Continued )
80
100
Magnetization (10-5emu)
Relative polarization,∆P (%)
LXII
Color Tables
High resistance state
Low resistance state
(b)
Figure 4.190 (a) Schematic representation of a
spin valve, a normal metal straddled by two
ferromagnetic metals. When the spins in
ferromagnetic metals on either end are aligned
parallel to each other, the system is in the lowresistance state top. When, for example, the spin
of the FM metal on the right is flipped by a
magnetic field, making the spins of the
ferromagnetic metals antiparallel, a high
resistance state is attained. (b) Schematic
representation of transport that is parallel to the
plane of a layered magnetic metal sandwich
structure for antialigned (upper figure – high
resistance) and aligned (lower figure – low
resistance) orientations.
(This figure also appears on page 1180.)
LXIII
j1
1
General Properties of Nitrides
Introduction
GaN as a representative of its binary cousins, InN and AlN, and their ternaries along
with the quaternary, is considered one of the most important semiconductors after Si.
It is no wonder that it finds ample applications in lighting and displays of all kinds,
lasers, detectors, and high-power amplifiers. These applications stem from the
excellent optical and electrical properties of nitride semiconductors. The parameters
are imperative in determining the utility and applicability of this class of materials to
devices, as will be made evident in this chapter and throughout the book.
In this chapter, the structural, mechanical, thermal, chemical, electrical, and
optical properties of GaN and its binary cousins as well as the substrates commonly
used for nitride epitaxy are treated in a general sense for quick reference. The detailed
properties associated with electrical and optical parameters and properties are
discussed in chapters dealing with transport and optical processes in GaN and
related alloys. Because GaN is used in the form of a thin film deposited on foreign
substrates, meaning templates other than GaN, a discussion of heteroepitaxial thin
films is of paramount importance. Consequently, the properties of nitride films
intricately depend on substrates, inclusive of the inherent properties such as lattice
constants and thermal expansion coefficients, and on the process-induced characteristics such as surface preparation and chemical and physical interactions at the
surface. These too are discussed in the book.
1.1
Crystal Structure of Nitrides
Group III nitrides can be of crystalline structures: the wurtzite (Wz), zinc blende (ZB),
and rock salt. Under ambient conditions, the thermodynamically stable structure is
wurtzite for bulk AlN, GaN, and InN. The zinc blende structure for GaN and InN has
been stabilized by epitaxial growth of thin films on {0 1 1} crystal planes of cubic
substrates such as Si [1], SiC [2], MgO [3], and GaAs [4]. In these cases, the intrinsic
tendency to form the Wz structure is overcome by the topological compatibility.
Handbook of Nitride Semiconductors and Devices. Vol. 1. Hadis Morkoc
Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 978-3-527-40837-5
j 1 General Properties of Nitrides
2
However, Wz structure could very likely be present at the extended defect sites. The
rock salt form is possible only under high pressures and, therefore, is laboratory form
of exercise.
Let us now discuss the space groups for the various forms of nitrides. The rock salt,
or NaCl, structure (with space group Fm3m in the Hermann–Mauguin notation and
O5h in the Schoenflies notation) can be induced in AlN, GaN, and InN under very high
pressures. The reason for this is that the reduction of the lattice dimensions causes
the interionic Coulomb interaction to favor the ionicity over the covalent nature. The
structural phase transition to rock salt structure was experimentally observed at the
following estimated pressure values: 22.9 GPa (17 GPa in other estimates) for AlN [5],
52.2 GPa for GaN [6], and 12.1 GPa for InN [7]. Rock salt III nitrides cannot be
produced by any epitaxial growth.
The space grouping for the zinc blende structure is F43m in the Hermann–
Mauguin notation and T 2d in the Schoenflies notation. The zinc blende structure has a
cubic unit cell, containing four group III elements and four nitrogen elements.
(Although the term zinc blende originated in compounds such as ZnS, which could
be in cubic or hexagonal phase, it has been used ubiquitously for compound
semiconductors with cubic symmetry. The correct term that should be used for the
cubic phase of GaN is actually sphalerite. However, to be consistent with the usage
throughout the literature, even at the expense of accuracy, the term zinc blende has
been used in this book). The position of the atoms within the unit cell is identical to
the diamond crystal structure. Both structures consist of two interpenetrating facecentered cubic sublattices, offset by one quarter of the distance along a body diagonal.
Each atom in the structure may be viewed as positioned at the center of a tetrahedron,
with its four nearest neighbors defining the four corners of the tetrahedron. The
stacking sequence for the (1 1 1) close-packed planes in this structure is AaBbCc.
Lowercase and uppercase letters stand for the two different kinds of constituents.
The wurtzite structure has a hexagonal unit cell and thus two lattice constants, c
and a. It contains six atoms of each type. The space grouping for the wurtzite
structure is P63mc in the Hermann–Mauguin notation and C46v in the Schoenflies
notation. The point group symmetry is 6 mm in the Hermann–Mauguin notation
and C6v in the Schoenflies notation. The Wz structure consists of two interpenetrating hexagonal close-packed (hcp) sublattices, each with one type of atom, offset along
the c-axis by 5/8 of the cell height (5c/8). The wurtzite and zinc blende structures are
somewhat similar and yet different. In both cases, each group III atom is coordinated
by four nitrogen atoms. Conversely, each nitrogen atom is coordinated by four group
III atoms. The main difference between these two structures lies in the stacking
sequence of closest packed diatomic planes. The Wz structure consists of alternating
biatomic close-packed (0 0 0 1) planes of Ga and N pairs, thus the stacking sequence of
the (0 0 0 1) plane is AaBbAa in the (0 0 0 1) direction.
Although the main interest is in Wz GaN as opposed to zinc blende GaN, a
description of stacking sequence of both GaN polytypes with the accepted Ramsdel
notation is warranted, so is the stacking order of SiC polytypes that are relevant to
GaN because they are used for substrates in GaN epitaxy. Therefore, a generic
description of stacking in Wz semiconductors is given below. A comprehensive
description of the tetrahedrally coordinated structures is imperative for a clear picture
1.1 Crystal Structure of Nitrides
j3
of nitride semiconductors, particularly the extended defects that are discussed in
detail in Chapter 4. The bonds describe a tetrahedron denoted by T, which has one
atom species at each of the three corners and the other atom species in its center [8,9].
The basal plane of this structure is defined by one face of the tetrahedron and the
bond perpendicular to this plane defines the c-axis. A rotation of 180 around the caxis produces a twin variant denoted by T0 as shown in Figure 1.1a (left).
(a)
Atom a
Atom b
[0001]
T
T'
⟨1120⟩
{1100}
In-plane bonds
Out-of-plane bonds
(b)
A
T3´
b
B
T1
a
A
T3´
b
B
Figure 1.1 Representation of the tetrahedrally coordinated
materials in the Ramsdel notation. (a) The two possible
0
0
tetrahedra. (b) The T1, T3, T1, T3, tetrahedral stacking composing
the 2H sequence. Courtesy of Pierre Ruterana [9].
T1
j 1 General Properties of Nitrides
4
The two variants (twins T and T0 ) are related to one another by mirror symmetry
about one of the {1 1 0 0} m-planes. A tetrahedron can occupy one of the three
possible positions in the basal plane. The representation of the tetrahedrally
coordinated materials in the Ramsdel notation is shown in Figure 1.1a for two
possible tetrahedra, one is the mirror image twin of the other with respect to the
(1 1 0 0) m-plane. The single bonds are on the (1 1 2 0) plane, called the a-plane. The
0
0
0
layers of the tetrahedra can then be denoted by T1, T2, T3, and by T1 ; T2 ; T3 for its
0
0
twin. An example of T1 ; T ; T1 ; T3 stacking order representing 2H ordering as in
wurtzitic GaN is shown in Figure 1.1b. The structure of nitride semiconductors and
most relevant polytypes of SiC can be completely described by a combinatorial
stacking of the aforementioned six tetrahedra layers. Naturally, not all the stacking
sequences must obey the following two rules to keep a corner sharing structure, as
such not all stacking orders are allowed:
(i) A tetrahedron T can be followed by another one of the same kind with the
0 0 0
following subscript: T1T2T3, and inversely for the twin variant: T3 T2 T1 .
(ii) A tetrahedron T1 must be followed by the twin variant of the preceding subscript:
0
0
T1 T3 , and inversely for its twin variant: T1 T2 .
In the Ramsdel notation, the stacking order for the wurtzite structure corresponding to various polytypes can be denoted as
.
.
.
.
0
0
0
T1 T3 or T2 T1 or T3 T2 for the 2H polytype, which is also applicable to Wz nitride
semiconductors;
0 0
T1T2T1 T3 for the 4H polytype;
0 0 0
T1T2T3T2 T1 T3 for the 6H polytype;
0 0 0
T1T2T3 or T3 T2 T1 for the 3C polytype.
The 3C, 4H, and 6H stacking sequences as well as 2H sequence on 6H sequence are
discussed in Chapter 3.
Recall that GaN crystallizes in the cubic structure (zinc blende or sphalerite, the
latter being the correct term and the former being the one used universally) or in the
more stable hexagonal structure (wurtzite). The anions (N3) form an hcp structure
in which the cations (Ga3þ) occupy half of the tetrahedral sites. The structure of a unit
cell of GaN projected along [0 0 0 1] is depicted schematically in Figure 1.2. The open
symbols represent g sites that are occupied by nitrogen atoms; the Ga atoms are in the
tetrahedral sites, b. These latter sites can either be at heights (3/8)c above (b1) or below
(b2) N site, depending on the crystal polarity.
A stick-and-ball representation of Ga-polarity and N-polarity Wz structure is
depicted in Figure 1.3. The Wz and zinc blende structures differ only in the bond
angle of the second nearest neighbor (Figure 1.4). As clearly shown, the stacking
order of the Wz along the [0 0 0 1] c-direction is AaBb, meaning a mirror image but no
in-plane rotation with the bond angles. In the zinc blende structure along the [1 1 1]
direction, there is a 60 rotation that causes a stacking order of AaBbCc. The point
with regard to rotation is illustrated in Figure 1.4b. The nomenclature for various
commonly used planes of hexagonal semiconductors in two- and three-dimensional
versions is presented in Figures 1.5 and 1.6. The Wz group III nitrides lack an
1.1 Crystal Structure of Nitrides
a
γ1
β1
γ1
γ1
*
γ1
β2
c
β1
γ2
β2
β1
γ1
u
*
β2
β1
β2
β1
γ1
β2
γ1
γ1
Figure 1.2 Schematic diagram showing the b1 and b2 tetrahedral
sites of GaN unit cell. Starting with the assumption that N
occupies the g sites, only one family of b sites can be
simultaneously occupied by Ga atoms. Courtesy of Pierre
Ruterana [9].
inversion plane perpendicular to the c-axis; thus, nitride surfaces have either a group
III element (Al, Ga, or In) polarity (referred to as Ga-polarity) with a designation of
(0 0 0 1) or (0 0 0 1)A plane or a N-polarity with a designation of (0 0 0 1) or (0 0 0 1)B
plane. We will use the former notations for each. The distinction between these two
directions is essential in nitrides because of their implications for the polarity of the
polarization charge. Three surfaces and directions are of special importance in
Ga-polarity
N-polarity
[0001]
[0001]
Ga
N
N
Ga
N
Ga
Ga
N
Ga
N
Ga
N
Ga
N
N
N
Ga
Ga
Ga
Ga
Ga
N
N
Ga
N
Ga
Ga
Ga
N
N
N
Ga
Ga
Ga
N
N
N
N
Ga
Ga
N
N
Ga
Ga
N
Ga
Ga
N
N
N
N
Ga
Ga
Ga
N
N
N
N
Ga
Figure 1.3 A stick-and-ball diagram of a hexagonal structure.
N
N
Ga
Ga
Ga
j5
j 1 General Properties of Nitrides
6
C
View normal to
[0001] and [111]
B
B
A
A
Ga
N
Wurtzitic
Zinc blende
View along
[0001] and [111]
(a)
Figure 1.4 A stick-and-ball stacking model of
crystals with (a, both top and bottom) 2H
wurtzitic and (b, both top and bottom) 3C
zinc blende polytypes. The bonds in an A-plane
(1 1 2 0) are indicated with heavier lines to
accentuate the stacking sequence. The figures on
top depict the three-dimensional view. The
(b)
figures at the bottom indicate the projections on
the (0 0 0 1) and (1 1 1) planes for wurtzitic and
cubic phases, respectively. Note the rotation in
the zinc blende case along the h1 1 1i direction.
(Please find a color version of this figure on the
color tables.)
nitrides, which are (0 0 0 1) c-, (1 1 2 0) a-, and (1 1 0 0) m-planes and the directions
associated with them, h0 0 0 1i, h1120i, and h1100i as shown in Figure 1.7. The
(0 0 0 1), or the basal plane, is the most commonly used surface for growth. The other
two are important in that they represent the primary directions employed in
reflection high-energy electron diffraction (RHEED) observations in molecular beam
epitaxial growth, apart from being perpendicular to one another. They also represent
the direction of stripes employed in the epitaxial lateral overgrowth (ELO), details of
which are discussed in Section 3.5.5.2.
The cohesive energy per bond in the wurtzite form is 2.88 eV (63.5 kcal mol1),
2.2 eV (48.5 kcal mol1), and 1.93 eV (42.5 kcal mol1) for AlN, GaN, and InN,
respectively [10]. The calculated energy difference DEW-ZB between wurtzite
and zinc blende lattice is small [11]: DEW-ZB ¼ 18.41 meV/atom for AlN,
DEW-ZB ¼ 11.44 meV/atom for InN, and DEW-ZB ¼ 9.88 meV/atom for GaN.
Wurtzite form is energetically preferable for all three nitrides compared to zinc
blende, although the energy difference is small.
The Wz structure can be represented by lattice parameters a in the basal plane and c
in the perpendicular direction, and the internal parameter u, as shown in Figure 1.8.
1.1 Crystal Structure of Nitrides
〈1100〉
〈1120〉
(m)
(a )
o
1010
30
(m
)
o
30
r
n
n
n
n
c
1100
0110
r
r
n
2110
1120
n
0111
s
(c)
1210
a
57
o
0114
d
1213
n
1012
r
2113
n
1101
s
1104
d
0001
c
1210
1213
n
o
.6
61
1123
n
c
(n)
r
(r)
r
n
n
1102
r
32.4
n
o
1100
m
a
a
a
1014
d
2113
n
0112
r
0110
1123
n
1011
s
(m)
2110
1120
1010
n
n
r
n
Common crystallographic
planes in sapphire
d
Plane Miller
name index spacing
a
m
c
r
n
s
(1120)
(1010)
(0001)
(1102)
(1123)
(1011)
2.379 Å
1.375 Å
2.165 Å
1.740 Å
1.147 Å
1.961 Å
Angles between
common planes
(0001) ^ (1102)
(0001) ^ (1123)
(0001) ^ (1011)
(0001) ^ (1121)
(0001) ^ (1120)
(0001) ^ (1010)
(1120) ^ (1010)
c^r
c^n
c^s
c^
c^a
c^m
a^m
57º 35'
61º 11'
72º 23'
79º 37'
90º 00'
90º 00'
30 00'
Figure 1.5 Labeling of planes in hexagonal symmetry ( for sapphire).
The u parameter is defined as the anion–cation bond length (also the nearest
neighbor distance) divided by the c lattice parameter. The c parameter depicts the
unit cell height. The wurtzite structure is a hexagonal close-packed lattice, comprising vertically oriented X–N units at the lattice sites. The basal plane lattice
parameter (the edge length of the basal plane hexagon) is universally depicted by a
and the axial lattice parameter, perpendicular to the basal plane, is universally
described by c. The interatomic distance in the basic unit is described by the
internal parameter u. In an ideal wurtzite structure represented by four touching
hard spheres,
pffiffiffiffiffiffiffiffi the values of the axial ratio and the internal parameter are
c=a ¼ 8=3 ¼ 1:633 and u ¼ 3/8 ¼ 0.375,
The crystallographic
!
pffiffiffi respectively.
pffiffiffi
!
vectors of wurtzite are a ¼ að1=2; 3=2; 0Þ, b ¼ að1=2; 3=2; 0Þ, and
!
the basis atoms are (0, 0, 0), (0, 0, uc),
c ¼ að0;
pffiffiffi0; c=aÞ. In Cartesian coordinates,
pffiffiffi
a(1=2; 3=6; c=2a), and a(1=2; 3=6; ½u þ 1=2c=a).
Table 1.1 tabulates the calculated structural parameters a, c/a, and e1 ¼ u uideal
for the III–V nitrides by three different groups [12–14]. In the case of Bernardini
et al. [12], they optimized the structure within both the generalized gradient
j7
j 1 General Properties of Nitrides
8
(tuvw) coordinate system
1010
v
2110
1120
0110
0111 s
1123 n
1012 r
2113 n
0114 d
1100
1101s
1104 d
1213 n
1213 n
1210 a
1210
0001 c
t
0112 r
1102 r
2113 n
1100 m
1014 d
1123 n
0110
1011 s
1120
2110
u
1010
Figure 1.6 A magnified view of labeling of planes
in hexagonal symmetry in the (tuvw) coordinate
system with w representing the unit vector in the
c-direction. The lines are simply to show
the symmetryonly. If the lines connecting m-points
among each other and a-points among each other
were to be interpreted as the projection of those
planes on the c-plane, the roles would be switched
in that the lines connecting the m-points would
actually represent the a-planes and lines
connecting the a-points would actually represent
the m-planes that are normal to the plane of the
page.
(1120) a-plane
[1120]
v
(1100) m-plane
[0110]
[1010]
[1210]
[1100]
[2110]
[2110]
t
[1010]
[1100]
[0110]
Ga
N
u
[1210]
m-planes
a-planes
[1120]
Figure 1.7 The orientations which are commonly used in nitrides,
namely the (1 1 2 0) and (1 1 0 0) planes and associated directions
are shown as projections on the (0 0 0 1) basal plane.
1.1 Crystal Structure of Nitrides
a
M
M
M
α
b1
c
N
b=u x c
M
M
β
b'
N
[0001]
N
b '2
M
N
b 3'
N
N
1
M
M
M
N
M
Figure 1.8 Schematic representation of a wurtzitic metal nitride
structure with lattice constants a in the basal plane and c in the
basal direction, u parameter, which is expressed as the bond
length or the nearest neighbor distance (b) divided by c (0.375
in ideal crystal), a and b (109.47 in ideal crystal) are the bond
0
0
0
angles, and b 1, b 2, and b 3, represent the three types of second
nearest neighbor distances.
approximation (GGA) and local density approximation (LDA). The experimental data
are from Leszczynski et al. [15].
In all Wz III nitrides, experimentally observed c/a ratios are smaller than ideal and
it has been postulated that not being so would lead to the zinc blende phase [16]. There
are two avenues that can lead to a deviation from ideal: changing the c/a ratio or
changing the u value. It should be pointed out that a strong correlation exists between
the c/a ratio and the u parameter so that when c/a decreases, the u parameter
increases in a manner to keep the four tetrahedral distances nearly constant through a
distortion of tetrahedral angles. For equal bond length to prevail, the following
relation must hold:
uð1=3Þða2 =c 2 Þ þ 1=4:
ð1:1Þ
Table 1.1 Structural parameters for GaN reported by Bechstedt,
Großner, and Furthm€
uller (BGF) [13] and by Wei and Zunger
(WZ) [14] using the local density approximation (LDA).
BGF
WZ
BFV (LDA)
BFV (GGA)
Experimental data
a (Å)
c/a
e1 (103c/a)
3.150
3.189
3.131
3.197
3.1890
1.6310
1.6259
1.6301
1.6297
1.6263
6.5
1.8
1.6
1.9
2.0
However, Bernardini, Fiorentini, and Vanderbilt [12] employed both the LDA and GGA methods.
Lattice constant a is given in Å and e1 in 103c/a.
j9
j 1 General Properties of Nitrides
10
The nearest neighbor bond length along the c-direction (expressed as b in
Figure 1.8) and off c-axis (expressed as b1 in Figure 1.8) can be calculated as
sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
2
1 2
1
a þ
u c2 :
b ¼ cu and b1 ¼
ð1:2Þ
3
2
In addition to the nearest neighbors, there are three types of second nearest
0
0
neighbors designated in Figure 1.8 as b 1 (one along the c-direction), b 2 (six of them),
0
and b 3 (three of them), which are given as [17]
sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
2
qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
4 2
1
0
0
0
2
a þ c2
u :
b 1 ¼ cð1 uÞ; b 2 ¼ a2 þ ðucÞ ; and b 3 ¼
3
2
ð1:3Þ
The bond angles, a and b, are given by [17]
"qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi #
1
a ¼ p=2 þ arccos
1 þ 3ðc=aÞ2 ð u þ 1=2Þ2
;
"qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
b ¼ 2 arcsin
2
4=3 þ 4ðc=aÞ ð u þ 1=2Þ
2
1
ð1:4Þ
#
:
Table 1.2 tabulates the calculated as well as experimentally observed structural
parameters discussed above, inclusive of the lattice parameters, the nearest and
second nearest neighbor distances, and the bond angles for three end binaries, GaN,
AlN, and InN. The distances are in terms of Å.
The lattice parameters are commonly measured at room temperature by X-ray
diffraction (XRD), which happens to be the most accurate one, using the Bragg law. In
ternary compounds, the technique is also used for determining the composition;
however, strain and relevant issues must be accounted for as the samples are in the
Table 1.2 Calculated (for ideal crystal) and experimentally
observed structural parameters for GaN, AlN, and InN [17].
GaN
u
a (Å)
c/a
b (Å)
b1 (Å)
0 Â
b 1 ðeÞ
0 Â
b 2 ðeÞ
0 Â
b 3 ðeÞ
a
b
AlN
InN
Ideal
Exp.
Ideal
Exp.
Ideal
Exp.
0.375
3.199
1.633
1.959
1.959
3.265
3.751
3.751
109.47
109.47
0.377
3.199
1.634
1.971
1.955
3.255
3.757
3.749
109.17
109.78
0.375
3.110
1.633
1.904
1.904
3.174
3.646
3.646
109.47
109.47
0.382
3.110
1.606
1.907
1.890
3.087
3.648
3.648
108.19
110.73
0.375
3.585
1.633
2.195
2.195
3.659
4.204
4.204
109.47
109.47
0.379
3.585
1.618
2.200
2.185
3.600
4.206
4.198
108.69
110.24
1.1 Crystal Structure of Nitrides
form of epitaxial layers on foreign substrates. The accuracy of X-ray diffraction and
less than accurate knowledge of the elastic parameters together allow determination of
the composition to only within about 1% molar fraction. In addition to composition,
the lattice parameter can be affected by free charge, impurities, stress (strain), and
temperature [18]. Because the c/a ratio correlates with the difference of the electronegativities of the two constituents, components with the greatest differences show the
largest departure from the ideal c/a ratio [19]. For GaN, the c/a ratio and the value of u
are measured as 1.627 (1.634 in Ref. [17]) and 0.377, respectively, which are close to the
ideal values [20]. AlN deviates significantly from the ideal parameters: c/a ¼ 1.601
(1.606 in Ref. [17]) and u ¼ 0.382. Although the data for InN are not as reliable, values of
u ¼ 0.379 and c/a ¼ 1.601 have been reported [17]. Inhomogeneities, strain, partial
relaxation of strain, and high concentration of structural defects may distort the lattice
constants from their intrinsic values and cause a wide dispersion among the reported
values. Table 1.3 lists a comparison of measured and calculated lattice parameters
reported for AlN, GaN, and InN crystallized in the wurtzite structure in more detail in
terms of the specifics of the sample used for measurements and complements. The
dispersion is even a greater concern in ternary and quaternaries, as compositional
inhomogeneities, in addition to the aforementioned issues, cause an additional
dispersion. The particulars of the ternaries are discussed in Section 1.5.
The wurtzite polytypes of GaN, AlN, and InN form a continuous alloy system
whose direct bandgaps range, according to data that adorned the literature for years,
from 1.9 eV for InN, to 3.42 eV for GaN, and to 6.2 eV for AlN. A revisit of the InN
bandgap indicates it to be about 0.78 eV [30] and the same for AlN is about 6 eV in
which case the energy range covered would be about 0.7–6 eV. Thus, the III–V
nitrides could potentially be fabricated into optical devices, which are active at
wavelengths ranging from the red well to the ultraviolet. The bandgaps of nitrides,
Table 1.3 Measured and calculated lattice constants of wurtzite AlN, GaN, and InN.
Compound
Sample
a (Å)
c (Å)
AlN
Bulk crystal [21]
Powder [22]
Epitaxial layer on SiC [23]
Pseudopotential LDA [24]
FP-LMTO LDA [25]
Bulk crystal [18]
Relaxed layer on sapphire [26]
Powder [29]
Relaxed layer on sapphire [27]
GaN substrate – LEO [28]
Pseudopotential LDA [24]
FP-LMTO LDA [25]
Powder [29]
Pseudopotential LDA [24]
FP-LMTO LDA [25]
3.1106
3.1130
3.110
3.06
3.084
3.189
3.1892
3.1893
3.1878
3.1896
3.162
3.17
3.538
3.501
3.53
4.9795
4.9816
4.980
4.91
4.948
5.1864
5.1850
5.1851
5.1854
5.1855
5.142
5.13
5.703
5.669
5.54
GaN
InN
LDA: local density approximation; FP-LMTO: full-potential linear muffin–tin orbital.
j11
j 1 General Properties of Nitrides
12
Figure 1.9 The bandgaps of nitrides, substrates commonly used
for nitrides, and other conventional semiconductors versus
their lattice constants. (Please find a color version of this figure
on the color tables.)
substrates commonly used for nitrides, and other conventional semiconductors are
shown in Figure 1.9 with respect to their lattice constants.
All III nitrides have partially covalent and partially ionic bonds. The concept of
fractional ionic character (FIC) is useful in interpreting many physical phenomena in
the crystals [31,32]. The FIC may be defined for a binary compound AB as
FIC ¼ jQ A Q B j=jQ A þ Q B j, where Q A and Q B are effective charges on atoms A
and B. The FIC values range from zero for a covalent compound (each atom has four
electrons) to 1 for an ionic compound (all eight electrons belong to the anion).
Figure 1.10 displays the charge distribution along the AB bond for all three compounds. The arrow along the bond charge indicates the atomic boundaries in the
crystals that are not always at the minimum of the line charge along the bond AB. This
should be expected taking into account the partial covalent bond of the compounds,
because only in the ionic crystals, the atomic boundary is clearly defined. Table 1.4 lists
the calculated effective radii, rIII and rN, the effective charges, and FIC for AlN, GaN,
and InN. The ionicity of AlN is high. This may explain the difficulties with AlN doping.
It is well known that only covalent semiconductors or semiconductors with a large
covalent component can form hydrogen-like shallow levels in the bandgap by
substitution of a host atom with a neighbor with one more or one less electron. GaN
and InN have a smaller than AlN but nearly equal ionicity. GaN was doped both p- and
n-type. Thus, one can expect that InN can also be doped n- and p-type. To date, only
n-type InN has been obtained because of high volatility of nitrogen and easiness of
nitrogen vacancy formation that acts as a donor in this compound.
1.1 Crystal Structure of Nitrides
2.0
AlN
1.5
1.0
0.5
0.0
0.0
0.5
1.0
r(Al-N)(λ)
1.5
2.0
1.5
2.0
2.0
ρ(r) (electrons/a.u.3)
GaN
1.5
1.0
0.5
0.0
0.0
0.5
1.0
r(Ga-N)(λ)
2.0
InN
1.5
1.0
0.5
0.0
0.0
0.4
0.8
1.2
1.6
2.0
r(In-N)(λ)
Figure 1.10 Charge density along the III–N bond in III nitride semiconductors.
The III-nitrides are commonly grown on mismatched substrates because of the lack
of suitable native substrates. Thus, the epitaxial layers are strained during cool down, if
they are sufficiently thick for them to relax at the growth temperature. The mechanical
forces related to strain dramatically change the band structure of the epitaxial layers.
The pressure dependence of the bandgap energy Eg can be expressed as Eg ¼ Eg(0)
gP þ dP2, where Eg(0) is the bandgap of stress-free semiconductor, g and d are the
Table 1.4 Calculated ionic radii (Å), effective charges (electrons),
and the fractional ionic character (FIC) for III nitrides [32].
Compound
rIII (Å)
rN (Å)
QIII (e)
QN (e)
FIC
AlN
GaN
InN
0.8505
0.9340
1.0673
1.0523
1.0119
1.0673
1.12
1.98
1.83
6.88
6.02
6.17
0.72
0.51
0.54
j13
j 1 General Properties of Nitrides
14
3.8
Energy gap (eV)
3.7
3.6
3.5
3.4
3.3
-2
0
2
4
6
8
10
Pressure (GPa)
Figure 1.11 Pressure dependence of the GaN energy gap,
showing the typical sublinear character. Solid line represents the
calculations of Christiansen and Gorczyca [35], which have been
rigidly upshifted by 0.82 eV for a better fit with experiments,
and the squares represent experimental results [6].
pressure coefficients, and P is the pressure. For GaN, g and d parameters are 4.2 · 103
and 1.8 · 105, respectively [33,34]. The bandgap is in terms of eVand the pressure is
in terms of kbar. The pressure dependence has, in general, a sublinear character. The
variation of the GaN energy gap with pressure, both theoretical [35] and experimental [6], is shown in Figure 1.11. The calculated pressure coefficients for III nitrides are
given in Table 1.5.
Parameters associated with mechanical properties of GaN in wurtzitic phase are
tabulated in Tables 1.6 and 1.7, the latter dealing with the sound wave velocity. The
same parameters for the zinc blende phase of GaN are tabulated in Tables 1.8 and 1.9.
Table 1.5 Calculated pressure coefficients for III nitrides including
wurtzitic, zinc blende, and rock salt phases (g in units of
meV GPa1 and d in units of meV GPa2) [35].
Zinc blende
polytype
Wurtzite polytype
Rock salt polytype
Compound
G
d
g
d
g
d
AlN
GaN
InN
40
39
33
0.32
0.32
0.55
42
40
16
0.34
0.38
0.02
43
39
41
0.18
0.32
0.08
It should be noted that rock salt phases cannot be synthesized and exist only under high pressures
beyond the phase transition point. g and d parameters with values of Eg ¼ Eg(0) þ gP þ dP2.
1.1 Crystal Structure of Nitrides
Table 1.6 Parameters related to mechanical properties of wurtzitic GaN (in part after Ref. [36]).
Wurtzite polytype GaN
Parameter value/comments
Group of symmetry
Molar volume, Vc (cm3 mol1)
Molecular mass (g mol1)
Density (g cm3)
C 46v (P63mc)
13.61
83.7267
6.11 or 6.15
Number of atoms in 1 cm3
Lattice constants
8.9 · 1022
a ¼ 3.1893 Å for powder,
c ¼ 5.1851 Å for powdera
210 [38]b or 20.4 · 1011 dyn
cm2 (204 GPa)
4
150
0.23 0.06 (0.198–0.37)
from C parameters
15.5
1200–1700
Bulk modulus B (GPa)
(compressibility1)
dB/dP
Youngs modulus (GPa)
Poissons ratio, n or s0
(n ¼ C13/(C11 þ C12))
Knoops hardness (GPa)
Surface microhardness
(kg mm2)
Nanoindentation hardness (GPa)
Yield strength (GPa)
Deformation potential, Eds
C11 (GPa)
C12 (GPa)
C13 (GPa)
C33 (GPa)
C44 (GPa)
10.8 at 300 K
0.1 at 1000 K
8.54 eV unscreened, 12 eV
screened
390 15, 29.6 · 1011 dyn
cm2 (296 GPa)
145 20, 13.0 · 1011 dyn
cm2 (130 GPa)
106 20, 15.8 · 1011 dyn
cm2 (158 GPa)
Comments/
references
The latter by
Bougrov et al. [37]
The latter by
Bougrov et al. [37]
[39]
At 300 K
300 K, using
Knoops pyramid
test [36,40,41]
[38,42]
The second set is
from Ref. [43]
The second set is
calculated from
the mean square
displacement of
the lattice atoms
measured by
X-ray diffraction
398 20, 26.7 · 1011 dyn
cm2 (267 GPa)
105 10, 2.41 · 1011 dyn
cm2 (241 GPa)
nh0 0 0 1i ¼ (Da/arelaz)/(Dc/crelax) or nh0 0 0 1i ¼ (Da/a0)/(Dc/c0) with Da ¼ ameas arelax and
Dcmeas crelax. Conversion: 1 dyn cm2 ¼ 0.1 Pa (i.e., 1 GPa ¼ 1010 dyn cm2). For details of
elastic constants and piezoelectric constants, see Tables 2.27 and 2.28, and at 300 K
Bs ¼ 210 10 GPa, Bs ¼ [C33(C11 þ C12) 2(C13)2]/[C11 þ C12 þ 2C33 4C13].
a
See Section 1.2.2 for details and lattice parameter for GaN on different substrates
b
Average of Voigt and Reuss bulk modulus.
j15
j 1 General Properties of Nitrides
16
Table 1.7 Wave propagation properties in wurtzitic GaN [36].
Wave
propagation
direction
[1 0 0]
[0 0 1]
Wave character
VL (longitudinal)
VT (transverse, polarization
along [0 0 1])
VT (transverse, polarization
along [0 1 0])
VL (longitudinal)
VT (transverse)
Expression for
wave velocity
Wave velocity
(in units of
105 cm s1)
(C11/r)1/2
(C44/r)1/2
7.96
4.13
(C11 C12)/2r)1/2
6.31
(C33/r)1/2
(C44/r)1/2
8.04
4.13
Parameters associated with thermal properties of GaN in wurtzitic and zinc blende
phases (expected to be identical or nearly identical – treated to be identical here) are
tabulated in Table 1.10.
The parameters associated with electrical and optical properties of wurtzitic GaN
are tabulated in Table 1.11. The same parameters associated with the zinc blende
phase of GaN are tabulated in Table 1.12.
Table 1.8 Parameters related to mechanical properties of zinc blende GaN (in part after Ref. [36]).
Zinc blende polytype GaN
Parameter value/comments
Group of symmetry
Molar volume, Vc, na,
or O (cm3 mol1)
Molecular mass (g mol1)
Density (g cm3)
Number of atoms in 1 cm3
Lattice constant (Å)
Bulk modulus, B (GPa)
2
Tp
d ðF43mÞ
ffiffiffi
ð 3a2 cÞ=4 ¼ 2:28310 23 cm3
dB/dP
Youngs modulus (GPa)
1.936 · 1023
6.15
8.9 · 1022
a ¼ 4.511 4.52
Bs ¼ 204 [36], 201
(theory) [45], 237 [46],
200 [47]
3.9, 4.3
181 [36]
Shear modulus, C 0 (GPa)
Poissons ratio, n or s0
67 [36]
0.352 [36]
Knoops hardness
Surface microhardness
Nanoindentation hardness
Yield strength
Deformation potential, Eds
C11 (GPa)
C12 (GPa)
C44 (GPa)
293
159
155
Bs ¼ [C33(C11 þ C12) 2(C13)2]/[C11 þ C12 þ 2C33 4C13] or Bs ¼
Comments/references
2
C12 Þ 2ðC 13 Þ
Bs ¼ CC3311ðCþ11C12þ þ
2C 33 4C13
Y0 ¼ (C11 þ 2C12)
(C11 C12)/(C11 þ C12)
C0 ¼ (C11 C12)/2
n or s0 ¼ C13/
(C11 þ C12)
[42]
C 33 ðC 11 þ C 12 Þ 2ðC13 Þ2
.
C11 þ C 12 þ 2C33 4C 13
1.1 Crystal Structure of Nitrides
Table 1.9 Wave propagation properties in zinc blende GaN (after Ref. [36]).
Wave propagation
direction
Wave character
Expression for
wave velocity
Wave velocity
(in units of 105 cm s1)
[1 0 0]
VL (longitudinal)
VT (transverse)
(C11/r)1/2
(C44/r)1/2
6.9
5.02
[1 1 0]
VL (longitudinal)
Vt//(transverse)
V t? (transverse)
[(C11þCl2þ2C44)/2r]1/2
Vt// ¼ VT ¼ (C44/r)1/2
[(C11 C12)/2r]1/2
7.87
5.02
3.3
[1 1 1]
V l
0
V l
[(C11 þ 2C12 þ 4C44)/3r]1/2
[(C11 C12 þ C44)/3r]1/2
8.17
3.96
0
For the crystallographic directions, see Ref. [44].
Table 1.10 Parameters related to thermal properties of GaN,
wurtzitic, and zinc blende phases are expected to be the in this
respect with the exception of the first two parameters, which are
for the wurtzitic phase (in part Ref. [36]).
GaN
Parameter value/comments
Comments/references
Temperature coefficient
(eV K1)
Thermal expansion (K1)
dEg/dT ¼ 6.0 · 104
Wurtzite structure only
Da/a ¼ 5.59 · 106, a|| ¼ aa
¼ 5.59 · 106 (wurtzite
structure) [48]
Dc/c ¼ 3.17 · 106; for a plot
versus temperature, see
Ref. [49] (wurtzite structure
only)
For low dissociation material
(106 cm2)
Thermal conductivity k
(W cm1 K1)
Debye temperature (K)
Melting point ( C)
Specific heat (J g1 C1)
Thermal diffusivity (cm2 s1)
Heat of formation, DH298
(kcal mol1)
Heat of atomization, DH298
(kcal mol1)
Heat of sublimation
(kcal mol1)
Heat capacity (J mol1 K1)
Specific heat (J mol1 K1)
(298 K < T < 1773 K)
Enthalpy, DH0 (kcal mol1)
Standard entropy of formation,
DS0 (cal mol1 K1)
11.9 at 77 K, 2.3 at 300 K, 1.5
at 400 K
600
>1700 (at 2 kbar), 2500
(at tens of kbar)
0.49
0.43
26.4
[50]
[37]
[37]
203
72.4 0.5
35.4 at 300 K
Cp ¼ 38.1 þ 8.96 · 103T
[51]
37.7
32.43
The specific heat Cp of Wz GaN at constant pressure for 298 K < T < 1773 K is
Cp ¼ 38.1 þ 8.96 · 103T (J mol1 K1) [51].
j17
j 1 General Properties of Nitrides
18
Table 1.11 Parameters related to electrical and optical properties
of Wz GaN (in part after Refs [36,44]).
Wurtzite polytype GaN
Parameter value/comments
Bandgap energy, Eg (eV),
direct
Breakdown field (cm1)
Electron affinity (eV)
Energy separation
between G and M–L
valleys (eV)
3.42 at 300 K, 3.505 at 1.6 K
Energy separation
between M–L valleys
degeneracy (eV)
Energy separation
between G and A valleys
(eV)
Energy separation
between A valley
degeneracy (eV)
Index of refraction
Dielectric constants
(static)
Dielectric constants (high
frequency)
Optical LO phonon energy (meV)
A1-LO, nA1(LO) (cm1)
Comments/
references
3–5 · 106 at 300 K
4.1
1.9 at 300 K
[53]
[37]
[37]
1 at 300 K
0.6 at 300 K
[52]
[37]
0.6 at 300 K
1.3–2.1 at 300 K
[52]
[37]
2 at 300 K
1 at 300 K
[52]
[37]
0.2 at 300 K
n (1 eV) ¼ 2.35 or 2.3
2.29, n (3.42 eV) ¼ 2.85 at 300 K
(extrapolated to 0 eV), E?c interference
method (the value for E||c is 1.5(2)%
lower at 500 nm); also see energy
dependence and long-wavelength
value [54]
10.4 (E||c)
9.5 (E?c)
8.9 in c-direction (E||c) at 300 K
5.35
5.8 (E||c) at 300 K
5.35 (E?c) at 300 K
5.47 (E||c)
91.2
[52]
[55]
[55]
[37]
[37]
[56]
[55]
[37]
710–735
[57]
744
A1-TO, nA1(TO||) (cm1)
E1-LO, nE1(LO? ) (cm 1)
533–534
741–742
[34]
[58]
533
746
E1-TO, nE1(TO? ) (cm 1)
E2 (low) (cm1)
E2 (high) (cm1)
556–559
143–146
560–579
[59]
559
Reflectivity
[55]
Raman [56]
Reflectivity
[55]
Raman [60]
1.1 Crystal Structure of Nitrides
Table 1.11 (Continued)
Wurtzite polytype GaN
Parameter value/comments
Energy of spin–orbital
splitting, Eso (meV)
11 (þ5, 2) at 300 K calculated from
the values of energy gap Eg,dir (given in
this table)
40 at 300 K
Energy of crystal-field
splitting, Ecr (meV)
Effective electron mass,
==
me or me
Effective electron mass,
me? or m?
e
Effective hole mass
Effective hole masses
(heavy), mhh
Effective hole masses
(light)
Effective hole masses
(split-off band), ms
Effective mass of density
of state, mv
Effective conduction
band density of states
(cm3)
Effective valence band
density of states (cm3)
Electron mobility
(cm2 V1 s1)
Hole mobility
(cm2 V1 s1)
n-doping range (cm3)
22 (2), calculated from the values of
energy gap Eg,dir (given in this table)
0.20m0 at 300 K
Comments/
references
[61]
[37]
[61]
[37]
0.20m0
0.27m0 by Faraday rotation
0.138–0.2
0.20m0, 300 K; fit of reflectance spectrum
[62]
[52]
0.15–0.23m0
0.8m0 at 300 K
mhh ¼ 1.4m0 at 300 K
[52]
[64]
Calculated
==
mhhz ¼ mhh ¼ 1:1m0 at 300 K
mhh? ¼ m?
hh ¼ 1:6m0 at 300 K
==
mhh ¼ 1:1 2:007m0
m?
hh ¼ 1:61 2:255m0
mlh ¼ 0.3m0 at 300 K
==
mlhz ¼ mlh ¼ 1:1m0 at 300 K
m?
lh ¼ mlh? ¼ 0:15m0 at 300 K
==
mlh ¼ 1:1 2:007m0
mlh? ¼ 0:14 0:261m0
msh ¼ 0.6m0 at 300 K
==
mshz ¼ mch ¼ 0:15m0 at 300 K
msh? ¼ m?
ch ¼ 1:1m0 at 300 K
==
msh? ¼ mch ¼ 0:12 0:16m0
m?
¼
0:252
1:96m0
ch
1.4m0
[63]
[15]
[70]
[52]
[52]
Calculated
[15]
[70]
[52]
[52]
Calculated
[36]
[70]
[52]
[52]
[37]
2.3 · 1018 at 300 K
4.6 · 1019 at 300 K
1400 experimental at 300 K
<20
50 000 at
20 K [65]
At 300 K
1016 cm3–high 1019
(Continued )
j19
j 1 General Properties of Nitrides
20
Table 1.11 (Continued)
Wurtzite polytype GaN
Parameter value/comments
Comments/
references
p-doping range (cm3)
Diffusion coefficient for
electrons (cm2 s1)
Diffusion coefficient for
holes (cm2 s1)
1016 cm3–mid 1018
25
[36]
5, 26, 94
[36,53]
The details of the energies of high symmetry points compiled by Fritsch et al. [52] are given
in Table 2.1. Dependence of the bandgap on hydrostatic pressure: Eg ¼ Eg(0) þ gP þ dP2, where
Eg(0) is the bandgap of stress-free GaN. The values for g parameter are 39–42 meV GPa1 and
the same for d parameter are 0.18 to 0.32 meV GPa2. For others, see Table 1.5.
The phonon energies are discussed later on in Table 1.27 in detail and effective
masses are discussed in Chapter 2. More details of effective masses can be found in
Table 2.9.
More should be said about the dielectric constants. Electromagnetic theory indicates
that for any longitudinal electromagnetic wave to propagate, the dielectric function
e(o) must vanish. Doing so leads to [66]
eðwÞ w2LO w2
;
¼
eð¥Þ w2TO w2
ð1:5Þ
where oLO and oTO represent the transverse optical (TO) and longitudinal optical (LO)
phonon frequencies and e(o) and e(1) represent the low and high (optical) frequency
dielectric constants. The phonon branches associated with a wurtzitic symmetry are
discussed in Section 1.2.2 dealing with the mechanical properties of GaN. When
o ¼ oLO, the dielectric function vanishes, e(oLO) ¼ 0. Equation 1.5 can be expanded to
the directional dependence of the dielectric function in nitrides in general and GaN in
particular. In the direction parallel to the c-axis or the z-direction, from the G point to
the A point, in the k-space, (with x, y representing the in-plane coordinates), the lowand high-frequency dielectric functions are related each with the help of A1(LO) and
E1(TO) phonons through [67]
e== ðwÞ ¼ e¥?
w2 w2== ðLOÞ
w2 w2== ðTOÞ
:
ð1:6Þ
Likewise, Equation 1.5 can be expanded in the direction perpendicular to the c-axis or
in the basal plane or the (x, y) plane, the z-direction (in k-space between the G point and
M (1/2, 0, 0) or K (1/3, 1/3, 0) points), the low- and high-frequency dielectric functions
are related each with the help of A1(TO) and E1(LO) phonons through
e? ðwÞ ¼ e¥?
w2 w2? ðLOÞ
;
w2 w2? ðTOÞ
ð1:7Þ
where ? and // indicate in the basal plane and along the c-direction, respectively.
1.1 Crystal Structure of Nitrides
Table 1.12 Parameters related to electrical and optical properties
of zinc blende GaN (in part after Refs [36,44]).
Zinc blende polytype GaN
Parameter/comments
Comments/references
Bandgap energy (eV)
Breakdown field (V cm1)
Index of refraction
Dielectric constant (static)
3.2–3.28 at 300 K
5 · 106
n (at 3 eV) ¼ 2.9, 2.3
9.7 at 300 K
==
9.2 by ð2e?
0 þ e0 Þ=3 of
wurtzitic form
5.3 at 300 K
3.302 at low temperature
[36]
Dielectric constant (high
frequency)
Energy separation between G and
X valleys, EG (eV)
Energy separation between G and
L valleys, EL (eV)
Spin–orbit splitting in valence
band, Dso or Eso (eV)
Effective electron mass, me
Effective hole masses (heavy)
1.4
1.1
1.6–1.9
2
0.02
0.017
0.13m0
0.14m0
mhh ¼ 1.3m0,
½110
mhh ¼ 1:52m0
m[100] ¼ 0.8m0,
[37]
[52]
Using Lyddane–Sachs–Teller
relation (e0/ehigh ¼ o2LO/
o2TO)
[36]
[52]
[36]
[52]
At 300 K [36]
[69]
At 300 K [37]
[52]
At 300 K
[36,70]
½100
mhh ¼ 0:84m0
m[111] ¼ 1.7m0,
½111
mhh ¼ 2:07m0
Effective hole masses (light)
Effective hole masses (split-off
band), ms, mch, or mso
Effective conduction band
density of states
Effective valence band density of
states
Electron mobility (cm2 V1 s1)
Hole mobility (cm2 V1 s1)
Diffusion coefficient for
electrons (cm2 s1)
Diffusion coefficient for holes
(cm2 s1)
Electron affinity
Optical LO phonon energy (meV)
Second set of figures are from
Ref. [52], which are deemed
more reliable
mlh ¼ 0.19m0,
½110
mlh ¼ 0:20m0
m[100] ¼ 0.21m0,
½100
mlh ¼ 0:22m0
m[111] ¼ 0.18m0,
½111
mlh ¼ 0:19m0
msh ¼ 0.33m0,
mso ¼ 0.35m0
m[100] ¼ 0.33m0
m[111] ¼ 0.33m0
1.2 · 1018 cm3
At 300 K [37]
4.1 · 1019 cm3
At 300 K [37]
1000 at 300 K
350 at 300 K
25
[36]
[36]
[36]
9, 9.5, 32
[36]
4.1 eV
87.3
[37]
At 300 K
The details of the energies of high symmetry points are given in Table 2.1.
j21
j 1 General Properties of Nitrides
22
For wurtzitic GaN, the various directional components of phonon frequencies
are o?(LO) ! E1(LO) ¼ 91.8 meV, oz(LO) ¼ o//(LO) ! A1(LO) ¼ 91 meV, o?(TO) !
E1(TO) ¼ 69.3 meV, and oz(TO) ¼ o//(TO) ! A1(TO) ¼ 66 meV. In the z-direction
(along the c-direction) and perpendicular to the z-direction (in basal plane), LO and
TO phonons are not mixed. For any direction other than the in-plane and out-of-plane
configurations, the LO and TO phonons mix and hybridize. For a given propagation
direction with an angle y relative to the c-axis (0z), one finds three phonon branches.
One is an ordinary TO phonon mode with atomic displacement in the (0xy) plane.
The other two branches have a mixed TO and LO character and their dielectric
functions are given by the solutions of [68]
e== cos2 q þ e? sin2 q ¼ 0:
ð1:8Þ
Using the above relationship, the phonon energy as a function of the angle can easily
be calculated. Doing so leads to the conclusion that the upper branch (LO-like)
remains between A1(LO) and E1(LO) energies, whereas the lower branch (TO-like)
remains between A1(TO) and E1(TO) energies. Therefore, the dispersion remains
small compared to the LO–TO separation, owing to the relatively small cell asymmetry and the large ionicity of atomic bonds. A more important consequence of
LO–TO mixing is that the TO-like mode becomes coupled to carriers whereas in the
c-direction A1(LO) mode and in the basal plane E1(LO) phonons couple to the carriers.
For the special case o ¼ 0 (or very small frequencies compared to the LO and TO
phonon frequencies), the relationship between the optical and static dielectric
constants reduces to the well-known Lyddane–Sach–Teller relationship
eðwÞ w2LO
¼
;
eð¥Þ w2TO
ð1:9Þ
which will be used to determine the optical frequency dielectric constant from the
knowledge of A1(LO) and A1(TO) phonon frequencies along the c-direction and
E1(LO) and E1(TO) in the basal plane. This relationship is used very often.
Parameters related to the energy bandgap, carrier mass, and mechanical properties
of AlN have been determined [71–76]. Extensive data on all the binary and ternary
band structure parameters can be found in Chapter 2. For example, additional
parameters on the critical point energies for Wz GaN, AlN, and InN are given in
Tables 2.1–2.3, respectively. Tables 2.4–2.6 list the critical point energies for ZB GaN,
AlN, and InN, respectively. Effective masses and other band parameters for Wz GaN
are listed in Table 2.9. Table 2.10 tabulates the Luttinger band parameters for ZB GaN.
Table 2.14 lists the effective band parameters for Wz AlN, whereas the effective
masses and band parameters for Wz AlN are tabulated in Table 2.15. Luttinger
parameters for ZB AlN are listed in Table 2.16. The band parameters and effective
masses for Wz InN are tabulated in Tables 2.19 and 1.20, respectively.
Returning to the content of this chapter, parameters associated with the mechanical properties of AlN in wurtzitic and zinc blende phases are tabulated in Tables 1.13
and 1.14, respectively. The parameters related to the sound wave velocity in wurtzitic
AlN are listed in Table 1.15.
1.1 Crystal Structure of Nitrides
Table 1.13 Parameters related to mechanical properties of wurtzitic AlN (in part after Ref. [36]).
Wurtzite AlN
Parameter/comments
C46v (P63 mc)
9.58 · 1022
12.47
40.9882
3.28
3.255 g cm3 by X-ray
3.23 g cm3 by X-ray
Lattice constants
a ¼ 3.112 Å, c ¼ 4.979–4.982 Å
Bulk modulus, B (GPa)
159.9–210.1, 21 · 1011 dyn cm2
(210 GPa) (Bs ¼ 210)
dB/dP
5.2–6.3
Youngs modulus, E or Y0 (GPa)
374, 308
0.18–0.21
Poissons ratio, n or s0
Poissons ratio s0 along the
{0001}, c-plane
different crystallographic
f1 1 2 0g, a-plane
ðl ¼ h0001i; m ¼ h1 1 0 0iÞ
directions
f1 1 2 0g, a-plane
ðl ¼ h1 1 0 0i; m ¼ h0001iÞ
Knoops hardness (GPa)
10–14 at 300 K
Nanoindentation hardness (GPa) 18
Yield strength (GPa)
0.3 at 1000 C
Surface microhardness on basal
800 kg mm2 by 300 K, using
plane (0 0 0 1)
Knoops pyramid test
C11 (GPa)a
410 10
149 10
C12 (GPa)a
C13 (GPa)a
99 4
389 10
C33 (GPa)a
C44 (GPa)a
125 5
Velocity of the longitudinal sound 10 127 m s1
waves, vl
Velocity of the shear waves, vs
6333 m s1
Comments/references
Group of symmetry
Number of atoms in 1 cm3
Molar volume, Vc (cm3 mol1)
Molecular mass (g mol1)
Density (g cm3)
Longitudinal elastic modulus, Cl
Shear elastic modulus, Cs
[77]
[78]
[78]
The latter from Ref. [79]
0.287 Ref. [80]
0 Ref. [80]
0.216 Ref. [80]
[81]
[82]
[36,40,41]
[42,83]
Refer to Table 1.29 as well
Refer to Table 1.29 as well
Refer to Table 1.29 as well
Refer to Table 1.29 as well
[79]
The sound velocities and
related elastic module
(experimental data)
334 GPa
131 GPa
Conversion: 1 dyn cm 2 ¼ 0.1 Pa (i.e., 1 GPa ¼ 1010 dyn cm2). See Table 1.29 for more details.
For details of elastic constants and piezoelectric constants, see Tables 2.27 and 2.28. This
expression is given already in conjunction with Table 1.6 and it is also Bs ¼ [C33(C11 þ C12) 2
(C13)2]/[C11 þ C12 þ 2C33 4C13]. Temperature derivatives of the elastic module: dln Cl/
dT ¼ 0.37 · 104 K1; dln Cs/dT ¼ 0.57 · 104 K1; dln Bs/dT ¼ 0.43 · 104 K1.
a
See Table 1.29 for a more in-depth treatment of these parameters.
Parameters associated with thermal properties of wurtzitic AlN are tabulated in
Table 1.16.
Parameters associated with electrical and optical properties of wurtzitic AlN are
tabulated in Table 1.17. The same range of parameters associated with the zinc blende
j23
j 1 General Properties of Nitrides
24
Table 1.14 Parameters related to mechanical properties of zinc blende AlN.
Not much is known
about the zinc
blende phase
Zinc blende AlN
Lattice constant (Å)
Bandgap (eV)
E Xg ðeVÞ
E Lg ðeVÞ
Bulk modulus, B (GPa)
Youngs modulus (GPa)
a ¼ 4.38
5.4, indirect
4.9
9.3
228
Shear modulus (GPa)
Poissons ratio, n or s0
C11 (GPa)
348
304
C12 (GPa)
C44 (GPa)
me ðGÞ
ml ðXÞ
mt ðXÞ
168
135
0.25
0.53
0.31
160
193
Comments/references
[84]
[84]
[84]
Bs ¼ (C11þ2C12)/3
Y0 ¼ (C11 þ 2C12) ·
(C11 C12)/(C11 þ C12)
C ¼ (C11 C12)/2
n or s0 ¼ C12/(C11þC12)
The latter figures are from
Ref. [84]
[84]
[84]
[84]
See Table 2.18 for more details.
phase of GaN is tabulated in Table 1.18. Mechanical, phonon, properties of epitaxial
AlN (deposited on silicon and sapphire substrates at 325 K by ion beam assisted
deposition (IBAD)) have been investigated by Ribeiro et al. Raman scattering
measurements revealed interesting features related to the atomic composition and
structure of the films [94]. Vibrational modes corresponding to 2TA(L) at 230 cm1,
2TA(X) at 304 cm1, 2TA(S) at 435 cm1, TO(G) at 520 cm1, TA(S) þ TO(S) at
615 cm1, accidental critical points at 670 and 825 cm1, 2TO(D) at 950 cm1,
2TO(L) at 980 cm1, 2TO(G) at 1085 cm1, 2TA(X) þ 2TO(G) at 1300 cm1, and
3TO(G) at 1450 cm1 have been observed. While identifying the vibrational modes,
one should be wary of the peak at 2330 cm1 caused by the molecular nitrogen on
Table 1.15 Acoustic wave propagation properties in wurtzite AlN [36].
Wave propagation
direction
[1 0 0]
[0 0 1]
Wave character
VL (longitudinal)
VT (transverse, polarization
along [0 0 1])
VT (transverse, polarization
along [0 1 0])
VL (longitudinal)
VT (transverse)
For the crystallographic directions, see Ref. [44].
Expression for
wave velocity
(C11/r)1/2
(C44/r)1/2
(C11-C12)/2r)1/2
(C33/r)1/2
(C44/r)1/2
Wave velocity
(in units of 105 cm s1)
11.27
6.22
6.36
10.97
6.22
1.1 Crystal Structure of Nitrides
Table 1.16 Parameters related to thermal properties of wurtzitic AlN (in part after Ref. [36]).
Wurtzite polytype AlN
Value
Comments/references
Thermal expansion (K1)
Da/a ¼ a|| ¼ aa ¼ 4.2 · 106,
Dc/c ¼ aort ¼ ac ¼ 5.3 · 106
Da/a ¼ 2.9 · 106,
Dc/c ¼ 3.4 · 106
aort ¼ ac ¼ 5.27 · 106,
a|| ¼ aa ¼ 4.15 · 106
[48,85–87]
Thermal conductivity
(W cm1 K1)
Thermal diffusivity
(W cm1 C1)
Debye temperature (K)
Melting point (K)
Specific heat (J g1 C1)
Thermal diffusivity (cm2 s1)
Heat of formation, DH298
(kcal mol1)
Heat of atomization, DH298
(kcal mol1)
Free energy, DG298 (kcal mol1)
k ¼ 2.85–3.2
2.85 at 300 K
950, 1150
3273
3023 (between
100 and 500 atm
of nitrogen)
3487 (2400 C at 30 bar)
0.6
[88]
T ¼ 20–800 C. X-ray,
epitaxial layers, by Sirota
and Golodushko [89],
also see Ref. [48]
[90]; later results by
Slack et al. [91]
[90]
[92]
[78]
See Figure 1.29 and the
expressions below this table
1.47
64
209.7
68.15
For 293 < T < 1700 K, Da/a300 ¼ 8.679 · 102 þ 1.929 · 104T þ 3.400 · 107T2 7.969 ·
1011T3. For 293 < T < 1700 K, Dc/c300 ¼ 7.006 · 102 þ 1.583 · 104T þ 2.719 · 107T2
5.834 · 1011T3. The specific heat Cp of AlN for constant pressure: for 300 < T < 1800 K,
Cp ¼ 45.94 þ 3.347 · 103T 14.98 · 105T2 (J mol1 K1); for 1800 < T < 2700 K,
Cp ¼ 37.34 þ 7.866 · 103T (J mol1 K1). After Ref. [93]. Optical emission measurements
indicate the FXA transition at 6.023 with an associated binding energy of 63 meV, which sets the
bandgap of Wz AlN at 6.086.
the surface of c-Si [95]. It is worth noting that, owing to the extremely weak Raman
signal usually presented by AlN films, it is not uncommon to ascribe some of the
features erroneously to AlN [96].
Conduction band first- and second-order pressure derivatives [36]:
Eg ¼ Eg(0) þ 3.6 · 103P 1.7 · 106P2 (eV) [121]
EM ¼ EM(0) þ 7.5 · 104P þ 1.0 · 106P2 (eV)
EL ¼ EL(0) þ 8.0 · 104P þ 6.9 · 107P2 (eV)
Ek ¼ Ek(0) þ 6.3 · 104P þ 1.7 · 106P2 (eV)
where P is pressure in kbar.
j25
j 1 General Properties of Nitrides
26
Table 1.17 Parameters related to optical and electrical properties
of wurtzitic AlN [97–102] (in part from Ref. [36]).
Wurtzite polytype AlN
Parameter
Comments/references
Bandgap energy (eV)
From the dichroism of the
absorption edge, it follows
that the G1 0 state lies slightly
above the G6 state (transition
E||c (G1 0 v Glc) at lower
energy than transition E ? c
(G6v Glc)), both states being
split by crystal field
interaction [105]
6.026 at 300 K
6.2 eV at 300 K
[103,104]
Excitonic contribution
near direct edge [105]
6.23 at 77 K
Excitonic contribution
near direct edge [105]
Excitonic edge assuming
exciton binding energy of
75 meV [106]
With a free exciton binding energy of 63 meV
[107,108]
6.28 at 300 K
6.086 at 5 K
Breakdown field (V cm1)
dEg/dP (eV bar1)
Conduction band energy
separation between G and
M–L valleys (eV)
Conduction band energy
separation between G and
M–L valleys
Conduction band energy
separation between M–L
valleys degeneracy (eV)
Conduction band energy
separation between G and K
valleys (eV)
Conduction band K valley
degeneracy (eV)
Valence band energy of
spin–orbital splitting,
Eso (eV)
6.0 at 300 K
6.1 at 5 K
1.2–1.8 · 106
3.6 · 103
0.7
1
[36]
[109,110]
[78]
[52]
0.6
[78]
0.2
[52]
1.0
[78]
0.7
2
[52]
[78]; empirical pseudopotential calculations of
Fritsch et al. [52] do not
show degeneracy at this
critical point
0.019 at 300 K
0.036
[108]
1.1 Crystal Structure of Nitrides
Table 1.17 (Continued)
Wurtzite polytype AlN
Parameter
Comments/references
Valence band energy of
crystal field splitting, Ecr (eV),
G7 on top of G9
Effective conduction band
density of states (cm3)
Effective valence band
density of states (cm3)
Index of refraction
Dielectric constant (static)
0.225
[108]
6.3 · 1018
[78]
4.8 · 1020
[78]
Dielectric constant (high
frequency)
4.68
4.77
4.84 at 300 K
4.35 for E//c (modeling)
4.16 for E?c (experiment)
2.1–2.2 at 300 K
Infrared refractive index
Effective electron mass, me
Effective hole masses (heavy)
==
For kz direction mhz or mhh
For kx direction mhx or m?
hh
Effective hole masses (light)
==
For kz direction mlz or mlh
For kx direction mlx or m?
lh
Effective hole masses (splitoff band)
==
For kz direction msoz or mch
For kx direction msox or
n (3 eV) ¼ 2.15 0.05
9.14 at 300 K
7.34
8.5 0.2 at 300 K
9.32 for E//c (modeling)
7.76 for E?c (experiment)
4.6 at 300 K
m?
ch
1.9–2.1 at 300 K
1.8–1.9 at 300 K
3 for E//c (modeling)
2.8 for E?c (experiment)
0.27 and 0.35m0
0.25–0.39m0
0.4m0 at 300 K
==
me ¼ 0:231 0:35m0
m?
e ¼ 0:242 0:25m0
==
mhh ¼ 3:53m0 at 300 K
2.02–3.13m0 at 300 K
By reflectivity [111]
[78,112]
[113]
[113]
[111]
[78]
Reflectivity
[113]
[113]
Epitaxial films and
monocrystal
Polycrystalline films
Amorphous films [87]
[113]
[113]
[14,114,115]
[107]
[52]
m?
hh ¼ 10:42m0 at 300 K
==
mhh ¼ 1:869 4:41m0
m?
hh ¼ 2:18 11:14m0
3.53m0, 0.24m0
==
mlh ¼ 1:869 4:41m0
m?
lh ¼ 0:24 0:350m0
0.25m0 at 300 K
[116]
[107]; from Mg binding
energy
[116]
[52]
[52]
At 300 K [116]
[52]
[52]
[116]
3.81m0 at 300 K
==
mch ¼ 0:209 0:27m0
[52]
m?
ch
[52]
¼ 1:204 4:41m0
(Continued )
j27
j 1 General Properties of Nitrides
28
Table 1.17 (Continued)
Wurtzite polytype AlN
Parameter
Comments/references
Effective mass of density of
state, mv
Optical phonon energy
(meV)
nTO(E1) phonon wave
number (cm1)b
7.26m0 at 300 K
[116]
nLO(E1) phonon wave
number (cm1)
nTO(A1) phonon wave
number (cm1)
nLO(A1) phonon wave
number (cm1)
n(E2) phonon wave number
(cm1)
nTO(E1) phonon wave
number (cm1)
nTO(A1) phonon wave
number (cm1)
nLO(E1) phonon wave
number (cm1)
nLO(A1) phonon wave
number (cm1)
n(1)(E2) phonon wave number
(cm1)
n(2)(E2) phonon wave number
(cm1)
99.2
895
614
608
671.6
821
888.9
888
514
667.2
659.3
663
909
303a
426
657–673
First column [117];
second column [118];
third column [113]
See Table 1.30 for more
details
[87,111,119,120]
607–614 or 659–667
895–924
888–910
241–252
655–660
The details of the energies of high symmetry points are given in Table 2.2. See Table 1.30 for
additional details for phonon wave numbers. More details of effective masses can be found in
Table 3.15. Temperature dependence of energy gap: Eg ¼ Eg(0) – 1.799 · 103T2/(T þ 1462) (eV)
by Guo and Yoshida [103].
a
Room-temperature Raman, tentative.
b
For more details regarding vibrational modes, refer to Section 1.3.1.
Phase transition from the wurtzite phase to the rock salt structure (space
group O5h ; lattice parameter 4.04 Å) takes place at the pressure of 17 GPa (173 kbar)
[109,110].
Parameters associated with the electrical and optical properties of zinc blende AlN
are listed in Table 1.18. For details regarding the Luttinger parameters for the valence
band in zinc blende AlN, refer to Table 2.16. Parameters associated with the
mechanical properties of wurtzitic InN are tabulated in Table 1.19.
For wurtzite crystal structure, the surfaces of equal energy in G valley should be
ellipsoids, but effective masses in the z-direction and perpendicular directions are
estimated to be approximately the same.
1.1 Crystal Structure of Nitrides
Table 1.18 Parameters related to optical and electrical properties of zinc blende AlN.
Zinc blende polytype of AlN
Value
Comments/references
Bandgap energy (eV)
4.2; 6.0
5.8
All (theory)
9.56
4.46
All at 300 K
[52]
Dielectric constant (static)
Dielectric constant
(high frequency)
Energy separation between
G and X valleys EG (eV)
Energy separation between
G and L valleys EL (eV)
Spin–orbit splitting in valence
band, Dso or Eso (eV)
Deformation potential (eV)
Effective electron mass, me
Effective hole masses (heavy)
Effective hole masses (light)
Effective hole masses
(split-off band), ms, mch,
or mso
Luttinger parameter c1
Luttinger parameter c2
Luttinger parameter c3
a
a
a
0.7
0.5
2.3
3.9
0.019
[52]
[84]
[52]
[84]
[84]
9
0.23m0
[84]
[52]
½100
mhh ¼ 1:02m0
½111
mhh
¼ 2:64m0
½110
mhh
¼ 1:89m0
mlh
½100
¼ 0:37m0
½111
mlh
¼ 0:30m0
½110
mlh
¼ 0:32m0
[52]
[52]
0.54m0
[52]
1.85
0.43
0.74
[52]
C. Persson, and A. Ferreira da Silva, Linear optical response of zinc-blende and wurtztie III-N
(III ¼ B, AI, Ga, and In), Journal of Crystal Growth 305 pp. 408–413 (2007)
The parameters associated with thermal properties of wurtzitic InN are tabulated
in Table 1.20.
The specific heat Cp of InN at constant pressure for 298 K < T < 1273 K [51] is
Cp ¼ 38.1 þ 1.21 · 102T (J mol1 K1). Refer to Table 1.31 for a detailed treatment of
mechanical properties of InN.
The parameters associated with electrical and optical properties of wurtzitic InN
are tabulated in Table 1.21.
Available parameters associated with the mechanical properties of zinc blende
InN, primarily calculated, are tabulated in Table 1.22. Other parameters dealing with
electrical and optical properties of zinc blende InN, primarily calculated, are listed in
Table 1.23.
j29
j 1 General Properties of Nitrides
30
Table 1.19 Parameters related to mechanical properties of wurtzitic InN (in part after Ref. [36]).
Wurtzite InN
Value
Group of symmetry
C 46v (P63 mc)
Molar volume (cm3 mol1)
18.49
Molar mass (g mol1)
128.827
Density (g cm3)
6.89
6.98
6.81
a ¼ 3.548
a ¼ 3.5446
a ¼ 3.533
c ¼ 5.760
c ¼ 5.7034
c ¼ 5.693
165
140
Lattice constants (Å)
Bulk modulus B (GPa)
dB/dP
3.8
Nanoindentation hardness(GPa)
11.2
Comments/references
Measured by displacement
X-ray, 298.15 K [122]
Epitaxial layers, X-ray [123];
300 K [124]
Epitaxial layers, X-ray [123];
300 K [124]
[124]
[125]
Youngs modulus (GPa)
Poissons ratio, n or s0
Knoops hardness (GPa)
Deformation potential, Eds
C11 (GPa)
C12 (GPa)
C13 (GPa)
C33 (GPa)
C44 (GPa)
Can be calculated using S
parameters and Equation 2.10
0.82, 0.68
7.10 eV
223
115
92
224
48
C31 ¼ 70
205
Estimate
[42]
[42]
[42]
[42]
[42]
Eg ¼ Eg(0) þ 3.3 · 102P (eV), where P is pressure in GPa [35,126]. For details of elastic constants
and piezoelectric constants, see Table 2.19. Bs ¼ [C33(C11 þ C12) 2(C13)2]/
[C11 þ C12 þ 2C33 4C13].
1.2
Gallium Nitride
Despite the fact that GaN has been studied far more extensively than the other group
III nitrides, further investigations are still needed to approach the level of understanding of technologically important materials such as Si and GaAs. GaN growth
often suffers from large background n-type carrier concentrations because of native
defects and, possibly, impurities. The lack of commercially available native substrates
1.2 Gallium Nitride
Table 1.20 Parameters related to thermal properties of wurtzitic InN (in part after Ref. [36]).
Wurtzite polytype InN
Value
Temperature coefficient
Thermal expansion
dEg/dT ¼ 1.8 · 104 eV K1
Da/a ¼ 2.70 · 106 K1;
Dc/c ¼ 3.40 · 106 K1
Da/a ¼ 2.85 · 106 K1;
Dc/c ¼ 3.75 · 106 K1
Da/a ¼ 3.15 · 106 K1;
Dc/c ¼ 4.20 · 106 K1
Da/a ¼ 3.45 · 106 K1;
Dc/c ¼ 4.80 · 106 K1
Da/a ¼ 3.70 · 106 K1;
Dc/c ¼ 5.70 · 106 K1
aa ¼ 3.8 · 106 K1;
ac ¼ 2.9 · 106 K1
0.8 0.2 W cm1 K1
0.45 W cm1 C1
1.76 W cm1 C1,
300 K (estimate for ideal InN)
4.6
Thermal conductivity
Heat of formation,
DH298 (kcal mol1) (Wz)
Heat of atomization,
DH298 (kcal mol1)
Melting point
Debye temperature
Specific heat (J mol1 K1)
Heat capacity, Cp (cal mol1 K1)
Entropy, S0 (cal mol1 K1)
TSFCw at formation, DH0f
(kcal mol 1)
TSFCa at formation, DS0f
(kcal mol 1 K 1)
TSFCw at formation,
DG0f (kcal mol 1)
TSFCw at fusion, DHm (kcal mol1)
TSFCw at fusion, DSm (cal mol1 K1)
N2 equilibrium vapor pressure
Comments/references
At 190 K
At 260 K
At 360 K
At 460 K
At 560 K
[124]
Estimate [127]
175
1373 K
2146 K, vapor pressure 105
bar at 1100–1200 C
660 K at 300 K
370 K at 0 K
Cp ¼ 38.1 þ 1.21 · 102T
9.1 þ 2.9 · 103T
10.4
34.3, 30.5
[92]
[124]
[128]
[51]
298–1273 K
298.15 K
Experimental 298.15 K
25.3
Experimental 298.15 K
22.96
Experimental 298.15 K
14.0
10.19 cal mol1 K1
1 atm
105 atm
Theoretical
Theoretical
800 K
1100 K
a
TSFC: thermodynamic state function changes.
exacerbates the situation. These, together with the difficulties in obtaining p-type
doping, and the arcane fabrication processes caused the early bottlenecks stymieing
progress. Information available in the literature on many of the physical properties of
GaN is in some cases still in the process of evolution, and naturally controversial. This
j31
j 1 General Properties of Nitrides
32
Table 1.21 Parameters related to electrical and optical properties
of wurtzitic InN (in part after Ref. [36]).
Wurtzitic InN
Value
Comments/references
Bandgap energy, Eg (300 K)
1.89 eV, 1.5 eV, 0.78 eV
See Section 1.3.1 for an expan
ded discussion
15.3
e0,ort ¼ 13.1
300 K [124]
300 K [128]
e0, || ¼ 14.4,
300 K [128]
8.4
300 K, using the Lyddane–
Sachs–Teller relation
(e0 =ehigh ¼ w2LO =w2TO ) [129,130]
Heavily doped film, infrared
reflectivity [131]
[132]
At 300 K [124]
At 300 K and l ¼ 1.0 mm,
interference method;
n ¼ 3–1020 cm3 [131]
At l ¼ 0.82 mm [131]
At l ¼ 0.66 mm [131]
Electron affinity
Dielectric constant (static)
Dielectric constant (static,
ordinary direction)
Dielectric constant (static,
extraordinary direction)
Dielectric constant (high
frequency)
9.3
Infrared refractive index
Energy separation
between G and M–L valleys
(eV)
Energy separation
between M–L valleys
degeneracy
Energy separation
between G and A valleys (eV)
Energy separation
between A valley degeneracy
Energy separation
between G and G1 valleys (eV)
Energy separation
between G1 valley
degeneracy (eV)
Effective conduction band
density of states
Effective valence band
density of states
Valence band crystal field
splitting, Ecr
5.8
2.9
2.56
2.93
3.12
Reported range: 2.80–3.05
2.9–3.9
300 K [124]
4.8
0.6
[52]
300 K [124]
0.7
0.7–2.7
[52]
300 K [124]
4.5
1
[52]
300 K [124]
0.6
1.1–2.6
[52]
300 K [124]
1
300 K [124]
9 · 1017 cm
300 K [124]
5.3 · 1019 cm3
300 K [124]
0.017 eV
300 K [124]
1.2 Gallium Nitride
Table 1.21 (Continued)
Wurtzitic InN
Value
Comments/references
Valence band spin–
orbital splitting, Eso
Index of refraction
0.003 eV
300 K [124]
2.9 at 300 K
2.56 at 300 K (interference
method; n ¼ 3–1020 cm3,
l ¼ 1.0 mm)
2.93
3.12
0.11m0
==
me ¼ 0:1 0:138m0
m?
e ¼ 0:1 0:141m0
1.63m0 at 300 K
0.5m0 at 300 K
==
mhh ¼ 1:350 2:493m0
m?
hh ¼ 1:410 2:661 m0
0.27m0 at 300 K
==
mlh ¼ 1:350 2:493m0
m?
lh ¼ 0:11 0:196m0
0.65m0 at 300 K
==
mch ¼ 0:092 0:14m0
m?
ch ¼ 0:202 3:422
1.65m0 at 300 K
[124]
[131]
[131]
[131]
[133]
[52]
[52]
[32,134,135]
[136]
[52]
[52]
[32,134,135]
[52]
[52]
[32,134,135]
[52]
[52]
[134,135]
73 at 300 K
[124]
Effective electron mass, me
Effective hole masses
(heavy), mh
Effective hole masses (light),
mlp
Effective hole masses
(split-off band), ms
Effective mass of density of
state, mv
Optical LO phonon
energy (meV)
The details of the energies of high symmetry points are given in Table 2.3. More details of effective
masses can be found in Table 2.19.
Table 1.22 Available parameters for mechanical for zinc blende InN.
Zinc blende InN
Value
Lattice constant
Density (g cm3)
Bulk modulus (GPa)
dB/dP
Youngs modulus (GPa), Y0 or E
a ¼ 4.98 Å
6.97
138–155, 145.6 [42]
Shear modulus (GPa)
Poissons ratio, n or s0
C11 (GPa)
C12 (GPa)
C44 (GPa)
187
125
86
Comments/references
Derived from X-ray data
Bs ¼ (C11 þ 2C12)/3
3.9–4.0
Y0 ¼ (C11 þ 2C12)
(C11 C12)/(C11 þ C12)
C ¼ (C11 C12)/2
n or s0 ¼ C12/(C11 þ C12)
See Ref. [42]
See Table 1.31 for details
j33
j 1 General Properties of Nitrides
34
Table 1.23 Available electrical and optical properties of zinc blende InN, primarily calculated.
Zinc blende InN
Value
Comments/references
Bandgap energy, Eg (300 K)
2.2 eV
In the absence of any
reliable data, the bandgap to a
first extent can be assumed to be
similar to that for Wz InN.
See Ref. [137] for a detailed
treatment
Dielectric constant
1.5–2.1 (theory)
0.2 eV below the
Wz polytype
8.4
2.88 0.30
2.90 0.30
2.90
3.05 0.30
2.65
6.97
3
LWL
==
ð2e?
0 þ e0 Þ
of wurtzitic
By
3
form (the spur)
Using Lyddane–Sachs–Teller
relation (e0 =ehigh ¼ w2LO =w2TO )
Theory
—
Transmission interference
Transmission interference
NIRSR
Derived from X-ray data
[52]
2.6
[52]
0.006
[84]
0.13m0
[52]
12.45
Dielectric constant (high frequency)
Refractive index at LWL
at 600–800 nm
at 900–1200 nm
at 900–1200 nm
at 620 nm
Density (g cm3)
Energy separation between
G and X c1 valleys EG (eV)
Energy separation between
G and L valleys EL (eV)
Spin–orbit splitting in valence
band, Dso or Eso (eV)
Effective electron mass, me
Effective hole masses (heavy)
Effective hole masses (light)
Effective hole masses
(split-off band), ms or mch or mso
[138]
½110
mhh
½100
mhh
½111
mhh
½110
mlh
½100
mlh
½111
mlh
¼ 2:12 m0
[52]
¼ 1:18 m0
¼ 2:89 m0
¼ 0:20 m0
[52]
¼ 0:21 m0
¼ 0:19 m0
0.36m0
[52]
LWL: long-wavelength limit; NIRSR: normal incidence reflectance of synchrotron radiation.
is in part a consequence of measurements being made on samples of widely varying
quality. For this book, when possible we have disregarded the spurious determination. However, measurements are too few to yield a consensus, in which case the
available data are simply reported.
1.2 Gallium Nitride
The burgeoning interest in nitrides has led to substantial improvements in the
crystal growth and processing technologies, thus overcoming many difficulties encountered earlier. Consequently, a number of laboratories consistently obtained highquality GaN with room-temperature background electron concentrations as low as
5 · 1016 cm3. The successful development of approaches leading to p-type GaN has led
to the demonstration of excellent p–n junction LEDs in the UV, violet, blue, green, and
even yellow bands of the visible spectrum with brightness suitable for outdoor displays,
CW lasers, and UV detectors, including the ones for the solar blind region. Moreover,
power modulation doped field effect transistors (MODFETs) also generically referred to
as heterojunction field effect transistors (HFETs) have been developed. What follows
reports on the state of knowledge regarding the physical properties of GaN.
1.2.1
Chemical Properties of GaN
Since Johnson et al. [139] first synthesized GaN in 1932, a large body of information
has repeatedly indicated that GaN is an exceedingly stable compound exhibiting
significant hardness. It is this chemical stability at elevated temperatures combined
with its hardness that has made GaN an attractive material for protective coatings.
Moreover, owing to its wide energy bandgap, it is also an excellent candidate for device
operation at high temperatures and caustic environments. Although the hardness may
have initiated the interest in GaN, it is the excellent semiconducting features that have
piqued the attention of researchers. While the thermal stability of GaN allows freedom
of high-temperature processing, the chemical stability of GaN presents a technological
challenge. Conventional wet etching techniques used in semiconductor processing
have not been as successful for GaN device fabrication. For example, Maruska and
Tietjen [140] reported that GaN is insoluble in H2O, acids, or bases at room
temperature, but does dissolve in hot alkali solutions at very slow rates. Pankove [141]
noted that GaN reacts with NaOH forming a GaOH layer on the surface and
prohibiting wet etching of GaN. To circumvent this difficulty, he developed an
electrolytic etching technique for GaN. Low-quality GaN has been etched at reasonably
high rates in NaOH [142,143], H2SO4 [144], and H3PO4 [145–147]. Although these
etches are extremely useful for identifying defects and estimating their densities in
GaN films, they are not as useful for the fabrication of devices [148]. Well-established
chemical etching processes do help for the device technology development, and the
status of these processes in the case of GaN can be found in Volume 2, Chapter 1.
Various dry etching processes reviewed by Mohammad et al. [149] and Pearton
et al. [150] are promising possibilities and are discussed in Volume 2, Chapter 1.
1.2.2
Mechanical Properties of GaN
GaN has a molecular weight of 83.7267 g mol1 in the hexagonal wurtzite structure. The
lattice constant of early samples of GaN showed a dependence on growth conditions,
impurity concentration, and film stoichiometry [151]. These observations were
j35
j 1 General Properties of Nitrides
36
attributed to a high concentration of interstitial and bulk extended defects. A case in
point is that the lattice constants of GaN grown with higher growth rates were found to
be larger. When doped heavily with Zn [152] and Mg [153], a lattice expansion occurs
because at high concentrations the group II element begins to occupy the lattice sites of
the much smaller nitrogen atom. At room temperature, the lattice parameters of GaN
platelets [18] prepared under high pressure at high temperatures with an electron
concentration of 5 · 1019 cm3 are a ¼ 3.1890 0.0003 Å and c ¼ 5.1864 0.0001 Å.
The freestanding GaN with electron concentration of about 1016 cm3, originally
grown on sapphire (0 0 0 1) by hydride vapor phase epitaxy (HVPE) followed by liftoff,
has lattice constants of a ¼ 3.2056 0.0002 Å and c ¼ 5.1949 0.0002 Å . For GaN
powder, a and c values are in the range of 3.1893–3.190 and 5.1851–5.190 Å,
respectively. Experimentally observed c/a ratio for GaN is 1.627, which compares
well with 1.633 for the ideal case, and the u parameter calculated using Equation 1.1 is
0.367, which is very close to the ideal value of 0.375.
For more established semiconductors with the extended defect concentration from
low to very low, such as Si, GaAs, and so on, the effect of doping and free electrons on
the lattice parameter has been investigated rather thoroughly. In bulk GaN grown by
the high-pressure technique, the lattice expansion by donors with their associated free
electrons has been investigated [18]. However, large concentration of defects and
strain, which could be inhomogeneous, rendered the studies of this kind less reliable
in GaN layers. In spite of this, the effect of Mg doping on the lattice parameter in thin
films of GaN has been investigated. Lattice parameters as large as 3.220–5.200 Å for a
and c values, respectively, albeit not in all samples with similar hole concentrations,
have been reported [154]. For GaN bulk crystals grown with high-pressure techniques
and heavily doped (a small percentage) with Mg, the a and c lattice parameters were
measured to be 3.2822–5.3602 Å [155]. Suggestions have been made that the c
parameter of implanted GaN layers increases after implantation and languishes after
annealing [156]. However, the a parameter could not be precisely measured because
sharp off-normal diffraction peaks are needed to determine this parameter accurately.
For the zinc blende polytype, the calculated lattice constant, based on the measured
GaN bond distance in Wz GaN, is a ¼ 4.503 Å. The measured value for this polytype
varies between 4.49 and 4.55 Å, while that in Ref. [18] is 4.511 Å, indicating that the
calculated result lies within the acceptable limits [157]. A high-pressure phase
transition from the Wz to the rock salt structure has been predicted and observed
experimentally. The transition point is 50 GPa and the experimental lattice constant
in the rock salt phase is a0 ¼ 4.22 Å. This is slightly different from the theoretical
result of a0 ¼ 4.098 Å obtained from first-principles nonlocal pseudopotential
calculations [158].
Tables 1.6 and 1.10 compile some of the known properties of Wz GaN. Parameters
associated with electrical and optical properties of Wz GaN are tabulated in Table 1.11.
The same parameters associated with the zinc blende phase of GaN are tabulated in
Table 1.12.
The bulk modulus of Wz GaN, which is the inverse of compressibility, is an
important material parameter. Various forms of X-ray diffraction with the sample
being under pressure can be used to determine the lattice parameters. Once the
1.2 Gallium Nitride
lattice parameters are determined as a function of pressure, the pressure dependence
of the unit cell volume can be obtained and fitted with an equation of state (EOS), such
as the Murnaghans EOS [159], and based on the assumption that the bulk modulus
has a linear dependence on the pressure:
0 ! 1=B
BP
1þ
;
B0
0
V ¼ V0
ð1:10Þ
where B0 and V0 represent the bulk modulus and the unit volume at ambient pressure,
respectively, and B0 the derivative of B0 versus pressure. X-ray diffraction leads to the
determination of the isothermal bulk modulus, whereas the Brillouin scattering
leads to the adiabatic one. Nevertheless, in solids other than molecular solids, there is
no measurable difference between the two thermodynamic quantities [160].
The bulk modulus (B) of Wz GaN has been calculated from first principles [161]
and the first-principle orthogonalized linear combination of atomic orbitals (LCAO)
method [158], leading to the values of 195 and 203 GPa, respectively. Another
estimate for B is 190 GPa [158]. These figures compare well with the value of
194.6 GPa estimated from the elastic stiffness coefficient [79] and a measured value
for 245 GPa [6].
The bulk modulus is related to the elastic constants through
B¼
ðC 11 þ C12 ÞC33 2C 213
C11 þ C12 þ 2C33 4C13
ð1:11Þ
and the range of bulk modulus values so determined is from about 173 to
245 GPa [160].
Using the room-temperature elastic constants of single-crystal GaN calculated by
Polian et al. [38] yields an adiabatic bulk modulus, both Voigt and Reuss averages, of
210 GPa [91].
Earlier experimental investigations of the elastic constants of Wz GaN were carried
out by Savastenko and Sheleg [162] using X-ray diffraction in powdered GaN crystals.
The estimates of the Poissons ratio from the early elastic coefficients (n ¼ C13/C11 þ
C12) [162] and its measured [26] values of 0.372 (for nh0 0 0 1i ¼ (Da/arelax)/(Dc/crelax))
and 0.378 (for nh0 0 0 1i ¼ (Da/a0)/(Dc/c0)), respectively, are in good agreement (to
avoid confusion the R value is defined as R ¼ 2C31/C33). The experiments were
performed on GaN layers on sapphire substrates because of X-ray diffraction.
However, the results obtained later point to a Poissons ratio of more near 0.2 as
tabulated in Table 1.6 and depend on crystalline direction. The Poissons ratio for the
ZB case can be calculated from the elastic coefficients for that polytype as n or
s0 ¼ (C12/C11 þ C12) leading to values of about 0.352 as tabulated in Table 1.8. The
Poissons ratio varies along different crystalline directions as tabulated in Table 1.13
for AlN. It should be noted that there is still some spread in the reported values of
elastic stiffness coefficients, as discussed in detail in the polarization sections of
Section 2.12. More importantly, Kisielowski et al. [39] pointed out that expression
ðn 1Þh0 0 0 1i ¼ ðDa=arelaz Þ=ðDc=c relax Þ;
ð1:12Þ
j37
j 1 General Properties of Nitrides
38
where Da ¼ ameas arelax and Dc ¼ cmeas crelax, should be used to calculated the
Poissons ratio, n. Doing so leads to a Poissons coefficient of nGaN ¼ 0.2–0.3.
Chetverikova et al. [163] measured the Youngs modulus and Poissons ratio of their
GaN films. From the elastic stiffness coefficients, Youngs modulus Eh0001i is
estimated to be 150 GPa [157,162]. Sherwin and Drummond [164] predicted the
elastic properties of ZB GaN on grounds of values for those Wz GaN samples
reported by Savastenko and Sheleg [162]. The elastic stiffness coefficients and the
bulk modulus are compiled in Table 1.24. Considering the wide spread in the
reported data more commonly used figures are also shown.
Wagner and Bechstedt [178] calculated the elastic coefficients of Wz GaN using a
pseudopotential plane wave method and pointed out the discrepancies among the
results from different calculations and measurements tabulated in Table 1.24. It is
argued that reliable values produce 2C13/C33 ¼ 0.50–0.56 and n ¼ 0.20–0.21 [178].
The agreement between ab initio calculations [42,178] and some measure-
Table 1.24 Experimental and calculated elastic coefficients (Cii),
bulk modulus (B) and its pressure derivative (dB/dP), and
Youngs modulus (E or Y0) and (in GPa) of Wz GaN and ZB GaN
(in part from Ref. [160]).
Technique
C11
C12
C13
C33
C44
B0
X-ray [162]
XAS
EDX
ADX
Brillouin [38]
Brillouin
Brillouin [166]
Brillouin [28]
Brillouin [169]
Brillouin [170]
Ultrasonic [171]
Ultrasonic [165]
Single crystal X-ray
Most commonly
used values
PWPP [42]
FP-LMTO [165]
Kim [168]
PWPP (Wagner)
ZB GaN
296
130
158
267
24.1
195
245
188
237
210
180
204
192
175
192
173
208
207
390
374
365
373
315
373
377
370
145
106
135
141
118
141
160
145
380
110
367
396
431
C11
253–264
135
144
109
515–C11
153–165
106
70
114
80.4
96
80
114
110
398
379
381
387
324
387
209
390
105
101
109
94
88
94
81.4
90
B0
E
150
4
3.2
4.3
356
329
362
281
362
161
343
105
103
100
64
104
405
392
476
414
95
91
126
60–68
202
207
201
207
200–237
363
355
461
373
3.9–4.3
The room-temperature elastic constants of single-crystal GaN have been determined by Polian
et al. [38] yielding an adiabatic bulk modulus, both Voigt and Reuss, averages, of 210 GPa. The
term B0 ¼ dB/dP represents the derivative of B0 versus pressure. EDX: energy dispersive X-ray;
ADX: angular dispersive X-ray diffraction; XAS: X-ray absorption spectroscopy; PWPP: plane
wave pseudopotential; FP-LMTO: full-potential linear muffin–tin orbital.
1.2 Gallium Nitride
ments [38,165,166] is satisfactory. However, several calculations [167,168] and measurements [28,169–171] suffer from deviations in one or more of the values of elastic
constants. The results from Savastenko and Sheleg [162] show excessive deviation for
all the elastic constants and, therefore, should be avoided completely. The results
from surface acoustic wave measurements of Deger et al. [165] on epitaxial epilayers
have been corrected for piezoelectric stiffening and, therefore, are among the most
reliable.
The vibrational properties of nitrides can best be described within the realm of
mechanical properties. These vibrations actually serve to polarize the unit cell [172].
Phonons can be discussed under mechanical and optical properties. Here an arbitrary
decision has been made to lump them with the mechanical properties of the crystal.
Using GaN as the default, a succinct discussion of vibrational modes, some of which
are active Raman modes, some are active in infrared (IR) measurements, and some are
optically inactive called the silent modes, is provided [173]. Vibrational modes, which
go to the heart of the mechanical properties, are very sensitive to crystalline defects,
strain, and dopant in that the phonon mode frequencies and their frequency broadening can be used to glean very crucial information about the semiconductor. The
method can also be applied to heterostructures and strained systems. Electronic
Raman measurements can be performed to study processes such as electron–phonon
interaction in the CW or time-resolved schemes. Time-resolved Raman measurements as applied to hot electron and phonon processes under high electric fields have
important implication regarding carrier velocities. A case in point regarding GaN is
treated in this context in Volume 3, Chapter 3.
The wurtzite crystal structure has the C46v symmetry and the group theory
predicts the existence of the zone center optical modes A1, 2B1, E1, and 2E2. In
a more simplified manner, one can consider that the stacking order of the Wz
polytype is AaBb while that for the ZB variety is AaBbCc. In addition, the unit cell
length of the cubic structure along [1 1 1] is equal to the width of one unit bilayer,
whereas that for the hexagonal structure along [0 0 0 1] is twice that amount.
Consequently, the phonon dispersion of the hexagonal structure along [0 0 0 1]
(G ! A in the Brillouin zone) is approximated by folding the phonon dispersion for
the ZB structure along the [1 1 1] (G ! L) direction [174], as shown in Figure 1.12.
Doing so reduces the TO phonon mode at the L point of the Brillouin zone in the
zinc blende structure to the E2 mode at the G point of the Brillouin zone in the
hexagonal structure. This vibrational mode is denoted as EH
2 with superscript H
depicting the higher frequency branch of the E2 phonon mode. As indicated in the
figure there is another E2 mode at a lower frequency labeled as EL2 . This has its
genesis in zone folding of the transverse acoustic (TA) mode in the zinc blende
structure. It should be noted that in the hexagonal structure there is anisotropy in
the macroscopic electric field induced by polar phonons. As a result, both the TO
and LO modes split into the axial (or A1) and planar (or E1) modes where atomic
displacement occurs along the c-axis or perpendicular to the c-axis, respectively.
This splitting is not shown in Figure 1.12 as it is very small, just a few meV, near
zone center; phonon dispersion curves for GaN including the splitting of the A1 and
E1 modes can be found in Volume 3, Figure 3.84.
j39
j 1 General Properties of Nitrides
40
Zinc blende
Wurtzitic
A1
LO
H
B1
H
TO
E1
A
[0001]
Phonon frequency
E2
Γ
L
B1
LA
L
E2
TA
Γ
A
[111]
L
Wave vector
Figure 1.12 Schematic depiction of the phonon
dispersion curves for ZB and Wz structures. Also
shown are the G and A points of the zone in
relation to the real space hexagonal structure.
Phonon branches along the [1 1 1] direction in
the ZB structure are folded to approximate those
of the wurtzite structure along the [0 0 0 1]
direction, because the unit cell length of the cubic
structure along the [1 1 1] direction is equal to the
width of one unit bilayer, while that for the
hexagonal structure along the [0 0 0 1] directions
is twice that amount. Patterned after Ref. [174].
As discussed below, in the context of hexagonal structures, group theory predicts
eight sets of phonon normal modes at the G point, namely 2A1 þ 2E1 þ 2B1 þ 2E2.
Among them, one set of A1 and E1 modes are acoustic, while the remaining six
modes, namely A1 þ E1 þ 2B1 þ 2E2, are optical modes. As shown in Figure 1.12, one
A1 and one B1 mode (BH
1 ) derive from a singly degenerate LO phonon branch of the
zinc blende system by zone folding, whereas one E1 and one E2 mode (EH
2 ) derive
from a doubly degenerate TO mode in the cubic system.
The first-order phonon Raman scattering is due to phonons near the G point zone
center, that is, with wave vector k 0, because of the momentum conservation rule in
the light scattering process. Raman measurements typically are employed to probe
the vibrational properties of semiconductors. When performed along the direction
perpendicular to the c-axis or the (0 0 0 1) plane, the nomenclature used to describe
this configuration is depicted as ZðXY; XYÞZ. Here, following Portos notation [175]
A(B, C)D is used to describe the Raman geometry and polarization, where A and D
1.2 Gallium Nitride
represent the wave vector direction of the incoming and scattered light, respectively,
whereas B and C represent the polarization of the incoming and scattered light. In
Raman scattering, all the above-mentioned modes, with the exception of B1 modes,
are optically active. Because of their polar nature, the A1 and E1 modes split into
longitudinal optical (A1-LO and E1-LO) meaning beating along the c-axis, and
transverse optical (A1-TO and E1-TO), meaning beating along the basal plane. To
reiterate, the A1 and B1 modes give atomic displacements along the c-axis, while the
others, E1 and E2, give atomic displacements perpendicular to the c-axis, meaning on
the basal plane. Here, the A1 and E1 modes are both Raman and IR active whereas the
two E2 modes are only Raman active and the two B1 modes are neither Raman nor IR
active, meaning silent modes. In the ZðX Y; X YÞZ configuration, only the E12 (or EL2 or
E2 low), E22 (or EH
2 or E2 high), and A1(LO) modes should be observable. In particular,
in ZðX ; XÞZ and ZðY; YÞZ geometries, all three modes are observable, while in
ZðX; YÞZ or ZðY; XÞZ geometries only E2 modes are detected [175]. The details of the
mode–Raman configuration relationship are tabulated in Table 1.25. Shown in
Figure 1.13 are the modes in the Raman backscattered geometries in relation to
hexagonal crystalline orientation that can be used to sense the various phonon modes
indicated.
The acoustic modes, which are simple translational modes, and the optical modes
for wurtzite symmetry are shown in Figure 1.14. The calculated phonon dispersion
curves [57] for GaN are shown in Figure 1.15. There is another way to describe the
number of vibrational modes in zinc blende and wurtzitic structures, which is again
based on symmetry arguments. In the wurtzite case [66], the number of atoms per
unit cell s ¼ 4, and there are total of 12 modes, the details of which are tabulated in
Table 1.26. This table also holds for the zinc blende polytypes with s ¼ 2. This implies
a total of six modes in zinc blende as opposed to 12 in wurtzite, three of which are
acoustical (1 LA and 2 TA) and the other three are optical (1 LO and 2 TO) branches.
These phonon modes for a wurtzite symmetry, specifically the values for wurtzite
GaN, are listed in Table 1.27 obtained from Refs [56,157,176,177] along with those
obtained from first-principles pseudopotential calculations [161,178]. Also listed are
TO and LO optical phonon wave numbers of ZB GaN [25,179].
Table 1.25 Raman measurement configuration needed to observe
the phonon modes in hexagonal nitrides.
Mode
Configuration
A1(TO), E2
A1(TO)
E1(TO)
E1(TO), E1(LO)
E2
E2
A1(LO), E2
X ðY; YÞX
X ðZ; ZÞX
X ðZ; YÞX
X ðY; ZÞY
X ðY; YÞZ
ZðY; X ÞZ
ZðY; YÞZ
j41
j 1 General Properties of Nitrides
42
Z(X,X)Z + Z(X,Y)Z
X(Z,Z)X
A1 (TO)
H
E2
X(Z,Y)X
A1 (TO)
L
E2
A1(LO)
Quasi-E1 (LO)
H
E2
Y
Y
Z
X
GaN
Substrate
GaN
Z
Substrate
c-axis
X
c-axis
Figure 1.13 Schematic representation of two Raman
configurations with incident and scattered light directions in the
backscattering geometry for ZðX ; X ÞZ þ ZðX; YÞZ configuration
to sense EL2 , EH
2 , and A1(LO) modes, X ðZ; ZÞX configuration
to sense A1(TO) and quasi-E1(LO) modes, and X ðZ; YÞX
configuration to sense A1(TO) and EH
2 modes.
Owing to the presence of elastic strain, thin epilayers have different phonon
energies compared to the bulk samples. In the general case, strain can give rise to a
shift and splitting of phonon modes. However, for uniaxial strain along the c-axis or
biaxial strain in the c-plane, the crystal retains its hexagonal symmetry resulting in
only a shift of the phonon frequencies. The strain effects on GaN optical phonon
energies have been studied experimentally [181] and theoretically [178]. Within a
perturbative approach, the change in the frequency of a given phonon l under
symmetry-conserving stress can be expressed in terms of the two strain components,
exx and ezz, representing perpendicular and parallel to the z-axis, respectively, as
DOl ¼ 2 alexx þ blezz, where al and bl are the corresponding deformation potential
constants. The derivation of strain values from the Raman measurements of phonon
frequencies is straightforward, once the phonon deformation potentials are known.
Davydov et al. [181] combined high-resolution X-ray diffraction (HRXRD) measurements and Raman spectroscopy results to determine the phonon deformation
potentials in GaN epitaxial layers grown on 6H-SiC. The strain components were
obtained by comparing the lattice constants of the epitaxial layers with those of the
strain-free GaN (a0 ¼ 3.1880 Å, c0 ¼ 5.18 561 Å). The Raman spectrum of a strain-free
300 mm thick GaN layer grown on sapphire was used as a reference. Also, using their
relation to the hydrostatic pressure coefficients [6] through the bulk elastic coefficients, the sets of phonon deformation potentials were derived for most of zone
center optical phonons. As seen in Table 1.27, except for the A1-TO mode, the phonon
energies and the deformation potentials reported by Davydov et al. [181] agree
well with the pseudopotential plane wave calculations reported by Wagner and
Bechstedt [178]. Wagner and Bechstedt argue that the published conversion factors
1.2 Gallium Nitride
A 1 mode
Z
Z
Z
B 1 (1) mode
N
N
N
Ga
Ga
Ga
N
N
N
Ga
Ga
Ga
X
X
Z
B 1 (2) mode
Z
E 1 mode
X
Z
E 2 (1) mode
N
E 2 (2) mode
N
N
Ga
Ga
Ga
N
N
Ga
N
Ga
Ga
X
(a)
X
X
A mode
1
L
Z
Z
B1H
Z
B1
N
N
N
Ga
Ga
Ga
N
N
N
Ga
Ga
Ga
X
X
L
E2
Z
Z
X
H
E2
N
N
Ga
Ga
N
N
Ga
(b)
X
Figure 1.14 Atomic vibrations in Wz GaN. The larger atom
represents Ga while the smaller one is for N. X ¼ (1 0 0),
Y ¼ (0 1 0), and Z ¼ (0 0 1) represent the optical polarization
directions: (a) for general wave vector and (b) for zone center
phonons.
Ga
X
j43
j 1 General Properties of Nitrides
44
800
100
E1(LO)
E1 (LO)
GaN
700 B
1
B1 A1(LO)
600 E2
E2 E (TO)
1
A1(TO)
A1(TO)
500
60
400
B1
B1
Energy (meV)
Frequency (cm–1)
80
40
300
200
E2
E2
20
100
0
0
Γ
K
M
Γ
A
H
L
A
DOS
Reduced wave vector coordinate
Figure 1.15 Calculated phonon dispersion curves and phonon
density of states for hexagonal bulk GaN. The solid and dashed
lines correspond to the L1(or T1) and L2 (or T2) irreducible
representations (following Ref. [180]), respectively. Note how
close the E1(LO) and A1(LO) modes are, making high-quality
samples with sharp modes imperative for their experimental
delineation [169].
Table 1.26 Acoustic and optical phonon modes in a crystal with
wurtzite symmetry such as GaN, AlN, and InN, where s represents
the number of atoms in the basis.
Mode type
Number of modes
LA
TA
Total acoustic modes
LO
TO
All optical modes
All modes
1
2
3
s1
2s 2
3s 3
3s
The s parameter for wurtzite symmetry is 4. This table is also applicable to the zinc blende case but
with s ¼ 2.
Wz [157,177]
556–559
533–534
741–741
710–735
143–146
560–579
Phonon mode
E1-TO
A1-TO
E1-LO
A1-LO
EL2
EH
2
BL1
BH
1
(a)
558.5
532.5
745.0
737.0
Wz
template [177]
558.8
531.8
741
734
144
567.6
[169]
Wz relaxed
568
540
757
748
142
576
337
713
Wz unstrained
(calculated) [178]
bk
680 50
1290 80
80 35
920 60
ak
820 25
630 40
115 25
850 25
Raman [181]
717
640
775
664
75
742
334
661
ak
Calculated [178]
Deformation potentials – Wz
by Akasaki and Amano [157] (Wz) and Huang et al. [177] (Wz template). Also shown are the calculated values. (b) Measured phonon
wave numbers (in units of cm1) for wurtzitic GaN. (c) Zinc blende phase phonon wave numbers for zinc blende GaN [179] (theory [59]).
Table 1.27 (a) Zone center optical phonon wave numbers (in units of cm 1) of GaN obtained from Raman scattering at 300 K compiled
591
695
703
695
4
715
275
941
bk
1.2 Gallium Nitride
j45
GaN on sapphire, about 50–70 mm thick, at 300 K.
LO(L) (cm1)
TO(L) (cm1)
LA(L) (cm1)
TA(L) (cm1)
TO (cm1)
LO (cm1)
LO(G) (cm1)
TO(G) (cm1)
LO(X) (cm1)
TO(X) (cm1)
LA(X) (cm1)
TA(X) (cm1)
748 [59]
562 [59]
639 [59]
558 [59]
286 [59]
207 [59]
Mode
Mode
(c)
E1-TO
A1-TO
E1-LO
A1-LO
EL2
EH
2
BL1
BH
1
Phonon mode
(b)
Table 1.27 (Continued)
675 [59]
554 [59]
296 [59]
144 [59]
558 [179]
730 [179]
558.8
531.8
741
741
144
567.6
560 [58]
750 [58]
Wz unstrained (measured) (collected in
Refs [174,178], but based on Refs [6,169])
46
j 1 General Properties of Nitrides
1.2 Gallium Nitride
between the luminescence or Raman shifts and the corresponding biaxial stress are
seldom directly measured data. They are either obtained using elastic constants or are
constructed from deformation potentials, which have been obtained by means of
additional hydrostatic pressure coefficients. Owing to these varying procedures and
different sets of parameters used to extract the conversion coefficients from the raw
experimental data, discrepancies in the experimental reports of deformation potentials are present.
1.2.3
Thermal Properties of GaN
The lattice parameter of semiconductors depends on temperature and is quantified
by thermal expansion coefficient (TEC), which is defined as Da/a or aa and Dc/c or ac,
for in-plane and out-of-plane configurations, respectively. It depends on stoichiometry, extended defects, and free-carrier concentration. As in the case of the lattice
parameter, a scatter exists in TEC particularly for nitrides as they are grown on foreign
substrates with different thermal and mechanical properties. Measurements
made over the temperature range of 300–900 K indicate the mean coefficient
of thermal expansion of GaN in the c-plane to be Da/a ¼ aa ¼ 5.59 · 106 K1.
Similarly, measurements over the temperature ranges of 300–700 and 700–900 K,
respectively, indicate the mean coefficient of thermal expansion in the c-direction
to be Dc/c ¼ ac ¼ 3.17 · 106 and 7.75 · 106 K1, respectively [140]. Sheleg and
Savastenko [49] reported a TEC near 600 K for perpendicular and parallel to the
c-axis of 4.52 0.5 · 106 and 5.25 0.05 · 106 K1, respectively. Leszczynski and
Walker [182] reported aa values of 3.1 and 6.2 106 K1 for the temperature ranges of
300–350 and 700–750 K, respectively. The ac values in the same temperature ranges,
in order, were 2.8 and 6.1 106 K1.
In a similar vein, GaN and other allied group III nitride semiconductors are grown
at high temperatures and also subjected to increased junction temperatures during
operation of devices such as amplifiers and light emitting devices. As such, the
structures are subjected to thermal variations as well. In this context, it is imperative
to have knowledge of the thermal expansion coefficients, which are termed as TEC.
Assuming that these figure remain the same with temperature, the linear expansion
coefficients for the a and c parameters are tabulated in Tables 1.10 and 1.28 for
heteroepitaxial GaN. However, it is instructive to know the temperature dependence
of these parameters, which is shown in Figure 1.16.
Being grown on various substrates with different thermal expansion coefficients
leads to different dependencies of the lattice parameter on temperature. Temperature
dependence of GaN lattice parameter has been measured for a bulk crystal (grown at
high pressure) with a high free-electron concentration (5 · 1019 cm3), a slightly
strained homoepitaxial layer with a low free-electron concentration (about 1017 cm3),
and a heteroepitaxial layer (also with a small electron concentration) on sapphire [88].
The results of such study are tabulated in Table 1.28.
It can be seen that the bulk sample with a high free-electron concentration exhibits
a thermal expansion that is about 3% higher as compared to the homoepitaxial layer.
j47
j 1 General Properties of Nitrides
48
7
Linear expansion coefficent, a (10–6 K–1)
GaN
6
a
5
a
4
3
2
200
100
300
400
500
600
700
800
Temperature (K)
Figure 1.16 Wz GaN coefficient of linear thermal expansion
versus temperature for basal plane (a||), a parameter, and out of
the basal plane (a?), c parameter, directions [49].
As for the case of the heteroepitaxial layer on sapphire, the thermal expansion of the
substrate affects the dependence of the lattice parameter on temperature.
Various spectroscopic techniques, such as Auger electron spectroscopy, X-ray
photoemission spectroscopy (XPS), and electron energy loss spectroscopy (EELS)
have been very useful for the study of surface chemistry of GaN. Building on earlier
investigations of the thermal stability of GaN by Johnson et al. [139] and employing
Table 1.28 Lattice parameters for GaN samples at various
temperatures (lattice parameters c were measured with accuracy
of 0.0002 Å, lattice parameter a with accuracy of 0.0005 Å) [88].
T (K)
20
77
295
500
770
GaN bulk,
n ¼ 5 · 1019 cm3
Homoepitaxial GaN
on conductive GaN
substrate
GaN on sapphire
c (Å)
a (Å)
c (Å)
a (Å)
c (Å)
a (Å)
5.1836
5.1838
5.1860
5.1885
5.1962
3.1867
3.1868
3.1881
3.1903
3.1945
5.1822
5.1824
5.1844
5.1870
5.1944
3.1867
3.1868
3.1881
3.1903
3.1945
5.1846
5.1865
5.1888
5.1952
3.1842
3.1859
3.1886
3.1941
1.2 Gallium Nitride
the aforementioned techniques, the thermal stability and dissociation of GaN have
been examined further. As indicated earlier, the materials characteristics depend, to a
large extent, on defects and impurities, which in turn depend somewhat on growth
conditions. Because of this the materials, obtained from various sources, studied in
various laboratories exhibit different characteristics. This led to inconsistent results
from different laboratories. While some experimental studies on GaN stability
conducted at high temperatures suggested that significant weight losses occur at
temperatures as low as 750 C, others contradicted this proposition and suggested
that no significant weight loss should occur even at a temperature of 1000 C. Sime
and Margrave [183] followed the investigation by Johnson et al. [139] by studying the
evaporation of GaN and Ga metal in the temperature range 900–1150 C under
atmospheric pressure in N2, NH3, and H2 environments with an emphasis on the
formation and decomposition equilibrium.
The heat of evaporation was determined and the existence of (GaN)x polymers in the
gas phase was suggested. Morimoto [184] and Furtado and Jacob [185] observed that
GaN is less stable in an HCl or H2 atmosphere than in N2. Some controversy exists
regarding the process steps that dictate the decomposition of GaN. Using mass
spectroscopy, Gordienko et al. [186] noted that (GaN)2 dimers are the primary
components of decomposition. Others [187,188] found only N2 þ and Ga þ to be the
primary components in the vapor over GaN. On the basis of measurements of the
apparent vapor pressure, Munir and Searcy [189] calculated the heat of sublimation of
GaN to be 72.4 0.5 kcal mol 1. Thurmond and Logan [190] determined the equilibrium N2 pressure of GaN as a function of temperature by measuring the partial
pressure ratios existing in a (H2CNH3) gas mixture streaming over Ga and GaN.
Thermal stability of GaN was taken up later by Karpinski et al. [191] with a detailed
investigation of the problem at high temperatures and under pressure up to 60 kbar by
employing a tungsten carbide anvil cell activated by a gas pressure technique.
The bond strength in gallium nitride is high with bonding energy of
9.12 eV/molecule [192], particularly as compared to the more conventional semiconductors such as GaAs, which has a bonding energy of 6.5 eV/atom pair. As a result,
the free energy of GaN is very low in relation to the reference states of the free N and
Ga atoms. However, the N2 molecule is also strongly bonded with 4.9 eV/atom.
Therefore, the free energies of the constituents of GaN (Ga and N2) at their normal
states are close to that of the GaN crystal as illustrated in Figure 1.17, where the free
energy of GaN (1 mol) and the free energy of the sum of its constituents (Ga þ 1/2N2)
are shown as a function of temperature and N2 pressure. As the temperature
increases, the Gibbs free energy, G(T), of the constituents decreases faster than G
(T) of the GaN crystal. More importantly, GaN becomes thermodynamically unstable
at high temperatures. The crossing of G(T) curves determines the equilibrium
temperature where GaN coexists with its constituents at a given N2 pressure. The
application of pressure increases the free energy of the constituents more than G(T)
of the GaN crystal, which causes the equilibrium point to shift to higher temperatures, increasing the range of GaN stability.
The data on phase diagrams of GaN are limited and contradictory by reason of high
melting temperatures (Tm) and high nitrogen dissociation pressures (Pdis
N2 ). Dissociation
j49
j 1 General Properties of Nitrides
50
–600
Ga(l)+(1/2)N2(g)
Ga(s) +
(1/2)N2(g)
p = 1 bar
p = 1 kbar
G(kJ mol–1)
p = 10 kbar
–800
p = 20 kbar
GaN(s)
–1000
–1200
0
400
800
1200
1600
2000
T(K)
Figure 1.17 Gibbs free energy of GaN and its constituents as a
function of temperature and pressure [192].
pressure of MN, where M stands for Al, Ga, and In, and N for nitrogen, is defined
as the nitrogen pressure at the thermal equilibrium of the reaction [193]: MN(s)
¼ M(l) þ 1/2N2(g), where s, l, and g stand for solid, liquid, and gas states, respectively. Reported values for Pdis
N2 for GaN [193] show large discrepancies [191,194].
Specifically, in the high-pressure range, the partial pressure, p, versus the inverse
temperature, 1/T, curve of Karpinski et al. deviates markedly from the linear
dependence proposed by Thurmond and Logan as shown in Figure 1.18. Despite
the discrepancies, there is a good agreement in the Gibbs free energy with DG0
32.43T 3.77 · 104 700 cal mol1 for GaN synthesis between the two references.
The value of enthalpy DH0 (37.7 kcal mol1) is in good agreement as well with that
estimated by Madar et al. [194].
The stars in Figure 1.18 indicate the melting point of AlN at T M
AlN ¼ 3487 K, GaN
M
at T M
GaN ¼ 2791 K, and InN at T InN ¼ 2146 K. The GaN and InN melting points
so indicated may underestimate the real values, as perhaps a sufficient overpressure
was not maintained. Line fits correspond to 8.3 109 exp( 5.41 eV/kT), 1.5 · 1014
exp(3.28 eV/kT), and 7.9 · 1017 exp(2.78 eV/kT) bar for AlN, GaN, and InN,
respectively. The data over the larger temperature range are those compiled by
Ambacher [196]. The results of Madar et al. [194], Thurmond and Logan [190], and
Karpinski et al. [191] are also shown in a limited temperature range. For GaN (see
Figure 1.18), the nitrogen dissociation pressure equals 1 atm at approximately 850 C
and 10 atm at 930 C. At 1250 C, GaN decomposed even under pressure of 10 000 bar
of N2. The turning over of the partial pressure for GaN and InN at temperatures
approaching the melting point may need to be reexamined. What is clear, however, is
that GaN and particularly InN have very high partial pressures that make it imperative
to maintain high fluxes of N during growth. It should, therefore, come as no surprise
that the incorporation of nitrogen is not a trivial problem at high temperatures. For
the pressures below equilibrium at a given temperature, the thermal dissociation
occurs at a slow and apparently constant rate suggesting a diffusion-controlled
1.2 Gallium Nitride
Temperature (oC)
105
4700
1400
440
700
104
u
Th
103
rm
InN
on
Lo
2
n
ga
101
Ma
dis
d
an
PN (atm)
d
102
dar
et a
l.
AlN
Ka
100
n
rpi
et
ski
GaN
al.
10–1
10–2
10–3
2
4
6
10
8
104 /T ( K–1)
Figure 1.18 Equilibrium N2 pressure over the
MN(s) þ M(l) systems corresponding to GaN
(M ¼ Al, Ga, In) reported and/or compiled by
Slack and McNelly[195], Madar et al. [194],
Thurmond and Logan [190], Karpinski et al. [191],
and Ambacher [196]. The melting points of the
three binaries are indicated by stars. The
desorption activation energies, EMN, determined
by straight line fits to the data points are 3.5 eV
12
14
(336 kJ mol1), 3.9 eV (379 kJ mol1), and 4.3 eV
(414 kJ mol1) for InN, GaN, and AlN,
respectively. Caution should be exercised as
there is significant deviation from the activation
line for GaN and InN. This may simply be a
matter of not being able to maintain sufficient
pressure on GaN and InN at very high
temperatures to reach the real melting point.
process of dissociation. Expanded equilibrium vapor pressure data inclusive of GaAs
and InP in addition to the three nitride binaries reported by Matsuoka [197] are shown
in Figure 1.19. Melting points and other thermodynamic characteristics of III-N
compounds are given in Tables 1.10, 1.16, and 1.20 as compiled by Popovici and
Morkoc [198] as well as those collected from various sources as indicated in the
pertinent tables.
Investigations utilizing epitaxial thin films of GaN, as well as AlN and InN, have
been conducted by Ambacher et al. [199], who heated the samples in vacuum and
recorded the partial pressure of relevant gases with a quadrupole mass spectrometer.
Desorption spectra were then analyzed [200] to find the binding energies of various
desorbed species as well as the thermal stability of the sample for a given thermal
treatment. As expected, the nitrogen partial pressure increases exponentially above
TE ¼ 850 C for GaN underscoring the point that the decomposition temperature in
vacuum is much lower than the melting point shown in Figure 1.18. The rate of
j51
j 1 General Properties of Nitrides
52
Temperature ( oC)
Equilibrium vapor pressure (atm)
2000
1000
500
100
InN
10–2
10–4
GaN
AlN
GaAs
10–6
InP
10–8
0.0
0.5
1.0
1.5
2.0
1000/T (K–1)
Figure 1.19 Equilibrium vapor pressure of N2 over AlN, GaN
and InN, the sum of As2 and As4 over GaAs, and the sum of P2
and P4 over InP [197].
nitrogen evolution F(N) was set equal to the rate of decomposition, and the slope of ln
[F(N)] versus 1/T gives the effective activation energy of the decomposition in
vacuum as compared to those shown in Figure 1.18. The decomposition rate equals
the desorption of one monolayer every second (FN ¼ 1.5 · 1015 cm2 s1) at 970 C,
and the activation energy of the thermally induced decomposition is determined to be
EMN ¼ 3.9 eV (379 kJ mol1) for GaN.
Despite some disagreement, as mentioned above, investigations of the equilibrium
nitrogen overpressure versus temperature, PN2 –T, for GaN [92,190,201], including a
very complete and consistent set of data obtained by Karpinski et al. [191,202] have set
the stage for bulk template growth as well as setting benchmarks for growth of GaN by
nonequilibrium methods. Those authors employed direct synthesis and decomposition experiments and used the gas pressure technique (for pressures up to 20 kbar)
and the high-pressure anvil method beyond the reach of gas pressure technique
(up to 70 kbar). The results of these experiments are shown in Figure 1.20 [203,204].
The message in the form of N2 partial pressure is that one must stay below the
decomposition curve. This means that the selection of GaN synthesis temperature
directly depends on the pressure that the vessel can provide. For example, if a pressure
of 20 kbar is all that is available, then the temperature should be kept below about
1660 C. For a review of the stability of GaN as well as the growth GaN templates, the
reader is referred to Ref. [192].
As alluded to earlier, nitride semiconductors in general and GaN in particular are
considered for high-power/high-temperature electronic and optoelectronic devices
1.2 Gallium Nitride
Temperature (K)
3
2 × 103 1.5 × 103 1.25 × 10
105
1×10 3
Pressure limit
of 20 kbar
Pressure (bar)
104
103
GaN
Ga+1/2N 2
102
101
100
4
6
8
10
12
1/T (10 –4 K –1)
Figure 1.20 N2 partial pressure as a function of temperature GaN.
Ref. [192], originally in Refs [203,204].
where thermal dissipation is a key issue. Device applications assure that the thermal
conductivity (k) is an extremely important material property. Thermal conductivity is a
kinetic property determined by contributions from the vibrational, rotational, and
electronic degrees of freedom, and as such it is related to the mechanical properties of
the material. However, for convenience, this property is generally categorized under
the thermal properties of nitrides in this book. The electronic thermal conductivity
contribution is negligible for carrier concentrations 1019 cm3. The heat transport is
predominantly determined by phonon–phonon Umklapp scattering, and phonon
scattering by point and extended defects such as vacancies (inclusive of the lattice
distortions caused by them), impurities, and isotope fluctuations (mass fluctuation) as
elaborated on by Slack et al. [91]. For pure crystals, phonon–phonon scattering, which
is ideally proportional to T1 above the Debye temperature, is the limiting process.
The lattice contribution (phonon scattering) to the thermal conductivity, k, in a
pure solid is obtained from the kinetic theory as [205]
1
k lattice ðTÞ ¼ vs C lattice ðTÞLðTÞ;
3
ð1:13Þ
where T is the temperature, vs is the velocity of sound (nearly independent of
temperature), Clattice(T) is the lattice specific heat, and L(T) is the phonon mean free
length. In nearly all materials, the thermal conductivity, k(T), first increases with
temperature, reaches a maximum (kmax) at some characteristic temperature Tch, and
j53
j 1 General Properties of Nitrides
54
then decreases. At low temperatures, L is relatively long and is dominated by extrinsic
effects such as defects and/or finite crystal size and Clattice(T) (T/yD)3, where yD is
the Debye temperature. As the temperature increases, Clattice(T) begins to saturate
and the intrinsic temperature-dependent Umklapp processes become dominant,
leading to a decrease in L.
The other contribution, the electronic contribution, to the thermal conductivity is
negligible for carrier concentrations 1019 cm3. It can be expressed as [206]
k electr ðTÞ ¼
p2 nk2B Ttelectr
;
3mc
ð1:14Þ
where n is the carrier density, kB is the Boltzmann constant, telectr is the scattering
time of the electrons, and mc is the conduction band effective mass.
The first measurements of k of GaN were by Sichel and Pankove [207] on bulk
GaN (400 mm of material grown by HVPE) as a function of temperature (25–360 K):
k ffi 1.3 W cm1 K1 (along the c-axis) at 300 K. This room-temperature value measured is a little smaller than the value of 1.7 W cm1 K1 predicted in 1973 [77] and
much smaller than the k 4.10 W cm1 K1 calculated by Witek [208]. Using the
elastic constants reported by Polian et al. [38], Slack et al. [91] calculated a Debye
temperature of 650 K, which led to a more recent thermal conductivity at 300 K for
GaN of k ¼ 2.27 W cm1 K1, assuming that there is no isotope scattering in GaN.
This is very close to the measured values in high-quality freestanding GaN samples,
the electrical properties of which are discussed in Volume 2, Chapter 3 and optical
properties of which are discussed in Volume 2, Chapter 5.
Using a steady-state four-probe method and a high-quality freestanding GaN
template, Slack et al. [91] measured a value for k of 2.3 W cm1 K1 at room
temperature, which increased to over 10 W cm1 K1 at 77 K. The method holds
for four-probe thermal measurement, where the term four probe is analogous to
the four-probe electrical measurement method. Namely, a heater is attached on the
end of the sample – sandwiched in a copper clamp so that the heat flows through the
entire width of the sample, not just the surface –supplying a heat current Q
(analogous to an electrical current I). Two thermocouple junctions are attached
along the length of the specimen by two little copper clamps separated by a distance L,
the schematic representation of which is shown in Figure 1.21.
The heat current Q creates a temperature gradient of DT across the wafer. The k
value is calculated using k ¼ (P/DT)(L/A), where P is the power (¼voltage · current)
supplied to the heater and A represents the cross-sectional area of the sample.
Although the technique sounds simple, its accuracy depends very critically on
making sure that the heat conduction is through the specimen and along the
direction in which the temperature gradient is measured. To make certain that heat
is transferred in the said direction only, the radiation losses must be minimized as
well as making sure that the electrical wires used do not remove heat. To this end, the
sample is placed in a turbo pumped vacuum to eliminate conduction and convection
through the surrounding medium. Heat losses via conduction through the wires are
minimized using long (10 cm), thin (<100 mm) wires of low thermal conductivity,
typically chromel/constantan thermocouple and heater wires. Radiation losses are
1.2 Gallium Nitride
j55
Variable temperature
copper finger
Chromel
Thermocouple wires
Thermocouple 2
Constantan
Chromel
Δ
ΔT
Thermocouple 1
low
t f
a
He
Copper
Heater
resistor
le
mp
Sa
r te
de
un
st
Thermocouple for
absolute temperature
measurement
Figure 1.21 Schematic representation of the four-probe
thermoelectric measurement setup used to measure the thermal
conductivity of freestanding GaN [91].
minimized by surrounding the sample with a heat shield anchored thermally to the
cold tip of the cryostat. By carefully designing this shield, Slack et al. [91] were able to
reduce the total heat loss to the order of 1–2 mW K1 at room temperature. Because
the radiation losses follow T3 dependence, they die off rather quickly below room
temperature. For a sample with a thermal conductivity of 1 W cm1 K1, cross section
of 1 · 3 mm2, and thermocouple probe separation of 5 mm, the thermal conductance
is about 60 mW K1, so the heat losses are less than 5%. Fortunately, for wide
bandgap semiconductors such as GaN the thermal conductivities are high enough so
that the heat conduction is mainly through the sample, reducing the measurement
error. Just as a reference point, for samples of lower thermal conductance (either
lower conductivity or thinner), the heat losses can become important near room
temperature. Thus, the samples for lower thermal conductivity materials (e.g.,
glasses or thermoelectric alloys) usually need to be short with large cross-sectional
areas. The temperature-dependent thermal conductivity so measured for freestanding GaN is shown in Figure 1.22. From that temperature dependence and assuming
the heat dissipation is through acoustic phonons, a Debye temperature of yD 550 K
was deduced, which compares with 650 K reported by Slack et al. [91].
As can be seen in Figure 1.22, the measured thermal conductivity of GaN in the
temperature range of 80–300 K has a temperature power dependence of 1.22. This
slope is typical of pure adamantine crystals below the Debye temperature indicating
acoustic phonon transport where the phonon–phonon scattering is a combination of
j 1 General Properties of Nitrides
56
Thermal conductivity (W cm–1 K –1)
10
2
T –1.22
101
500 μm phonon
mean free path
100
Sichel and Pankove
10
–1
1
10
100
1000
Temperature (K)
Figure 1.22 The thermal conductivity of 200 mm thick freestanding
GaN sample (Samsung) as a function of temperature. The dashed
line indicates calculation using the boundary scattering limit for a
phonon mean free path of 500 mm. Also shown is the
T 1.22dependence between about 80 and 300 K, and earlier results
of Sichel and Pankove [207] measured using a 400 mm HVPE
sample. Courtesy of Slack and Morelli [91].
acoustic–acoustic and acoustic–optic interactions. This temperature dependence
strongly suggests that the thermal conductivity depends mainly on intrinsic phonon–phonon scattering and not on phonon–impurity scattering. Keep in mind that
the net electron concentration in the measured film is about 1016 cm3 and the hole
concentration is in the 1015 cm3 range. In addition, the dislocation density is low,
about 106 cm2. It should be pointed out that the thermal conductivity degrades with
increased dislocation density, particularly above 107 cm2, with a slope of 0.4 W
cm1 K1 per decade dropping down to slightly above 1 W cm1 K1 for a dislocation
density of mid-109 cm2. The dashed curve at low temperatures in Figure 1.22 has
been calculated for boundary scattering assuming a mean free path of 500 mm [91].
The mean free path is comparable with the average sample diameter, which indicates
that impurity scattering in this region is not dominant either.
The thermal conductivity of GaN is a tensor quantity and has two principal values
k? and k|| perpendicular and parallel to the c-axis, respectively, that is, in-plane and
out-of-plane values. The anisotropy in the sound velocity, which relates to the phonon
propagation velocity, has been calculated by Polian et al. [38] in the form of n? ¼ 5.56
· 105 cm s1 and n|| ¼ 5.51 · 105 cm s1 for in-plane and out-of-plane directions,
respectively. These values are smaller than the measured values, reported by
Deger et al. [165], of n? ¼ 8.02 · 105 cm s1 and n|| ¼ 7.79 · 105 cm s1 for in-plane
1.2 Gallium Nitride
and out-of-plane directions, respectively. The in-plane and out-of-plane sound
velocities reported in Ref [36] are tabulated in Tables 1.7 and 1.9 for wurtzitic and
zinc blende phases of GaN. Because the difference is negligible and the anharmonicity producing the phonon–phonon scattering is not discernible, one can conclude
that in-plane thermal conductivity measurements are a good representative of k in
GaN [91].
A newer method, named the scanning thermal microscopy (SThM) [209], has been
developed to measure thermal conductivity and is purported to provide nondestructive, absolute measurements with a high spatial/depth resolution of about 2–3 mm.
Thermal imaging is achieved using a resistive thermal element incorporated at the end
of a cantilever/AFM-type feedback as shown in Figure 1.23. The resistive tip forms one
element of a Wheatstone bridge as shown in Figure 1.24. The spatial/depth resolution
is estimated to be 2–3 mm for GaN and AlN. Upon contact with the sample, the tip
tends to cooldown due to heat conduction into the sample, which is related to its
thermal conductivity, k. The bridge circuit applies a compensating voltage (Uout) to
maintain its target operating temperature. The feedback signal for constant resistance
is a measure of the thermal conductivity of the material with which the tip is in contact,
specifically V 2out is proportional to k because power dissipation is the mechanism here.
Measurements of the absolute values of k are based on a calibration procedure. This
simply comprises calibrating the feedback signal, V 2out , for a constant thermal element
resistance against that for samples with known conductivities such as GaSb, GaAs,
InP, Si, and Al metal, as shown in Figure 1.25. The influence of the surface roughness
on the effective thermal conductivity is of concern. For a perfectly flat surface,
Figure 1.23 An artists view of the scanning thermal microscope.
Patterned after D.I. Florescu and F.H. Pollak. (Please find a color
version of this figure on the color tables.)
j57
j 1 General Properties of Nitrides
58
2
Vout ~ κ
R1
Tip
R2
Vout
R probe
Sample
R control
(a)
(b)
Figure 1.24 (a) Wheatstone bridge arrangement
in which the tip temperature is kept constant
before and after contact with the material whose
thermal conductivity is being measured. The
feedback signal Uout is related to thermal
conductivity, k. A calibration against known
samples such as Si, GaAs, GaP, and so on, leads
to absolute values of k. (b) Schematic diagram of
heat dissipation into the sample from the tip.
Courtesy of D.I. Florescu and F.H. Pollak.
the contact between the probe tip (radius of curvature 1 fm) and the sample surface
is very small. For rough surfaces, however, the tip could impinge on a valley- or
hillocklike feature with the valley/hillock leading to increased/decreased thermal
signal accompanied by a corresponding change in the measured effective thermal
conductivity.
0.68
Al
0.66
V 2out
0.64
0.62
0.60
Si
0.58
InP
0.56
GaAs
0.54
0.0
GaSb
0.5
1.0
1.5
2.0
–
1 –1
k (W cm K )
2.5
Figure 1.25 The feedback signal, V 2out, which is a measure of the
thermal conductivity of the material under test, for a constant
thermal element resistance for samples with known conductivities
such as GaSb, GaAs, InP, Si, and Al metal. Courtesy of D.I.
Florescu and F.H. Pollak.
1.2 Gallium Nitride
The SThM method has been applied to the measurement of the room-temperature
thermal conductivity on both fully and partially coalesced epitaxial lateral overgrown
GaN/sapphire (0 0 0 1) samples [209]. As expected, a correlation between low threading dislocation density and high thermal conductivity values was established. The
reduction in the thermal conductivity with increased dislocation density is expected
as threading dislocations degrade the sound velocity and increase the phonon
scattering in the material. In fact, due to the high defect concentrations in early
films, the thermal conductivity value measured was 1.3 W cm1 K1 [207]. Using this
method, the highest GaN k values, 2.0–2.1 W cm1 K1, were found in the regions of
the samples that were laterally grown and thus contained the lowest density of
threading dislocations. This compares with a value of 2.3 W cm1 K1 in a freestanding sample measured by the steady-state four-probe method discussed earlier. Even
then, it falls short of the predictions by Witek [208].
An explanation for the dramatic increase from to k 1.3 W cm1 K1 for the
early samples to 2.3 W cm1 K1for the freestanding sample, as iterated above is
most likely related to extended defect concentration (Dd) and the differences in
background doping. The effect of dislocation density on the thermal conductivity
has been calculated by Kotchetkov et al. [210]. The dislocation density in the thick
film measured by Sichel and Pankove was between 109 and 1010 cm2, while the
freestanding sample exhibited densities of less than 106 cm2 near the top surface
(Ga-polarity) and 107 cm2 near the bottom surface (N-polarity). Kotchetkov et al.
showed that k remains fairly independent of Dd up to some characteristic
after which it decreases about a factor of 2 for every decade of
value Dchar
d
increase in Dd.
The thermal conductivity has also been correlated to doping levels in HVPE
n-GaN/sapphire (0 0 0 1) by SThM on two sets of samples [211,212]. In both sets of
data, the thermal conductivity decreased linearly with log n, n being the electron
concentration, the variation being about a factor of 2 decrease in k for every decade
increase in n. Significantly, it was concluded that the decrease in the lattice
contribution to k, due to increased phonon scattering from impurities and free
electrons, predominates the increase in the electronic contribution. Also, a correlation between the film thickness and the improved thermal conductivity was found,
which is consistent with the observed general reduction of both extended (dislocations) and point defects with film thickness [212].
The k values at 300 K before and after plasma-induced effects on a series of n-GaN/
sapphire (0 0 0 1) samples fabricated by HVPE were also measured [213]. The sample
thicknesses were 50 5 mm and the carrier concentrations were 8 · 1016 cm3, as
determined by Hall effect measurements. The thermal conductivity before treatment
was found to be in the 1.70–1.75 W cm1 K1 range, similar to that previously
reported for HVPE material with this carrier concentration and thickness [211,212].
The k value was reduced, however, when the samples were processed under constant
Ar gas flow and pressure for a fixed period of time (5 min). The only variable
processing parameter was the DC bias voltage (125–500 V). After the initial 125 V
procedure, k exhibited a linear decrease with the DC voltage in the investigated range.
At 125 V, the thermal conductivity was only slightly less (k 1.65 W cm1 K1)
j59
j 1 General Properties of Nitrides
60
than the untreated case. The values of k had dropped to 0.3 W cm1 K1 for the
500 V case.
To a first extent, the temperature dependence of the specific heat of Wz GaN (Cp) at
constant pressure can be expressed by phenomenological expression [51]. In this
vein, the specific heat Cp of Wz GaN at constant pressure for 298 K < T < 1773 K can
be expressed as
CP ðTÞ ¼ 9:1 þ ð2:14 10 3 TÞðJ mol 1 K 1 Þ;
Cp ¼ 38:1 þ 8:96 10 3 T ðcal mol 1 K 1 Þ ð1 cal ¼ 4:186 JÞ:
ð1:15Þ
However, this expression is very simplistic, as will be seen below. As already
mentioned, free electrons (very effective at low temperatures), impurities, defects
(inclusive of point defects), and lattice vibrations contribute to specific heat. If GaN
with negligible free-electron concentration and defects were available, only the lattice
contribution would be considered, which is also the case in texts [214]. The specific
heat of Wz GaN has been studied by Koshchenko et al. [215] in the temperature range
of 5–60 K and also by Demidienko et al. [216] in the temperature range of 55–300 K
and discussed by Krukowski et al. [88,127]. The Debye expression for the temperature
dependence of specific heat in a solid at a constant pressure (Cp) can be expressed
as [214]
T
Cp ¼ 18R
qD
3 xðD
:
0
x 4 ex
ð ex
1Þ2
dx;
ð1:16Þ
where x D qD =T and R ¼ 8.3144 J mol1 K1 is the molar gas constant. The coefficient in front of the term R has been multiplied by 2 to take into account the two
constituents making up the binary GaN. By fitting the measured temperature-dependent heat capacity to the Debye expression, one can obtain the Debye temperature yD
specific to heat capacity. The experimental data of Demidienko et al. and Krukowski et
al. are plotted in Figure 1.26. Also shown in the figure is the calculated specific heat
using the Debye expression for Debye temperatures of 500, 600, 700, and 800 K. It is
clear that the quality of the data and/or sample prevents attainment of a good fit
between the experimental data and Equation 1.16. Consequently, a Debye temperature
with sufficient accuracy cannot be determined. It is easier to extract a Debye temperature using data either near very low temperatures or well below the Debye temperature
where the specific heat has a simple cubic dependence of temperature [214]:
3
T
:
ð1:17Þ
Cp ¼ 234R
qD
Unfortunately, the GaN samples contain large densities of free carriers and defects
that compromise the application of the Debye specific heat expression. Consequently,
a good fit to the data is not obtained and the Debye temperature so extracted is not as
dependable as desired.
There is a spread in the reported Debye temperatures for GaN, yD, in the range of
about 600 to 700 K. Slack [77] estimated a value of 600 K at 0 K by utilizing the more
established Debye temperatures for BeO and AlN. This compares with 550 K deduced
1.2 Gallium Nitride
50
q D = 800 K
40
–1
–1
Molar specific heat, Cp (cal mol K )
q D = 500 K
30
20
Cp data, GaN
q D = 500 K
q D = 600 K
10
q D = 700 K
q D = 800 K
0
0
200
400
600
800
1000
Temperature (K)
Figure 1.26 Molar specific heat at constant
pressure, Cp (cal mol1 K1), of GaN versus
temperature. Open circles represent the
experimental data. The solid lines are calculation
based on the Debye model for Debye
temperatures, yD, of 500, 600, 700, and 800 K.
Unfortunately, it is difficult to discern a Debye
temperature that is effective over a wide
temperature range because a large concentration
of defects and impurities is present in GaN.
However, a value of 600 K estimated by Slack is
used commonly. The data are taken from
Refs [215,216], as compiled in Ref. [88]. (Please
find a color version of this figure on the color
tables.)
from heat transfer due to acoustic phonons, as mentioned above. Because the samples
used in these measurements contained defects and large density of free electrons, the
dispersion among the data and Debye expression is attributed to defects at high
temperatures and free electrons at low temperatures. Elastic properties of GaN can also
be used to deduce the Debye temperature. In this vein, Raman scattering measurements yielded a Debye temperature of yD ¼ 650 K [38]. Calculations since the estimate
of Slack [77] yielded a range of 620–690 K [88].
Thermodynamic properties of Wz GaN have been reported by Elwell and
Elwell [217]. From the reaction
GaðsÞ þ 1=2N2 ðgÞ ¼ GaNðsÞ;
ð1:18Þ
the heat of formation of Wz GaN was calculated to be DH298 K ¼ 26.4 kcal mol1 [217],
or as the standard heat of formation DH ¼ 37.7 kcal mol1 [194]. The equilibrium
vapor pressure of N2 over solid GaN has been found to be 10 MPa at 1368 K and 1 GPa
at 1803 K [202]. A thorough description of the GaN phase diagram including the
equilibrium vapor pressure of N2 over GaN as well as AlN and InN has been
presented by Porowski and Grzegory [218] (1 cal ¼ 4.186 J).
j61
j 1 General Properties of Nitrides
62
1.3
Aluminum Nitride
AlN exhibits many useful mechanical and electronic properties. For example,
hardness, high thermal conductivity, resistance to high temperature and caustic
chemicals combined with, in noncrystalline form, a reasonable thermal match to Si
and GaAs in somewhat relaxed terms, make AlN an attractive material for
electronic packaging applications. The wide bandgap is also the reason for AlN to
be touted as an insulating material in semiconductor device applications. Piezoelectric properties make AlN suitable for surface acoustic wave device applications [219].
However, the majority of interest in this semiconductor in the context of electronic
and optoelectronic device arena stems from its ability to form alloys with GaN
producing AlGaN and allowing the fabrication of AlGaN/GaN and AlGaN/
InGaN-based electronic and optical devices, the latter of which is active from the
green wavelengths well to the ultraviolet. AlN also forms a crucial component of the
nitride-based AlInGaN quaternary, which makes tuning of the bandgap independent
of composition to some extent. This way, lattice-matched conditions to the underlying
epitaxial structure can be maintained while being able to adjust the bandgap.
AlN is not a particularly easy material to investigate because of the high reactivity of
aluminum with oxygen in the growth vessel. Early measurements indicated that
oxygen contaminated material can lead to errors in the energy bandgap and,
depending on the extent of contamination, in the lattice constant. Only recently
achieved contamination-free deposition environments coupled with advanced procedures have allowed researchers to consistently grow improved-quality AlN. Consequently, many of the physical properties of AlN have been reliably measured and
bulk AlN synthesized.
1.3.1
Mechanical Properties of AlN
When crystallized in the hexagonal wurtzite structure, the AlN crystal has a molar
mass of 40.9882 g mol1, restated for convenience. The cubic form is hard to obtain
and thus will be ignored. The point group symmetry for the wurtzite structure in the
Schoenflies notation is C46v (P63mc in the Hermann–Mauguin notation), restated for
convenience. Reported lattice parameters range from 3.110 to 3.113 Å for the a
parameter (3.1106 Å for bulk, 3.1130 Å for powder, and 3.110 Å for AlN on SiC), and
from 4.978 to 4.982 Å for the c parameter. The c/a ratio thus varies between 1.600 and
1.602. The deviation from that of the ideal wurtzite crystal (c/a ¼ 1.633) is probably
because of the lattice stability and ionicity. The u parameter for AlN is 0.3821, which is
larger than the calculated value of 0.380 using Equation 1.1. This means that the
interatomic distance and angles differ by 0.01 Å and 3 , respectively, from the
ideal [16].
Whereas the metastable zinc blende polytype AlN has a value of a ¼ 4.38 Å [220],
the rock salt structure has a value of a ¼ 4.043–4.045 Å at room temperature [5,221].
Table 1.13 summarizes some of the observed structural properties of AlN.
1.3 Aluminum Nitride
Early investigations of the elastic properties of AlN were carried out on sintered
polycrystalline specimens, owing to the unavailability of large single crystals. This,
however, paved the way to more refined measurements as single crystalline AlN
became available. The measured bulk modulus B, which is related to elastic stiffness
coefficients through Equation 1.11, and Youngs modulus Y0 or E are compiled in
Table 1.29 along with the entire set of elastic stiffness coefficients. The latter were
obtained by fitting the results of surface acoustic wave measurements made on
epitaxial AlN films and by Brillouin scattering measurements made on an AlN single
crystal [160]. Wagner and Bechstedt [178] suggested that reliable values for the elastic
constants should produce 2C13/C33 ¼ 0.5–0.6 and n ¼ 0.18–0.21. The ab initio
calculations [42,178] and some measurements [83,165] provide similar results.
However, the values from some of the calculations [165,222] and measurements [169,223] should be used with caution because of the large deviations in one
or more coefficients. Surface acoustic wave measurements of Deger et al. [165] are
very reliable because they include the correction for piezoelectric stiffening. Bulk
modulus values range from 159.9 GPa, measured by an ultrasonic method, to
237 GPa, measured by Brillouin scattering. The range for the same from calculations
is 111–239 GPa [160]. Youngs modulus is measured as 374 GPa for single-crystal
AlN [82] and 295 GPa for AlN thin films [224].
The hardness of AlN has been measured to be 12 GPa on the basal plane (0 0 0 1)
using a Knoop diamond indenter [225]. Some anisotropy in Knoops hardness has been
observed for the indent direction perpendicular to the c-axis with measured values in
Table 1.29 Experimental bulk modulus and elastic coefficients (in
GPa) of AlN (from Ref. [160] and references therein).
Method
C11
C12
C13
C33
C44
B
Ultrasonics [223]
Ultrasonic
Ultrasonic [165]
ADX
Brillouin [83]
Brillouin [169]
EDX
Hardness [82]
PWPP [42]
FP-LMTO [165]
HF [222]
PWPP [178]
345
125
120
395
118
201
159.9
209
207.9
210.1
237
185
220
207
218
231
210
Zinc blende
HF [222]
410
140
100
390
120
410.5
419
148.5
177
98.9
140
388.5
392
124.6
110
396
398
464
C11
FIX THIS
348
137
140
149
538
168
108
127
116
113
373
382
409
370
116
96
128
135
228
B0
E or Y0
5.2
334
308
354
6.3
354
326
5.7
374
329
322
365
322
Y0 ¼ (C11 þ 2C12) ·
(C11 C12)/
(C11 þ C12)
ADX: angular dispersive X-ray diffraction; EDX: energy dispersive X-ray; PWPP: plane wave
pseudopotential; FP-LMTO: full-potential linear muffin–tin orbital; HF: Hartree–Fock.
j63
j 1 General Properties of Nitrides
64
the range of 10–14 GPa [81]. More recent nanoindentation measurements on singlecrystal AlN revealed a hardness of 18 GPa [82].
The phonon structure of AlN has been the subject of numerous investigations. As
in the case of GaN, the phonon dispersion spectrum of Wz AlN has 12 branches,
3 acoustic, and 9 optical ones [226]. LO and TO phonon energies have been obtained
from fits to infrared reflectivity measurements, the results of which are tabulated in
Table 1.30. Raman-active optical phonon modes belong to the A1, E1, and E2 group
representations. Several Raman scattering studies on AlN have been conducted, and
the measured phonon energies are listed in Table 1.30 along with the calculated
values by Wagner and Bechstedt [178]. The shift of phonon energies with strain was
studied experimentally by Gleize et al. [227] and theoretically by Wagner and
Bechstedt [178]. Gleize et al. [227] investigated strained 500 nm thick AlN layers
grown on 6H-SiC. Using the strain values deduced from high-resolution X-ray
diffraction and phonon frequency shifts measured by micro-Raman spectroscopy,
the deformation potentials were obtained for most of the zone center optical phonons
of Wz AlN. The determination of the deformation potentials is based on the
knowledge of the ideal equilibrium state of the material from which strains and
phonon shifts are defined. Additionally, hydrostatic pressure coefficients and elastic
constants of the bulk material are also needed for extracting the deformation
potentials from the raw experimental data. The deviations in the published data
originate from the fact that different sets of parameters are typically used for this
purpose. Table 1.30 lists the phonon deformation potentials from Gleize et al. [227]
along with the results from pseudopotential plane wave calculations of Wagner and
Bechstedt [178], which produced slightly lower values.
The frequencies or energies of vibrational modes are very sensitive to the strain
state of the samples. Strain inhomogeneities and imperfections cause the linewidths
in Raman observable mode to be broad reducing the accuracy of central frequency
determination. Tischler and Freitas Jr [228] utilized freestanding and high-quality
AlN oriented along the (0 0 0 1) plane, as characterized by X-ray with full width half
maximum (FWHM) of 36–54 arcsec. Listed in Table 1.30 are also the data of Tischler
and Freitas Jr, which by virtue of the high quality of sample should be used as
standard. Tischler and Freitas Jr [228] also estimated the linewidths of the Raman
modes by fitting the data with Lorentzian peaks from which the phonon decay times
were deducedby relying on theuncertainty principle in the form of DE=
h ¼ G=
h ¼ 1=t,
where DE, G, and t represent the error bar for the energy and linewidths, and the
phonon decay time, respectively. The phonon decay times so deduced are in the
range of 0.7 ps for E1(LO) to 5.3 ps for E12.
To get a flavor of the range of experiments regarding the mechanical properties,
including vibrational phonon properties, of epitaxial AlN, a mention of the investigations on AlN deposited on silicon and sapphire substrates at 325 K by IBAD
undertaken by Ribeiro et al. [230] is made. Raman scattering measurements revealed
interesting vibrational features related to the atomic composition and structure of the
films. Features related to crystalline (c-) Si and corresponding to 2TA(L) at 230
cm1, 2TA(X) at 304 cm1, 2TA(S) at 435 cm1, TO(G) at 520 cm1, TA(S) þ TO
(S) at 615 cm1, accidental critical points at 670 and 825 cm1, 2TO(D) at
E1-TO
A1-TO
A1-LO
E1-LO
E12 ðE2 lowÞ
E22 ðE2 highÞ
Mode
82.8
75.4
75.4
112.8
30.5
81.2
(meV)
667.2
608.5
888.9
909.6
246.1
655.1
(cm 1)
Wz (Raman)
unstrained [228]
895–921
667–673
614–667
Ref. [157]
246
655
668
608
890
Ref. [229]
Wz (Raman range) (cm1)
Table 1.30 Optical phonon energies and phonon deformation potentials for AlN.
924
677
618
Wz unstrained
(calculated)
(cm 1) [178]
bk
901 145
904 163
ak
982 83
930 94
Raman [227]
744
394
808
835
776
867
bk
Calculated [178]
ak
Deformation potentials
1.3 Aluminum Nitride
j65
j 1 General Properties of Nitrides
66
1000
1000
800
800
600
600
400
400
200
200
0
Frequency(cm –1 )
Frequency(cm–1)
AlN
0
Γ
K
M
Γ
A
DOS
Figure 1.27 Phonon dispersion curves and phonon density of
states for Wz AlN reported in Ref. [234]. The data points have their
roots in Ref. [83] and are discussed and compared with the ab
initio calculations in Ref. [233].
950 cm1, 2TO(L) at 980 cm1, 2TO(G) at 1085 cm1, 2TA(X) þ 2TO (G) at
1300 cm1, and 3TO(G) at 1450 cm1 have been observed. A very narrow peak
seen at 2330 cm1 has been attributed to molecular nitrogen on the surface of
c-Si [231]. It is worth noting that because of the extremely weak Raman signal usually
presented by low quality AlN films, some of the previously reported features have
been erroneously identified [232]. Misidentification of some vibration modes could
lead to incorrect interpretations of the crystalline quality of AlN films.
As in the case of GaN, the acoustic modes are simple translational modes, whereas
the optical modes for wurtzite symmetry are more complex as shown in Figure 1.12.
The calculated phonon dispersion curves [233,234] along with the phonon density of
states for wurtzitic AlN are shown in Figure 1.27.
1.3.2
Thermal and Chemical Properties of AlN
Single crystalline forms of this compound either in the epitaxial form or bulk form
represent the focus of this treatment. In its most commonly available form, AlN is an
extremely hard ceramic material with a melting point higher than 2000 C. In single
crystalline form, the melting of AlN was measured to be 2750–2850 C at nitrogen
pressures of 100 and 200 atm (or bar) [235]. The melting temperatures for various
nitrides were also determined by Van Vechten [236], who made use of a semiempirical theory for electronegativity and concluded that the melting point of AlN is close to
3487 K. Slack and McNelly [195] calculated the N2 equilibrium pressures over liquid
Al to be 1, 10, and 100 bar at 2563, 2815, and 3117 C, respectively, as shown in
Figure 1.18 in the context of the GaN discussion.
1.3 Aluminum Nitride
6.00
AlN
Δa/a
Δ (×10 3 )
Δa/a, Δc/c
Δ
5.00
Δc/c
4.00
3.00
2.00
1.00
0.00
200
400
600
800
T (K)
1000
1200
1400
Figure 1.28 Variation of the thermal expansion coefficient of AlN
with temperature in the c-plane and in the c-direction [86,87,225].
Using X-ray techniques across a broad temperature range (77–1269 K), it was noted
by Slack and Bartram [85] that the thermal expansion of AlN is isotropic with a roomtemperature value of 2.56 · 106 K1. The thermal expansion coefficients of
AlN measured by Yim and Paff [237] have mean values of Da/a ¼ 4.2 · 106 K1
and Dc/c ¼ 5.3 106 K1. The dependence of the thermal expansion coefficient on
temperature in the c-plane and in the c-direction is shown in Figure 1.28, which can be
fitted by the following polynomials (for 293 < T < 1700 K):
Da=a0 ¼ 8:679 10 2 þ 1:929 10 4 T þ 3:400
10 7 T 2 7:969 10 11 T 3
ð1:19Þ
and
Dc=c 0 ¼ 7:006 10 2 þ 1:583 10 4 T þ 2:719
10 7 T 2 5:834 10 11 T 3 ;
ð1:20Þ
where a0 and c0 represent the 300 K lattice parameters. For AlN powder, Krukowski
et al. [88] reported the expansion coefficient for the a parameter to be 2.9 · 106 K1,
and the same for the c parameter to be 3.4 · 106 K1.
The specific heat of AlN has been discussed extensively. Mah et al. [238] approximated the specific heat Cp of AlN in the temperature interval 298–1800 K as
Cp ¼ 45:94 þ 3:347 10 3 T 14:98
10 5 T 2 J mol 1 K 1
ð1 cal ¼ 4:186 JÞ:
ð1:21Þ
j67
j 1 General Properties of Nitrides
68
The same for the higher temperature range of 1800–2700 K was approximated by
Glushko et al. [239] as
Cp ¼ 37:34 þ 7:86 10 3 T J mol 1 K 1
ð1 cal ¼ 4:186 JÞ
ð1:22Þ
relying on the following points: the specific heat Cp ¼ 51.5 J mol1 K1 at T ¼ 1800 K,
and an estimated value, Cp ¼ 58.6 J mol1 K1 at T ¼ 2700 K, as outlined by
Krukowski et al. [88].
Specific heat obtained from the above approximations coupled with the measured
values for a constant pressure from the literature has been tabulated [88], a plot of
which is shown in Figure 1.29 along with the calculated specific heat using
Equation 1.16 for Debye temperature values of 800–1100 K with 50 K increments.
The best fit between the data and the Debye specific heat expression for insulators
50
Specific heat, Cp (J mol–1 K–1)
40
30
20
Specific heat
AlN (J mol–1 K–1)
800 K
850 K
900 K
950 K
1000 K
1050 K
1100 K
10
0
0
200
400
600
800
1000
Temperature (K)
Figure 1.29 Molar specific heat at constant
pressure, Cp (J mol1 K1, 1 cal ¼ 4.186 J), of AlN
versus temperature. Open circles represent the
experimental data. The solid lines are calculation
based on the Debye model for Debye
temperatures, yD, in the range of 800–1100 K
with 50 K increments. The data can be fit with
Debye expression for yD ¼ 1000 K, which
compares with 950 K reported by Slack et al. The
data are taken from Ref. [88]. (Please find a color
version of this figure on the color tables.)
1.3 Aluminum Nitride
indicates a Debye temperature of 1000 K, which is in good agreement with 950 K
reported by Slack et al. [240]. Compared to the GaN figure, the Debye temperature so
obtained for AlN appears more dependable owing to a much better fit.
The equilibrium N2 vapor pressure above AlN is relatively low compared to that
above GaN that makes AlN easier to be synthesized. The calculated temperatures at
which the equilibrium N2 pressure reaches 1, 10, and 100 atm are 2836, 3088, and
3390 K, respectively [195]. Details of the thermodynamic properties of AlN can be
found in Refs [87,88].
Similar to GaN, albeit to a lesser extent, AlN exhibits inertness to many chemical
etches. A number of AlN etches have been reported in the literature. However,
molten salts such as KOH or NaOH at elevated temperatures such as 50–100 C,
lower than what is required for GaN by as much as 200 C, etch AlN at appreciable
rates. The surface chemistry of AlN has been investigated by numerous techniques
including Auger electron spectroscopy, XPS, ultraviolet photoemission spectroscopy
(UPS), ultraviolet photoelectron spectroscopy, and electron spectroscopy. One of
these investigations by Slack and McNelly [195] indicated that the AlN surface grows
an oxide 50–100 Å thick when exposed to ambient air for about a day. However, this
oxide layer was protective and resisted further decomposition of the AlN samples.
Details can be found in Refs [87,157].
The thermal conductivity k of AlN at room temperature has been predicted as
k ¼ 3.19 W cm1 K1 [77,241]. Values of k measured at 300 K are 2.5 [241] and
2.85 W cm1 K1 [85]. The predicted values are near 3.2 W cm1 K1 in an O-free
simulated material but are based on measurements in AlN containing O [77]. A more
recent prediction for the value in AlN is 5.4 W cm1 K1, which is much larger than
any measured value [208]. The measured thermal conductivity as a function of
temperature in bulk AlN containing some amount of O is plotted in Figure 1.30a and
b. Also shown is a series of samples with estimated concentrations of O showing an
overall reduction in the thermal conductivity with O contamination. The calculation
results of Slack et al. for impurity- and defect-free AlN are shown as well. In the
temperature range of interest where many of the devices would operate, the thermal
conductivity in the sample containing the least amount of O assumes a T 1.25
dependence.
In freestanding and 300–800 mm thick AlN samples grown by HVPE, originally on
Si(1 1 1) substrates, values in the range of 3.0–3.3 W cm1 K1 were measured by the
SThM method [242]. The dislocation density in these freestanding AlN templates was
about 108 cm2 [243].
1.3.3
Electrical Properties of AlN
Owing to the low intrinsic carrier concentration and the deep native defect and
impurity energy levels, the electrical characterization of AlN has usually been limited
to resistivity measurements. One such measurement by Kawabe and coworkers [244]
on transparent AlN single crystals yielded resistivities r ¼ 1011–1013 O cm, a value
consistent with other reports [245–247]. However, it was found that impure crystals,
j69
j 1 General Properties of Nitrides
70
103
"Pure AlN"-Theor
Thermal conductivity (W cm–1 K–1)
AlN
4 x 1019 cm–3
102
5 x 1019 cm–3
2 x 10 20 cm–3
3 x 10 20 cm–3
101
100
(J/mol-1 K-1)
10 –1
10 –2
10 –3
10 –1
(a)
10 0
10 1
10 2
Temperature (K)
10 3
10 4
102
"Pure AlN"
19
Thermalconductivity(W cm–1 K–1)
4 x 10
19
5 x 10
20
2 x 10
20
3 x 10
–3
cm
–3
cm
–3
cm
101
100
10
(b)
–3
cm
100
1000
Temperature (K)
Figure 1.30 (a) Thermal conductivity of singlecrystal AlN. The solid line alone indicates the
theory whereas the others represent
measurements of AlN with various
concentrations of O. The lower the O
concentration, the higher the thermal
conductivity. The theoretical data are from
Ref. [77], the data for the lowest O concentration
are from Ref. [241], and the data for the other
samples are from Ref. [91]. (b) The thermal
conductivity of AlN in a limited temperature
range of common interest that underscores the
detrimental effect of O on the thermal
conductivity [240].
1.3 Aluminum Nitride
which exhibited a bluish color possibly because of the presence of Al2OC, have
resistivities of r ¼ 103–105 O cm, much lower than those reported by Chu et al. [248],
who were purportedly able to obtain both n- and p-type AlN by introducing Hg and Se,
respectively. However, they failed to determine the net carrier concentrations owing
to very high resistivities. The n-AlN films grown by Rutz [249] had a quite low
resistivity (r ¼ 103 O cm), which is comparable to those of Kawabe et al. [81]. Although
Rutz [249] did not determine the source of the electrons, Rutz et al. [250] observed
an interesting transition in their AlN films in which the resistivity abruptly
decreased by two orders of magnitude with an increase in the applied bias. This
observation found applications to switchable resistive memory elements that are
operated at 20 MHz.
The insulating nature of these early films hindered meaningful studies of their
electrical transport properties. With the availability of refined growth techniques,
AlN, presently grown with much improved crystal quality, shows both n- and p-type
conductions. This has rejuvenated efforts to measure both electron and hole Hall
mobilities. Edwards et al. [251] and Kawabe et al. [252] carried out some Hall
measurements in p-type AlN producing a very rough estimate of the hole mobility
mp ¼ 14 cm2 V1 s1 at 290 K. The predictions for the Hall mobility in the entire range
of AlGaN alloy including the binary end points are treated in Volume 2, Chapter 3.
Not all the parameters needed for the calculations are known precisely, reducing the
confidence in predicted values somewhat. As in the case for GaN, the roomtemperature mobility is dominated by the polar optical phonon scattering.
1.3.4
Brief Optical Properties of AlN
Investigations of the optical absorption coefficient, a, of AlN at room temperature
were reviewed as early as 1976 by Slack and McNelly [195]. Harris and Youngman [253] have reviewed photoluminescence and cathodoluminescence characteristics of AlN. Because the AlN lattice has a very large affinity to oxygen, it is almost
impossible to eliminate oxygen contamination in AlN that affects observations.
Commercially available AlN powder is said to contain about 1–1.5 at.% oxygen. Some
oxygen is dissolved in the AlN lattice, with the remainder forming an oxide coating on
the surface of each powder grain.
After irradiation with ultraviolet light, AlN doped with oxygen was found to emit a
series of broad luminescence bands at near-ultraviolet frequencies at room temperature no matter whether the sample was powdered, single crystal, or sintered
ceramic. Pacesova and Jastrabik [254] observed two broad luminescence lines
centered in the vicinity of 3.0 and 4.2 eV and more than 0.5 eV wide for
samples contaminated at about 1 to 1.5 at.% oxygen. Youngman and Harris [255]
and Harris et al. [256] investigated the luminescence characteristics of polycrystalline-sintered AlN samples and noted a continuous shift of the peak position in the
ultraviolet luminescence line as a function of oxygen content up to a critical
concentration of about 0.75 at.%. The luminescence lines beyond this limit of
oxygen concentration remained stationary.
j71
j 1 General Properties of Nitrides
72
Epitaxial layers of about 1 mm or less in thickness on sapphire substrates led to
observation of donor bound excitons with phonon replicas and some deep emission [257]. Cathodoluminescence spectra obtained in reactive MBE (RMBE), using
ammonia as the nitrogen source, show a sharp band edge peak, which is tentatively
assigned to the optical recombination of a donor bound exciton (D0, X), sometimes
accompanied by weak one and two longitudinal optical phonon replicas. Also
observed is a broad band with maximum in the range 320–370 nm, although some
variation of this low-energy peak has been reported. The 6 K CL spectrum of the near
band edge region shows a sharp and strong peak at 2068 Å in addition to two weak
peaks on its low-energy side, which are not observed for all samples. The symmetric
[0 0 0 2] X-ray diffraction peak is narrow and the linewidth of the prominent near band
edge CL peak is 23 meV.
Bulk single crystalline AlN has also been investigated for its CL emission properties. In addition to the near band edge emission at about 6 eV, the ultraviolet oxygen
luminescence peak at 4.0 eV was also observed. In the investigation of Youngman and
Harris [255], 4.64 eV photons were used to specifically explore the below the band
transitions such as the peak near 4 eV. That investigation showed a blue shift with
increased O concentration in the peak under question. With the availability of highquality AlN substrates, presumably high-quality epitaxial AlN layers have been grown
on them and characterized for their optical properties by CL at 5 K [107]. In this
particular work, the CL measurements were carried out at different temperatures for
a fixed electron beam energy density value. The beam current was held at 5 mA and the
voltage at 10 kV. The energy density was about 700 W cm2. Some six emission lines
between the energies of 5.98 and 6.03 eV have been delineated in the lowtemperature near-band edge emission spectra of those AlN films. In addition to
unidentified neutral donor and possibly acceptor bound exciton lines, free exciton A
and its excited state, and free exciton B were observed. The availability of A exciton
ground and excited states led to a binding energy of 63 meV that gives a lowtemperature bandgap of 6.086 eV for this material. The provisionally accepted value
for the bandgap of this material is 6.2 eV (which is questionable now that the lower
value is supported by measurements performed in homoepitaxial layers).
In terms of absorption, Yim et al. [246] characterized AlN by optical absorption
determining the room-temperature bandgap to be direct with a value of 6.2 eV. It
should be pointed out that this early figure is not consistent with measurements
performed later on using high-quality samples as discussed in the previous paragraph.
Perry and Rutz [258] performed temperature-dependent optical absorption and
determined a bandgap of 6.28 eV at 5 K compared to their room-temperature value
of 6.2 0.1 eV. We should point again that it has been lowered to slightly over 6 eV at
low temperatures in high-quality samples. Several groups have reported comparable
values whereas others have produced questionable values considerably below 6.2 eV,
probably due to oxygen contamination or nonstoichiometry. In addition to the band
edge absorption, a much lower energy absorption peak at 2.86 eV (although some
variation in the peak position has been recorded from 2.8 to 2.9 eV) is likely owing to
nitrogen vacancies or nonstoichiometry as proposed by Cox et al. [245]. Yim et al. [237]
also observed a broad emission spectrum range of 2–3 eV with a peak at about 2.8 eV.
1.3 Aluminum Nitride
10
4
(0001) AlN
Single crystals
300 K
Nitrogen
vacancy
Absorption coefficient α (cm–1)
3
10
Oxygen
Pastrnak and
Roskovcova
2
10
1
10
0
10
1
2
4
3
Energy (eV)
5
6
Figure 1.31 Room-temperature absorption spectra of several AlN
films of varying thicknesses whose principal absorption edge
occurs at 6.2 eV. The bump near 4.5–4.8 eV in the data is attributed
to the oxygen absorption bands. The one at about 2.8 eV is
attributed to N vacancies [245–247,259–261]. The ones not
marked as Pastrnak and Roskovcova [259] are from Slack
et al. [91].
This peak does not correlate with the presence of oxygen. The oxygen absorption region
lies between 3.5 and 5.2 eV, as originally found by Pastrnak and Roskovcova [259–261].
The exact position of this particular peak appears to change with the oxygen content
from 4.3 eV at low oxygen content to 4.8 eV at high oxygen content. The results of the
studies on the room-temperature absorption coefficient, a, are shown in Figure 1.31
for three different crystals along with the results of Pastrnak and Roskovcova [259].
Low-temperature and room-temperature absorption data taken at 2 K in a thin film on
double side polished sapphire are shown in Figure 1.32.
In the only optical study of AlN impurities, Karel and coworkers [262–266] reported
on the luminescence of Mg and rare earth centers in AlN.
Measurements of the refractive index of AlN have been carried out in amorphous,
polycrystalline, and single-crystal epitaxial thin films. The values of the refractive
index, n, are in the range of 1.99–2.25 with several groups reporting n ¼ 2.15 0.05.
j73
j 1 General Properties of Nitrides
74
2 × 10 10
RT absorption2
1.5 × 1010
1 × 1010
5 × 10
9
0
1900
1950
2000
2050
2100
2150
2100
2150
Wavelength (Å)
(a)
3.5 × 10 9
3 × 10 9
2K absorption 2
2.5 × 10 9
2 × 10 9
1.5 × 10 9
1 × 10 9
5 × 10 8
0
1900
(b)
1950
2000
2050
Wavelength (Å)
Figure 1.32 Room-temperature (a) and 2 K (b) absorption
spectrum of a thin AlN film grown on double side polished c-plane
sapphire by RF MBE. The data were taken at the University of
Pittsburgh by Song Bay and W.J. Choyke using a sample prepared
in authors laboratory.
These values are found to increase with increasing structural order, varying between
1.8 and 1.9 for amorphous films, 1.9–2.1 for polycrystalline films, and 2.1–2.2 for
single-crystal epitaxial films. The spectral and polarization dependence of the index of
refraction has been measured and showed a near-constant refractive index in the
wavelength range of 400–600 nm. Some of these measurements also indicate that, in
the long-wavelength range, the dielectric constant of AlN (e0) lies in the range of
1.4 Indium Nitride
8.3–11.5, and that most of the values fall within e0 ¼ 8.5 0.2. Other measurements
in the high-frequency range produced dielectric constants of 4.68 and e1 ¼ 4.84. AlN
has also been examined for its potential for second harmonic generation.
Synchrotron radiation studies of AlN single crystals up to 40 eV have been
performed, which resulted in the observation of an 8 eV luminescence peak. The
same peak was also found in vacuum–ultraviolet reflection measurements.
1.4
Indium Nitride
InN forms the third binary anchor of the nitride family and was first synthesized in
1938 [267]. Compared to the other two, GaN and AlN, it is very difficult to form in high
quality because of an inherent reason. The disparity in size, electronegativity, and
very high vapor pressure of N over In imposes intractable tasks on the crystal
grower [197,268]. The very early attempts explored absorption properties of polycrystalline InN films grown by DC discharge [269], reactive cathodic sputtering [270], or
RF sputtering [271], and somewhat later RF deposition [272]. Inclusive properties of
InN grown by various methods have been discussed in a review [273]. Essentially, the
early results suggested a bandgap Eg ¼ 1.8–2.0 eV at room temperature. Those
techniques gradually gave way to more refined growth methods such as MBE and
OMVPE. The stoichiometry, however, has always been a pestering issue and will
always remain so, which appears to have been one of the sources of controversy as to
InNs true bandgap following the longstanding value of about 1.89 eV. An inordinate
number of reports adorned many reputable journals and filled the programs in
technical meetings wherein researchers in great numbers argued that the true
bandgap of InN is actually 0.7–0.8 eV. In fact, lower values such as 0.65 [274] and
0.67 eV [275] have also been reported. What appeared to be experimentally impeccable results made the theorists to reexamine and redebate their band structure
calculations [138,276]. The previously accepted larger value for the bandgap was
explained away by O contamination and Moss–Burstein shift [277] because of high
electron concentrations inherent to this semiconductor. Excellent reviews chronicling the developments in InN and controversy surrounding its bandgap are
available in the literature [278,279].
Owing in part to visceral difficulties touched upon above and its bandgap,
regardless of the controversy, InN has received nowhere near the attention given
to GaN and AlN. Reiterating, the problems with InN are difficulties in growing highquality crystalline InN samples, poor luminescence properties of InN, and the
existence of alternative well-characterized semiconductors such as AlGaAs and (Ga,
Al)AsP, which have energy bandgaps close to that of the old value of InN bandgap
(1.89 eV) and InGaAs close to that of the new InN bandgap. Setting the bandgap aside,
which is the holy grail of optoelectronic devices, InN possesses the largest roomtemperature mobility among all the nitride-based semiconductors. Predictions point
to InN being a superior channel layer with its higher mobility for field effect
transistors, a topic that will receive more coverage in the pages to follow.
j75
j 1 General Properties of Nitrides
76
Just when the data and calculations appear to indicate the bandgap of InN to be close
to 0.7–0.8 eV [30], there came the reports casting doubt on the accuracy of the small
bandgap. The controversy regarding the true bandgap of InN brought this semiconductor, from obscurity to controversy, in the words of Scott Butcher, who is one of the
pioneers involved in InN studies of and instrumental in determining the 1.89 eV
bandgap [278]. The controversy is actually not limited to just the bandgap. It spans the
whole gambit of its properties including the lattice constant, the effective electron mass,
not to mention the hole mass, which is simply left in the dark, the role of hydrogen and
oxygen, nonstoichiometry-induced defects, and point and extended defects. Returning
to the controversy surrounding the bandgap of InN, proponents of the smaller bandgap
argue that the measured larger bandgap from absorption data is most likely skewed by
Moss–Burstein shift and/orO contamination.Theyare alsoquick toargue that emission
near 1.8–1.9 eV has not been observed casting doubt on its accuracy. The opponents of
the larger bandgap put forward arguments ranging from Mie resonance owing to
scattering or absorption of light in InN-containing clusters of metallic In [280] to In-rich
nonstoichiometry-induced defects active near the 0.7–0.8 eV region [281]. In the efforts
of the authors of Ref. [280], microcathodoluminescencestudies coupled with imaging of
metallic In have shown that bright infrared emission at 0.7–0.8 eV arises in the close
vicinity of In inclusions and is likely associated with surface states at the metal/InN
interfaces. Employing thermally detected optical absorption measurements, Shubina
et al. [280] suggested that a true bandgap near 1.5 eV, reserving a more accurate figure for
the bandgap until after more detailed measurements are carried out. The presence of In
inclusions would also make suspect the mobility and doping level data published for this
material. In fact, metal inclusions placed by design in GaAs have been shown to skew the
electron mobility [282]. Setting this issue aside for now and referring the reader to
Chapter 2 for bandgap-related discussion and Chapter 3 for growth-related discussion,
let us segue into the discussion of InN properties.
As mentioned, the energy bandgap of InN corresponds to a portion of the
electromagnetic spectrum in which alternative and well-developed semiconductor
technologies are already available. Consequently, practical applications of InN are
more or less restricted to its alloys with GaN and AlN or related heterostructures. The
growth of high-quality InN and the enumeration of its fundamental physical
properties remain for the present a purely scientific enterprise except of course
their impact on the properties of the ternaries it makes with GaN and AlN. InN is not
different from GaN and AlN in the sense that it suffers from the same lack of a
suitable substrate material and, in particular, a high native defect concentration. All
these have hindered its progress. In addition, because of its rather poor thermal
stability InN cannot be grown at the high temperatures required by CVD growth
processes. As InN rapidly dissociates at high temperatures, even as low as 600 C, an
extraordinarily high nitrogen overpressure would be required to stabilize the material
up to the melting point, which is practically impossible. The large disparity of the
atomic radii of In and N is another factor that increases the difficulty in obtaining InN
of good quality. Notwithstanding the aforementioned characterization, the seminal
work of Tansley and Foley [271] first characterized many of the fundamental physical
properties of InN.
1.4 Indium Nitride
Tables 1.19–1.22 list the physical properties of InN. It has proved difficult to grow
high-quality single crystalline material that would enable detailed optical, structural,
and electrical measurements to be performed. Given the fact that the growth of bulk
single-crystal InN films using equilibrium techniques is unlikely, attention turned to
the deposition of thin films using nonequilibrium techniques. All of the early data
summarized below under various properties, unless otherwise specified, were obtained from highly conductive n-type polycrystalline InN grown by nonequilibrium
techniques.
There have been several studies that report rapid dissociation of InN at temperatures above 500 C. Because no high-quality InN has yet been grown, the resistance of
the high-quality material to chemical etching is unknown. Successful etching of
single crystalline InN films in a hot H3PO4 : H2SO4 solution has been measured as
was surface oxidation.
1.4.1
Crystal Structure of InN
To reiterate, indium nitride normally crystallizes in the wurtzite (hexagonal) structure. The zinc blende (cubic) form has been reported to occur in films containing both
polytypes. Because of the absence of good-quality single-crystal films, early studies
dealing with the crystal structure of InN were limited to mainly less than ideal thin
films, particularly the ordered polycrystalline films with crystallites in the thickness
range of 50–500 nm. Basically, the measurements confirm that, although InN
normally crystallizes in the Wz structure, it occasionally also crystallizes in the zinc
blende (cubic) polytype.
Thermal instability of InN forbids growth at high temperatures and a large lattice
mismatch with most available substrates inevitably leads to less than perfect
structural quality of the epitaxial films grown by any method although the electron
mobility in MBE-grown layers have improved considerably. A study based on XRD
analysis has provided important insights into the dependence of the structural
properties on the degree of lattice mismatch and film thickness of InN [283] on
three different kinds of substrates, namely sapphire, GaN, and AlN. Significant
improvement in the structural quality of the InN films, which do suffer from the
residual strain, was observed on GaN templates. Below a thickness of 1200 Å, the InN
film is composed of grain islands with different crystal orientations. Above this
thickness, screw dislocations are nucleated, relieving the strain and leading to a
reduction of the observed mosaicity in the surface morphology as grains with the
same orientation grow with film thickness exceeding 1200 Å.
1.4.2
Mechanical Properties of InN
The measured InN lattice parameters using powder technique are in the range of
a ¼ 3.530–3.548 Å and c ¼ 5.704–5.960 Å with a consistent c/a ratio of about
1.615 0.008. This c/a ratio is close to the more optimistic value of 1.633 determined
j77
j 1 General Properties of Nitrides
78
from layers especially grown under significant precautions, best possible growth
conditions, and presumably with reduced nitrogen vacancies [284]. Another value of
the c/a ratio of 1.612 has also been reported using powder diffractometry [29]. There
is no reliable experimental u parameter for InN. An examination of the reported data
indicates an unacceptably large scatter. This is possibly caused by nitrogen deficiency
because nitrogen atoms are closely packed in (0 0 0 1) planes. The single reported
measurement yields a lattice constant of a0 ¼ 4.98 Å in zinc blende (cubic) form InN
occurring in films containing both polytypes [284]. While the cubic polytype of InN
yields a molecular cell volume of 30.9 Å3, the hexagonal polytype gives a molecular
cell volume of 31.2 0.2 Å3.
The experimental density of InN deduced from Archimedean displacement
measurements is 6.89 g cm3 at 250 C [285]. This is comparable with 6.81 g cm3
estimated from X-ray data. In a hexagonal structure, the second-order elastic moduli
are C11, C12, C13, C33, and C44. The only report on InN elastic coefficients is by
Savastenko and Sheleg [162], but their results are lower than the values calculated by
the linear muffin–tin orbital (LMTO) and the plane wave pseudopotential (PWPP)
methods and are suggested to be completely unreliable [178]. Table 1.31 summarizes
the measured and the calculated elastic coefficients for both Wz and ZB InN. Because
these figures depend on the lattice constants that are within some 10%, values of
other nitrides can be used as a first approximation when absolutely needed [271,286].
The bulk modulus has been calculated from first principles by the local-density
approximation [287] and by the LMTO method [288] suggesting bulk modulus
B ¼ 165 GPa. The results of other calculations for bulk modulus are shown in
Table 1.31. Most of the properties of InN are tabulated in Tables 1.19–1.22.
As in the cases of Wz GaN and Wz AlN, Wz InN has 12 phonon modes at the zone
center (symmetry group: C6v), 3 acoustic and 9 optical ones with the acoustic
Table 1.31 Theoretical and experimental elastic coefficients and
bulk modulus (in GPa) of the various forms of InN [160].
Method
Structure
C11
C12
C13
C33
C44
B
X-ray [162]
LMTO [24]
PWPP [42]
PWPP [42]
PWPP
LMTO
PWPP
PWPP
ADX
LMTO
PWPP
PWPP
Wz
Wz
Wz
ZB
ZB
Wz
Wz
ZB
Wz
Wz
Wz
ZB
190
271
223
187
104
124
115
125
121
94
92
182
200
224
10
46
48
86
139
141
145.6
155
165
166
138
125.5
165
139
140
dB/dP ¼ B0
4
3.8
3.9
12.7
ADX: angular dispersive X-ray diffraction; PWPP: plane wave pseudopotential; LMTO: linear
muffin–tin orbital.
1.4 Indium Nitride
branches near zero at k ¼ 0. The infrared active modes are of the E1(LO), El(TO),
A1(LO), and A1(TO) type. Raman spectroscopy studies [289,290] have yielded four
optical phonons characteristic for InN with wave numbers 190 cm1 (E2), 400 cm1
(A1), 490 cm1 (E1), and 590 cm1 (E2) in InN layers grown by atomic layer epitaxy
(ALE). Moreover, a TO mode has been observed at 478 cm1 (59.3 meV) by reflectance and 460 cm1 (57.1 meV) by transmission measurements [286]. From other
reflectance data, the existence of a TO phonon mode at 478 cm1, consistent with
Ref. [286], and an LO mode at 694 cm1 was deduced [291].
1.4.3
Thermal Properties of InN
The linear thermal expansion coefficients measured at five different temperatures
between 190 and 560 K [292] indicate that both along the parallel and perpendicular
directions to the c-axis of InN these coefficients increase with increasing temperature. Thermal conductivity derived from the Leibfried–Schloman scaling parameter,
assuming that the thermal conductivity is limited by intrinsic phonon–phonon
scattering, is about 0.80 0.20 W cm1 K1.
Using InN microcrystals prepared by microwave nitrogen plasma, the specific heat
of InN has been measured with differential scanning calorimeter with a precision
better than 1% [88,293]. The data have been fit to the Debye equation, Equation 1.16,
as shown in Figure 1.33a. An expanded view of the experimental data over a
temperature range of 150–300 K is given in Figure 1.33b. Below 200 K, a Debye
temperature of 600 K appears to fit the data well, tabulated in Table 1.32. However,
above 200 K a Debye temperature of 700 K fits the data better. This paradox
indicates the poor quality of InN and significant contribution by nonvibrational
modes, as the Debye theory is developed for a perfect dielectric. Others [88] argue that
the Debye temperature of yD ¼ 660 K, albeit in a relatively small range, describes
InN best.
As shown in Figure 1.18, the melting point of InN, T M
InN , is 2146 K and the line fit to
the partial pressure data for N indicates a temperature dependence of 7.9 1017 exp
( 2.78 eV/kT) bar for InN. As can be seen in Figure 1.18, the nitrogen partial
pressure increases exponentially above TE ¼ 630 C for InN, illustrating that the
decomposition temperature in vacuum is much lower than the melting point
achievable under high pressures. To determine the effective decomposition activation energy more precisely, the nitrogen flux was calculated from the measured
nitrogen pressure [196,199]. The rate of nitrogen evolution F(N) is equal to the rate of
decomposition and the slope of ln[F (N)] versus 1/T in Figure 1.18 gives the effective
activation energy of decomposition in vacuum, EMN. The decomposition rate is that
corresponding to desorption of one monolayer in 1 s, in other words, FN ¼ 1.5 · 1015
cm2 s1, at 795 C. The activation energy of the thermally induced decomposition is
determined as EMN ¼ 3.5 eV (336 kJ mol1) for InN (M is for metal and N is for
nitrogen). This indicates temperature limits for high-temperature or high-power
devices. This together with the reported small values of InN bandgap does not bode
well for power devices based on InN alone. Of course, the picture is different if this
j79
j 1 General Properties of Nitrides
80
50
–1
Cp (J mol–1 K )
40
30
–1
–1
Cp (J mol K )
T = 600K
T = 700K
T = 800K
20
10
0
0
(a)
200
400
600
Temperature (K)
800
1000
–1
Specific heat, C p (J mo l K–1)
40
35
30
25
20
150
(b)
200
250
Temperature (K)
300
Figure 1.33 (a) The specific heat of InN with experimental data
points, albeit in a small range, and the 600, 700, and 800 K Debye
temperature fits. Data from Refs [88,127]. (b) Experimental
specific heat of InN in a temperature range of 150–300 K [88,127].
material is used in conjunction with GaN if the associated technological difficulties
can be overcome.
While the heat capacity of InN is 9.1 2.9 · 103 cal mol1 K1) at temperatures
between 298 and 1273 K, the entropy is 10.4 cal mol1 K1 at 298.15 K. The
1.4 Indium Nitride
Table 1.32 Specific heat, Cp, of InN at constant pressure [88,127].
T (K)
Cp (J mol1 K1)
153
163
173
183
193
203
213
223
233
243
253
263
273
283
293
25.38
26.54
27.96
29.12
30.15
31.18
31.95
32.59
33.50
34.26
35.17
35.81
36.97
37.61
38.65
equilibrium partial pressure of N2 above InN is about 1 atm at 800 K and it increases
exponentially with temperature to 105 atm at 1100 K.
1.4.4
Brief Electrical Properties of InN
It is fair to state that reliable experimental data for the true electron mobility in InN is
waiting to be obtained. As mentioned repeatedly, InN suffers from the lack of a
suitable substrate material and high native defect concentrations that limit its quality.
In addition, a large disparity of the atomic radii of In and N makes it more difficult to
obtain InN of high quality. As a result, nitrogen vacancies are thought to lead to large
background electron concentrations in InN. Because of all these factors, the electron
mobilities obtained from various films vary very widely, as can be inferred from
Table 1.33. The electron mobility in InN can be as high as 3000 cm2 V1 s1, perhaps
even much higher, at room temperature [294]. A study of the electron mobility of InN
as a function of the growth temperature indicates that the mobility of ultrahighvacuum electron cyclotron resonance radio-frequency magnetron sputtering (UHV
ECR-RMS) grown InN can be as much as four times the mobility of conventionally
grown (vacuum deposition) InN [295]. The dependence of the electron concentration
and mobility on the InN film thickness grown on AlN and GaN buffer layers by
plasma-enhanced MBE is shown in Figure 1.34.
Hall measurements in InN films grown on AlN buffer layers [300] that are in turn
grown on sapphire indicated electron mobility to be 1310 cm2 V1 s1 at room
temperature [307] for MBE-grown and 830 cm2 V1 s1 for OMVPE-grown material [304]. As discussed in Section 3.5 in conjunction with growth-related issues,
j81
j 1 General Properties of Nitrides
82
Table 1.33 A compilation of electron mobilities obtained in InN on
different substrates and for various deposition conditions in InN
as compiled in part in Ref. [296].
n-type carrier
concentration (cm3)
Carrier mobility
(cm2 V1 s1)
5–8 · 1018
250 50
1020
3–10 · 1018
20
20–50
1–200 · 1018
2–80 · 1020
5 · 1018
6 · 1020
6 · 1016
3
35–50
20
2
2
7–70 · 1016
3 · 1016 at 150 K
1020
730–3980
5000 at 150 K
10
4.8 · 1020
1–8 · 1020
1–10 · 1020
2–3 · 1020
38
50
50
20–60
2 · 1020
1020ae
2.0 · 1020
5.98 · 1018
100
220a
100
363
Sapphire
Sapphire
Sapphire
Sapphire, silicon,
mica
GaAs
GaAs
GaAs
Glass
5.0 · 1019
3.0 · 1018
700
542
Sapphire
Sapphire
8.8 · 1018
2–3 · 1018
1019
500
800
306
Sapphire
Sapphire
Glass, KBr
1.0 · 1019
4 · 1017
830
2100
Sapphire
Sapphire
1.4 · 1018
1420
Sapphire
5 · 1016
(3 · 1016 at 150 K)b
2700 (5000 at 150 K)b
a
b
Zinc blende polytype.
Have not yet been reproduced by others.
Substrate
Deposition technique
Sapphire, silicon,
various metals
Sapphire
Glass, fused
quartz
Fused quartz
Sapphire
Glass, NaCl
Fused quartz
Glass, silicon,
304 stainless steel
Glass, silicon
Glass, silicon
Glass
Reactive sputtering
Reactive evaporation
Reactive sputtering
Reactive sputtering
CVD
Reactive sputtering
Cathodic sputtering
RF ion plating
Reactive sputtering
Reactive sputtering
Reactive DC
magnetron sputtering
Magnetron sputtering
Plasma-assisted MOVPE
MOVPE
Reactive RF magnetron
sputtering
ECR-assisted MOMBE
Plasma-assisted MBE
ECR-MOMBE [297]
Reactive RF magnetron
sputtering [298]
MOVPE [299]
Migration-enhanced
epitaxy [300]
MOMBE [301]
MBE [302]
RF reactive ion
sputtering [303]
RF MBE [304]
MBE on thick HVPE
GaN [305]
Plasma-assisted
MBE [306]
Sputtering [308]
10
21
104
10
20
103
10
19
102
10
18
10
InN on GaN
mobility
electron conc.
InN on AlN
mobility
electron conc.
17
10
1
10
2
Mobility (cm2 V-1 s-1)
Electron density (cm–3)
1.4 Indium Nitride
101
100
3
10
Thickness (nm)
10
4
Figure 1.34 Electron density and mobility as a function of InN
thickness. Samples grown on a GaN buffer are indicated by
diamond and square symbols, and on an AlN buffer are indicated
by circles and inverted triangles. The diamond and inverted
triangle symbols indicate the electron mobility, whereas the
squares and circle symbols indicate the electron concentration, all
measured at room temperature. Courtesy of W.J. Schaff.
room-temperature mobilities increased to 2000 cm2 V1 s1 in MBE-grown InN
films. Some directions, such as the insertion of low-temperature intermediate layers
or AlN buffer layers have been pointed out for improvement. Very high inadvertent
donor concentrations, >1018 cm3, seem to be one of the major problems for further
progress. ON and SiIn have been proposed to be the likely dominant defects
responsible for high electron concentration based on their low formation energies.
Additionally, H has been proposed [307] as the dominant impurity candidate for the
state-of-the-art MBE-grown InN. Assuming that the measurements were performed
flawlessly and interpreted, there clearly seems to be some way to go to reach the goal
of a mobility of 2700 cm2 V1 s1 at an electron concentration of 5 · 1016 cm3
reported for RF reactive ion sputtered growth of InN nearly a few decades
earlier [308].
Ensemble Monte Carlo calculations have been the popular tool to investigate the
carrier velocity field characteristics theoretically. Although early application of this
method to InN was done by OLeary et al. [309], a more detailed calculation based
on the full details of the conduction band structure appeared later in a paper by
Bellotti et al. [310]. A peak electron drift velocity of 4.2 · 107 cm s1 has been
predicted at an electric field of 65 kV cm1, substantially higher than that in GaN,
j83
j 1 General Properties of Nitrides
84
with a noticeable anisotropy for field direction parallel or perpendicular to the basal
plane. The velocity is seen to decrease to 3.4 · 107 cm s1 with an increase of field to
70 kV cm1 and reach a value of 2.0 · 107–1.0 · 107 cm s1 at the onset of impact
ionization, for field directions parallel and perpendicular to the basal plane, respectively. The calculated low-field mobility is 3000 cm2 V1 s1. This study claims to
report the first calculation of high-field electron transport in InN. Electron–phonon
coupling would hurt the high-field velocity as it seems to be the case in GaN, see
Volume 3, Chapter 3 for details.
Another interesting aspect of electron transport is its transient behavior, which is
relevant to short channel devices with dimensions smaller than 0.2 mm, where a
significant overshoot is expected [311] to occur in the electron velocity over the steadystate drift velocity. In yet another calculation, Foutz et al. [311] found that of the three
III nitride binaries, GaN, InN, and AlN, this overshoot is most pronounced in InN
and occurs above a critical field of 65 kV cm1. The peak velocity at this field is
4.2 · 107 cm s1 and the velocity overshoot is retained over longer distances as
compared to that for GaN and AlN.
1.4.5
Brief Optical Properties of InN
As mentioned in the opening statements for Section 1.4, while the bandgap value
reported in the early stages of InN development dating back to as early as 1980s, a
controversy developed as to the true value of the InN bandgap developed. This is
detailed in Section 2.9.2. However, a brief treatment is provided here for completeness. Excellent reviews chronicle and detail the evolution of the controversy in
InN [278,279]. A number of groups have described optical measurements performed
on InN [25,313,314]. Early values of the room-temperature InN direct bandgap
ranged from 1.7 to 2.07 eV. A value of 1.89 eV was measured by optical absorption by
Tansley and Foley [308], who also measured the infrared absorption of InN and
observed an unidentified donor level approximately 50–60 meV below the conduction band edge. Reflectance spectroscopy on single-crystal material, using synchrotron radiation over the range 2–20 eV[315], later extended to 130 eV [316], was
performed to determine the optical parameters of InN.
A few studies of the interband optical absorption performed on InN thin films
deposited by sputtering techniques [271] and OMVPE [103], were found consistent
with a fundamental energy gap of about 2 eV. However, weak photoluminescence
peaks with energies ranging from 1.81 to 2.16 eV were observed for InN grown on Si
substrates later on [317]. In one such case, an emission centered at 1.86 eV at a
temperature below 20 K was seen, whereas the reflectance measurement showed a
strong plasma reflection at 0.7 eV, corresponding to an effective mass of 0.12m0.
Another branch of studies shows that in improved InN films a strong photoluminescence transition at energies around 1 eV [30,318,319] appears. Observing that the
position of the photoluminescence energy agrees with the onset of strong absorption,
the optical transition at about 1 eV has been attributed to the fundamental
bandgap [319].
1.4 Indium Nitride
4
Assuming large gap
Assuming small gap
E g (eV)
3
2
1
0
0
InN
20
40
60
80
Composition
100
In2 O3
Figure 1.35 Vegards law plot of InN–In2O3 pseudobinary alloy
system with both 0.7 and 1.9 eV bandgaps shown (dashed lines
and solid line, respectively) versus O composition reaching In2O3,
which has a bandgap of 3.75 eV reported in Ref. [320]. Also shown
is the calculated (using LCAO) bandgap with 10% oxygen if O-free
bandgap is 0.7 eV [278,322].
Following the wide acceptance of the above-mentioned data by the community, a
couple of crystal growth groups, often in collaborations with others who focused on
characterization, began to question the nearly 1.9–2.0 eV bandgap, because the newer
and supposedly more improved samples showed a strong emission at much smaller
energies, primarily around 0.7–0.8 eV. Those latter groups argued that the early
samples did not show efficient PL near the band edge and had to have been
contaminated with O to support the earlier measurements. One should, however,
keep in mind that In2O3 has a bandgap of 3.75 eV [320], and if the Vegards law is
applicable it would take some 35% O in InN to boost its bandgap from say 0.8 to
1.9 eV, which is substantial, as shown in Figure 1.35. Also shown is the bandgap of
InN for 10% O contamination assuming an O-free bandgap of 0.7 eV [321,322]. Not
only large amounts O is needed, but also that O must form an alloy with InN. Clearly,
the assignment of the 1.9 eV bandgap to O, on the premise that the bandgap for O-free
InN is 0.7–0.8 eV, requires large amounts of O, which is not supported by experiments, as discussed in detail in Section 2.10.1.
To account for the 1.9 eV measured bandgap, the proponents of the 0.7 eV bandgap
also suggested that the former result could be accounted for by Moss–Burstein blue
shift owing to high electron concentration. This effect relates to semiconductors
where the electron concentration is larger than the density of states and the Fermi
level is actually in the conduction band itself. The extent of the penetration naturally
depends on the electron concentration. Consequently, the measured apparent optical
gap is skewed upward. This possibility was suggested in 1974 by Trainor and
Rose [323] and has been the topic of an extensive study using In2O3 as the model
wide bandgap material [320]. What accompanies the Moss–Burstein shift is the
j85
j 1 General Properties of Nitrides
86
2.4
E G +E F (eV)
2.2
2.0
1.8
1.6
1.4
1.2
1.0
0.8
0.6
Moss–Burstein effect
0.4
10 16
10 17
10 18
10 20
10 19
a
b
c
d
e
f
g
h
i
10 21
–3
Carrier concentration (cm )
Figure 1.36 The measured bandgap of InN
(combination of the bandgap and the
Moss–Burstein shift) deduced from absorption
measurements versus the carrier concentration
reported by various groups. The solid line shows
the expected blue shift in the bandgap because of
Moss–Burstein shift. Solid and dashed lines
indicate nonparabolic and parabolic theories,
respectively. (a) Ref. [341]; (b) Ref. [324];
(c) Refs [274,318]; (d) Ref. [331]; (e) Ref. [271];
(f) Refs [303,325]; (g) Refs [131,272,326,327];
(h) Ref. [328]; (i) Ref. [329]. Collated by Butcher
and Tansley [278,279].
bandgap renormalization, which is a red shift, caused by tail states extending into the
bandgap and counters the former effect to some extent. Here too there is a
controversy in that the samples of older times did not indicate large Moss–Burstein
effect, which has been attributed by the proponents of the 0.7 eV bandgap, as having
been heavily reduced owing to band tail states.
Butcher [278] collated the apparent bandgap (presumably a combination of the
bandgap with its band tailing and Moss–Burstein effect) measured by optical
absorption and the electron concentration data for a large group of samples as
shown in Figure 1.36. Although an argument for Moss–Burstein effect can be made
for samples with high carrier concentrations, the same cannot be applied to a good
many samples exhibiting the large bandgap while having low carrier concentration.
To be thorough, one should recognize that many of the InN samples contained in
Figure 1.36 with low carrier concentrations are heavily compensated, casting some
amount of uncertainty as well. Turning the argument around and assuming that the
large bandgap data are more dependable, the low bandgap data could be explained
with some defect level in In-rich material or by Mie resonant absorption due to In
inclusions. These are discussed in Section 2.10.1.
After discussing the controversy regarding the bandgap and making many
references to the low bandgap measured in InN, let us briefly discuss the data
obtained in layers grown by MBE under In-rich conditions, which exhibit the
so-called small bandgap. The optical absorption data in InN, purportedly having
0.7–0.8 eV bandgap, show an onset at 0.78 eV not near 1.9–2.0 eV, as shown in
1.4 Indium Nitride
Figure 1.37a. The absorption coefficient increases gradually with increasing photon
energy and at the photon energy of 1 eV it reaches a value of more than 104 cm1. This
high value is consistent with an interband absorption in semiconductors. Moreover,
the integrated PL intensity increased linearly with excitation intensity over three orders
of magnitude, lending more credence to the notion that the observed nonsaturable
peak relates to the fundamental interband transition. The absorption squared versus
the energy plots used to obtain the apparent bandgap in a semiconductor with very high
carrier concentration underestimates the bandgap owing to band tailing. Briot
et al. [330,331] estimated a bandgap near 1.2 eV from the absorption data while taking
the large carrier concentration of 1019 cm3 into account. This, however, does not
account for the large discrepancy between the 0.7–0.8 eV group and 1.8–2.0 eV group.
The free-electron concentration in this sample was measured by Hall effect to be
5 · 1018 cm3. Figure 1.37a also shows that the samples exhibit intense roomtemperature luminescence at energies close to the optical absorption edge. Additionally, the 77 K photoreflectance (PR) spectrum exhibits a transition feature at 0.8 eV with
a shape characteristic for direct gap interband transitions. Consistent with the
absorption data, no discernible change in the PR signal near 2 eV is seen. The
simultaneous observations of the absorption edge and PL and PR features at nearly
the same energy led Wu et al. [30] to argue that this energy position of 0.78 eV is the
fundamental bandgap of InN. This value is very close to the fundamental gap for InN
reported by the other group, Davydov et al. [319], in favor of the smaller bandgap for
InN. Tsen et al. [332] studying nonequilibrium optical phonons in a high-quality
single-crystal MBE-grown InN with picosecond Raman spectroscopy reached the
conclusion that their results are not consistent with the large bandgap of InN. Using
the possible phonon emission allowed by energy and momentum conservations for a
range of excitation photon energies, they argued that their observations are consistent with the small bandgap of InN. The basis of the argument is that if the bandgap
energy were 1.89 eV, no nonequilibrium phonons could be observed, contradicting
the observation of Tsen et al. [332], details of which are discussed in Section 2.10.1.
Figure 1.37b shows the room-temperature electron mobility, the peak energy of PL,
and the transition energy determined by PR as functions of electron concentration,
showing that the transition energies increase with increasing free-electron concentration. This indicates that the transitions from higher energy occupied states in the
conduction band contribute significantly to the PL spectrum. The PL energy as a
function of pressure shows a linear pressure coefficient of 0.6 meV kbar1, which is
considerably smaller than that for other III–V compounds. For comparison, the
pressure coefficient of GaN is 4.3 meV kbar1 [333], AlxGa1xN is 4.1 meV kbar1 for
0.12 < x < 0.6 [334], and GaAs is 11 meV kbar1 [335], as compiled by Wu et al. [30].
The sapphire substrate on which the InN layer is grown has a larger bulk modulus
than InN, which will reduce hydrostatic pressure transmission to the InN film
providing that the film remains coherently strained. Using experimental elastic
constants for sapphire and theoretical elastic constants for InN, Wu et al. [30]
estimated the correction factor for coherently strained InN on sapphire to be
1.45, leading to between 0.6 and 0.9 meV kbar1 for the bandgap change. More
work is needed to shed light on this unusually low pressure dependence although
j87
j 1 General Properties of Nitrides
88
8
6
PL or PR signal
ab. at 300 K
5
4
3
2
PR at 77 K
Absorption coefficient (104 cm-1)
7
PL at 300 K
1
0.5
1
1.5
2
0
2.5
Energy (eV)
(a)
0.90
103
mobility at 300 K
Energy (eV)
102
0.80
PL at 300 K
- Mobility (cm2 V 1s 1)
0.85
101
PL at 12 K
0.75
PR at 77 K
100
0.70
1018
(b)
1019
1020
Electron concentration, n (cm–3)
Figure 1.37 (a) Optical absorption (300 K), PL
(300 K), and PR (77 K) spectra of a typical InN
sample. This sample is undoped with roomtemperature electron concentration of
5.48 · 1018 cm3. The spike on the PR spectrum
at 0.97 eV is an artifact due to the light source
used in the PR measurement. (b) Room-
temperature mobility, PL peak energy (300 and
12 K), and the critical energy determined by PR
(77 K) as a function of free-electron
concentration. The sample with
n ¼ 1 · 1019 cm3 (indicated by an arrow) is the
Ritsumeikan sample.
1.5 Ternary and Quaternary Alloys
studies of InGaN showing smaller pressure dependence as the InN concentration is
increased are consistent with data on InN [336].
Tyagai et al. [337] performed reflection and transmission measurements in InN
with electron concentration larger than 1020 cm3. They were able to estimate an
effective mass of me ¼ 0:11m0 and an index of refraction of n ¼ 3.05 0.05, which is
in reasonable agreement with the value measured by Tsen et al. [332]. The longwavelength limit of the refractive index was reported to be 2.88 0.15. The temperature dependence of the InN bandgap indicates a bandgap blue shift of 23 meV from
300 to 77 K [291,338]. Inushima et al. [132] reported the effective mass to be 0.24m0 in
InN grown by UV-assisted atomic layer epitaxy under atmospheric pressure. Using
infrared spectroscopic ellipsometry, Kasic et al. [339] arrived at a value of m ¼ 0.14m0
in an MBE-grown InN layer having an electron concentration of n ¼ 2.8 · 1019 cm3.
Using surface reflection of extrinsic semiconductors in the infrared region by the
free-carrier plasma, used earlier for GaN [340], Wu et al. [341] determined the
effective mass of the free carriers to be 0.07m0 at the bottom of the conduction band.
This method hinges on the knowledge of the plasma frequency, electron concentration, and optical dielectric constant through the relation
ne2
;
ð1:23Þ
m ¼
ee¥ w2P
where all the terms have their usual meanings and wP represents the plasma
frequency. Using e¥ ¼ 6:7 (compares with figures in the range of 5.8–9.3 tabulated
in Table 1.21) and utilizing infrared reflection measurements for a series of samples
with different carrier concentrations, Wu et al. [341] arrived at an electron effective
mass value of 0.07m0 at the bottom of the conduction band.
The temperature dependence of the bandgap of InN indicates a bandgap temperature coefficient of [342]
ðdE g =dTÞ ¼ 1:8 10 4 eV K 1 :
ð1:24Þ
1.5
Ternary and Quaternary Alloys
Many important GaN-based devices involve heterostructures as the primary means of
achieving an improved performance. Ternary alloys of wurtzite polytypes of GaN,
AlN, and InN have been obtained in the continuous alloy systems whose
direct bandgap ranges from the old value for InN of 1.9 eV (the new value is
approximately 0.7 eV according to Ref. [30]) for InN to 6.2 eV for AlN (the new value
is approximately 6 eV). For an in-depth understanding of the physical mechanisms that
underlie their operations, the properties of these alloys need to be extensively studied.
Many of these properties such as the energy bandgap, effective masses of the electrons
and holes, and the dielectric constant depend on the alloy composition. Although
measured data for these parameters in InGaN and InAlN have been obtained, they
are still not very precise (for AlGaN see Ref. [157] and for InGaN see Ref. [343]).
Yamasaki et al. [344] have reported on p-InGaN. More research is necessary to confirm
j89
j 1 General Properties of Nitrides
90
these conclusions regarding p-AlN and p-InGaN. AlN and GaN are slightly lattice
mismatched (2.4%). It has been noted that, for many devices, only small amounts of
AlN are needed in the GaN lattice to provide sufficient carrier and optical field
confinements.
1.5.1
AlGaN Alloy
The ternary alloys of wurtzite and zinc blende polytypes of GaN with AlN form
a continuous alloy system with a wide range of bandgap and a small change in
the lattice constant. An accurate knowledge of the compositional dependence of the
barrier as well as material is a requisite in attempts to analyze heterostructures in
general and quantum wells (QWs) and superlattices in particular. The barriers can be
formed of AlGaN or AlN, and while dependent on the barrier material, the wells
can be formed of GaN or AlGaN layers. The compositional dependence of the lattice
constant, the direct energy gap, and electrical and CL properties of the AlGaN
alloys were measured by Yoshida et al. [345]. A similar investigation followed a few
years later [346]. On the structural side, namely the calculated lattice parameter of
this alloy, predictions indicate that Vegards law applies [347] (also reviewed in
Ref. [17]):
aAlx Ga1 x N ¼ 3:1986 0:0891x Å and
c Alx Ga1 x N ¼ 5:2262 0:2323xÅ :
ð1:25Þ
By bringing to bear various tools such as HRXRD, the experimental data
for various AlGaN support the applicability of Vegards law in that the experimental data aAlx Ga1 x N ¼ ð3:189 0:002Þ ð0:086 0:004Þx Å and c Alx Ga1 x N ¼
ð5:188 0:003Þ ð0:208 0:005Þx Å are within about 2% of those predicted by
linear interpolation, Vegards law. However, the bond lengths exhibit a nonlinear
behavior, deviating from the virtual crystal approximation. Essentially, the nearest
neighbor bond lengths are not as dependent on composition as might beexpected from
the virtual crystal approximation.
The ensuing investigations to pin down the compositional dependence of the
bandgap of this important alloy continued with conflicting results. These are
discussed below following the presentation of an empirical expression used to relate
the bandgap to composition.
The compositional dependence of the principal bandgap of AlxGa1xN can be
calculated from the following empirical expression providing that the bowing
parameter, b, is known accurately:
E g ðxÞ ¼ xE g ðAlNÞ þ ð1 xÞE g ðGaNÞ bxð1 xÞ;
ð1:26Þ
where Eg(GaN) ¼ 3.4 eV, Eg(Al N) ¼ 6.1 eV, x is the AlN molar fraction, and b is the
bowing parameter.
1.5 Ternary and Quaternary Alloys
An earlier compilation by Amano et al. [348] already pinpointed the discrepancy in
the reported bowing parameters. For example, Yoshida et al. [349] concluded that, as
the AlN mole fraction increases, the energy bandgap of AlxGa1xN deviates upward,
implying a negative value for the bowing parameter b. This contrasts the data of
Wickenden et al. [350] that support a vanishing bowing parameter b. Koide et al. [351]
observed that the bowing parameter is positive and that the bandgap of the alloy
deviates downward indicating a positive value for the bowing parameter. To determine the bowing parameter accurately, as investigations expanded [352–362] so did
the dispersion in the bowing parameters ranging from 0.8 eV (upward bowing) to
þ2.6 eV (downward bowing), as compiled by Yun et al. [363].
Much of this spread emanates from the likely dispersion in the quality of
AlxGa1xN, thus erroneous determination of its bandgap and to a lesser extent its
lattice parameter. Because the genesis of PL transitions could be nonintrinsic, a
technique relying on absorption or modulated photoreflectance is more accurate in
the determination of bandgap energy. Using X-ray and surface analytical techniques,
such as secondary ion mass spectroscopy (SIMS) and Rutherford backscattering
(RBS), for determining composition, reflectance, and absorption for bandgap for
AlGaN layers spanning the entire compositional range, Yun et al. [363] revisited the
bowing parameter. The results of this study, shown in Figure 1.38 in the form of
AlGaN bandgap versus the composition, yield a bowing parameter of b ¼ 1.0 eV for
the entire range of alloy compositions. In this figure, the solid line represents a least
square fit to the data, which in turn are depicted by solid circles. X-ray diffraction
peaks generally tend to be wider for alloy compositions around the midway point that
is the most likely source of error in determining the bowing parameter. The situation
is exacerbated by the fact that the data points near the middle of the compositional
range determine the bowing parameter to a much larger extent, as near each of the
binary ends the compositional variation approaches the linear line. It is still possible
that as the quality of the films improves smaller bowing parameters could result. A
bowing parameter as low as 0.7 has been reported.
Hall measurements for n-Al0.09Ga0.91N demonstrated a carrier concentration of
5 · 1018 cm3 and a mobility of 35 cm2 V1 s1 at 300 K [364]. This measurement did
not reveal any temperature-dependent mobility of n-A10.09Ga0.91N. Other Hall
measurements [365] on Mg-doped p-Al0.08Ga0.92N grown by MOVPE, however,
addressed the temperature dependence of the mobility [365]. They indicate that the
hole mobility decreases with increasing temperature, reaching a value of about
9 cm2 V1 s1 for a doping density of 1.48 · 1019 cm3. This low mobility is ascribed
to a high carrier concentration and the intergrain scattering present in the samples.
While the lattice constant was studied, it was observed to be almost linearly
dependent on the AlN mole fraction in AlGaN.
Until recently, the resistivity of unintentionally doped AlGaN was believed to
increase so rapidly with increasing AlN mole fraction that AlGaN became
almost insulating for AlN mole fractions exceeding 20%. As the AlN mole fraction
increased from 0 to 30%, the n-type carrier concentration dropped from 1020 to
1017 cm3 and the mobility increased from 10 to 30 cm2 V1 s1. An increase in
j91
j 1 General Properties of Nitrides
92
6.5
Bangap of AIGaN (eV)
6.0
5.5
5.0
b=0
b=1
4.5
4.0
3.5
0
0.2
0.4
0.6
0.8
1
Al composition (x)
Figure 1.38 Experimental data of energy bandgap of AlGaN
(0 x 1), plotted as a function of Al composition (solid circle),
and the least squares fit (solid line) giving a bowing parameter of
b ¼ 1.0 eV. The dashed line shows the case of zero bowing. As the
quality of the near 50 : 50 alloy layers get better, giving rise to
sharper X-ray and PR data, smaller bowing parameters may ensue.
Bowing parameters as low as 0.7 have been reported.
the native defect ionization energies with increasing AlN may possibly be responsible
for this variation. Our knowledge of the doping characteristics of AlGaN is still
incomplete. For example, it is not known how the dopant atoms such as Si and Mg
respond to the variation of the AlN mole fraction in AlGaN. However, it was
suggested that as the AlN mole fraction increases, the dopant atom moves deeper
into the forbidden energy bandgap. AlGaN with an AlN mole fraction as high as
50–60% may be doped by both n- and p-type impurity atoms. The ability to dope a
high mole fraction AlGaN, especially when low-resistivity p-type material is required,
is important because it may otherwise restrict the overall characteristics of devices
such as laser diodes. A low AlN mole fraction in AlGaN has been considered
sufficient for acceptable optical field confinement. However, this must be addressed
before the potential of AlGaN with respect to the other wide bandgap semiconductors
is fully realized.
1.5.2
InGaN Alloy
The growth of high-quality InN and an enumeration of its fundamental physical
properties remain somewhat elusive as compared to the other alloy, AlGaN.
1.5 Ternary and Quaternary Alloys
Notwithstanding the difficulties in technology, InGaN is already an integral part of
important device designs. InxGa1xN (x is the InN mole fraction) is not any less
important than AlxGa1xN for the fabrication of electrical and optical devices, such as
LEDs and lasers, which can emit in the violet or blue wavelength range. It can be a
promising strained QW material for these devices, but added complexities such as
the phase separation and other inhomogeneities make the determination of the
bandgap of InGaN versus the composition a very difficult task.
The first growth of single crystalline InGaN by MOVPE was realized by Nagatomo
et al. in 1989 [366] and Matsuoka et al. [367], followed by Yoshimoto et al. in 1991 [368].
Since then, considerable effort has been expended worldwide on this material, as it is
responsible for emission in the near-UV, violet, blue, and green colors of the optical
spectrum. High-efficiency blue and green LEDs utilizing InGaN active layers are
commercially available. This material, however, is not as easy to grow because of the
high vapor pressure of N on In and also mismatch between the large In atom and the
small N atom. To mitigate this problem, V/III ratios in excess of 20 000, increasing
with InN mole fraction, as well as reduced growth temperatures are employed.
Matsuoka et al. [369] discovered that lowering the growth temperature to 500 C from
nominal temperatures such as 800 C increased the In content in the layers, but at the
expense of reduced quality. Efforts to increase the In concentration by raising the
indium precursor temperature or the carrier gas flow rate resulted in the degradation
of the structural and surface morphology so much that In droplets were formed on
the surface [370].
The great disparity between Ga and In could lead to issues such as phase separation
and instabilities. In this vein, Ho and Stringfellow [371] investigated the temperature
dependence of the binodal and spinodal boundaries in the InGaN system with a
modified valence force field model. The calculation of the extent of the miscibility gap
yielded an equilibrium InN mole fraction in GaN of less than 6% at 800 C [371]. In
the annealing experiments in argon ambient, the phase separation in an InxGa1xN
alloy with x 0.1 was observed at temperatures between 600 and 700 C [372],
pointing to the large region of solid immiscibility of these alloys. However, under
nonequilibrium growth conditions, InxGa1xN layers were grown in the entire range
of compositions. But, the decomposition into two phases upon annealing of the
InxGa1xN alloys (x ¼ 0.11 and x ¼ 0.29) at 600 and 700 C was observed pointing to
the existence of the miscibility gap. For some alloys with x ¼ 0.6, the phase separation
could not be observed at 600 C. Above 800 C, the alloy samples with x ¼ 0.1 actively
evaporated from the substrate. These results suggest that the solid solutions are
grown in metastable conditions and decomposed under annealing conditions.
Koukitu et al. [373] performed a thermodynamical analysis of InGaN alloys grown
by MOCVD. They found that in contrast to other III–III–V alloy systems where the
solid composition is a linear function of the molar ratio of the group III metalorganic
precursors at constant partial pressure of group V gas, the solid composition of
InGaN deviates significantly from a linear function at high substrate temperatures.
Kawaguchi et al. [374] reported a so-called InGaN composition pulling effect in
which the indium fraction is smaller during the initial stages of growth but increases
with increasing growth thickness. This observation was to a first extent independent
j93
j 1 General Properties of Nitrides
94
of the underlying layer, GaN or AlGaN. The authors suggested that this effect is
caused by strain caused by the lattice mismatch at the interface. They found that a
larger lattice mismatch between InGaN and the bottom epitaxial layers was accompanied by a larger change in the In content. What one can glean from this is that the
indium distribution mechanism in InGaN alloy is caused by the lattice deformation
because of the lattice mismatch. With increasing thickness, the lattice strain is
relaxed owing to the formation of structural defects and roughness, which weakens
the composition pulling effect.
Other substrates have also been used for InGaN growth. It has been reported that
the crystalline quality of InGaN is superior when grown with the composition that
lattice matches ZnO substrate to that grown directly on (0 0 0 1) sapphire substrate [368,369]. In the same investigations, it was observed that InGaN films grown
on sapphire substrates using GaN as buffer layers exhibited much better optical
properties than InGaN films grown directly on sapphire substrates [375]. For a given
set of growth conditions, an increase of InN in InGaN can be achieved by reducing the
hydrogen flow [376]. As in the case of AlGaN, the calculated lattice parameter of this
alloy follows Vegards law [347] (also reviewed in Ref. [17]):
aInx Ga1 x N ¼ 3:1986 þ 0:3862x Å
and
c Inx Ga1 x N ¼ 5:2262 þ 0:574x Å:
ð1:27Þ
By bringing to bear various tools such as HRXRD, the experimental data for
various AlGaN support the applicability of Vegards law in that the experimental data
aAlx Ga1x N ¼ ð3:560 0:019Þþð0:449 0:019Þx Å and c Alx Ga1x N ¼ ð5:195 0:002Þþ
ð0:512 0:006Þx Å are within about 2% of that predicted by linear interpolation,
Vegards law.
As in the case of AlGaN but to a larger extent, the bond lengths exhibit a nonlinear
behavior, deviating from the virtual crystal approximation. Essentially, the nearest
neighbor bond lengths are not as dependent on composition as might be expected
from the virtual crystal approximation.
The compositional dependence of InGaN bandgap is a crucial parameter in
designs of any heterostructure utilizing it. As such, the topic has attracted a number
of theoretical [377–382] and experimental (to be discussed below) investigations and
reports. Similar to the case of AlGaN, the energy bandgap of InxGa1xN over
0 x 1 can be expressed by the following empirical expression:
g
g
g
E Inx Ga1 x N ¼ xE InN þ ð1 xÞE GaN bInGaN xð1 xÞ
¼ 0:7x þ 3:4ð1 xÞ bInGaN xð1 xÞ eV;
g
g
ð1:28Þ
where E GaN ¼ 3:40 eV and E InN ¼ 1:9 eV or near 0.7 eV. For the nomenclature
GaxIn1xN, the terms x and 1 x in Equation 1.28 must be interchanged. Another
point of caution is that the sign in front of the bowing parameter is changed to positive in
some reports.When a comparison ismade, the sign of theb parametermust bechanged.
An earlier investigation of InGaN bowing parameter for alloys with small concentrations of InN by Nakamura et al. [383] led to a bowing parameter of 1.0, which is
in disagreement with the value of 3.2 reported by Amano et al. [348], who took into
1.5 Ternary and Quaternary Alloys
consideration the strain and piezoelectric fields as well. It should be mentioned that
these reports dealt with the Ga-rich side of the alloy. To obtain a fit over a large range of
compositions, a composition-dependent bowing parameter has been suggested. As
the InGaN is grown on GaN, there are many complicating factors, such as the
piezoelectric effect and the nonuniform strain; the impact of the former can be made
negligible by growing thick films. Moreover, compositional inhomogeneities due to
partial phase separation are present. If the strain caused by the lattice mismatch were
uniform, it would be compressive due to the InN lattice constant being 11% larger
than that of GaN with an accompanying blue shift of the band edge. Herein lies the
dilemma faced by the experimentalists. Growing thick films could minimize the
extent of strain and the piezoelectric effect. However, this is nearly an intractable
proposition. The relaxation value of the lattice constant and the origin of the optical
transitions must be known accurately to determine the bandgap versus composition
dependence. Absorption and/or reflection measurements, provided that the absorption edge is sharp, are more useful in determining the bandgap but again require
thick and/or high-quality films. Detailed X-ray reciprocal-space mapping undertaken
by Amano et al. [348] purportedly indicated that InGaN wells and even somewhat
thicker InGaN layers grown on GaN buffer layers are coherently strained; a conclusion reached by the observation that the in-plane lattice constants of GaN and InGaN
match. At the same time, though, the layer thicknesses well exceeded the calculated
critical values.
When a bandgap of 1.9 eV for InN is assumed as the end point value for InN in
regard to InGaN ternary, large and/or more than one bowing parameter is required to
fit the compositional dependence of the bandgap energy. For example, a bowing
parameter of 2.5 eV was obtained from optical absorption measurements and a value
of 4.4 eV was obtained from the position of the emission peaks [384]. Nagatomo
et al. [366] noted that the InxGa1xN lattice constant varies linearly with the In mole
fraction up to at least x ¼ 0.42, but it violates the Vegards law for x > 0.42, which may
be caused by erroneous determination of the composition and illustrates well the
problem at hand. Even additional investigations did not agree on the exact value of the
bowing parameter. For example, a value for bInGaN ¼ 3.9 0.5 eV was reported when
0.9 eV was used for the InN gap, but the bowing parameter had to be increased to
5.1 0.4 eV when 1.9 eV was used for the InN gap [385]. Using the bandgap determined by PL, a bowing parameter of 4.5 eV was also reported [386]. However, when
reflectivity measurements together with PL data corrected for Stokes shift were used,
bInGaN ¼ 2.5 0.7 eV was obtained for 0.9 eV bandgap of InN and bInGaN ¼ 3.5 0.7
eV for 1.9 eV InN bandgap. Optical transmission measurements led to a bowing
parameter of 8.4 eV [387]. At least one theoretical effort resulted in a bowing parameter
of 1.2 eV [377]. In fact, linear bandgap dependence on composition with a slope of
3.57 eV for up to 25% InN content has also been reported [388]. Linear dependence
with a slope of 4.1 eV for InN mole fraction, x < 0.12, has been reported in another
publication as well [389]. Wu et al. [390] visited the bandgap dependence of InGaN on
composition by considering 0.8 eV for the bandgap of the end binaryInN. Figure 1.39
shows the composition dependence of the bandgap of InGaN, determined by using
photomodulated transmission [391] and optical absorption [392] measurements, as a
j95
j 1 General Properties of Nitrides
96
Lattice constant (Å)
3 .2
Energy gap (eV)
4
3.3
3 .4
3 .5
3 .6
abs.
300 K PL
PT
Old data
Calorimetric
GaN
3
2
InN
1
0
0.0
0.2
0.4
0.6
0.8
1.0
Composition (x)
Figure 1.39 PL peak energy and bandgap of InGaN determined by
optical absorption as a function of composition, as compiled in
Ref. [390], including previously reported data for InN. The solid
curve shows the fit to the bandgap energies (determined by
absorption and phototransmission) using a bowing parameter
b ¼ 1.43 eV [390].
function of GaN fraction. The data near the GaN binary end include those reported by
Pereria et al. [392], Shan et al. [390], and ODonnel et al. [393].
Care was taken by observing the dependence of squared absorption coefficient on
light probe energy and seeing nearly a linear dependence to gain confidence on the
measured bandgap and also confirming the values by bandgaps determined by
photomodulated transmission measurements. The slight deviation from linearity
near the InN end of the ternary has been attributed to the nonparabolicity of the
conduction band caused by the k p interaction between the G6 symmetry conduction
band and the G8 symmetry valence bands. As shown by the solid curve in Figure 1.39,
the compositional dependence of the bandgap in the entire composition range can be
well fit by a bowing parameter of b ¼ 1.43 eV. Shown in Figure 1.39 with dashed line is
the fit to the empirical expression using energy of 1.9 eV for InN and bowing
parameter of 2.63 eV to demonstrate that it does represent the Ga-rich side of the
compositions well. However, the bowing parameter of 1.43 eV that is good for the
entire compositional range is the one that utilizes 0.77 eV for the bandgap of InN.
In an investigation with a different set of objectives, Yoshimoto et al. [368] studied
the effect of growth conditions on the carrier concentration and transport properties
of InxGa1xN. They observed that if the deposition temperature is increased from 500
to 900 C, InxGa1xN grown on sapphire with x 0.2 suffers from a reduction in
carrier concentration from 1020 to 1018 cm3, but gains from an increase in the
carrier mobility from less than 10 to 100 cm2 V1 s1. The same group later noted that
this trend does not change if the films are grown on ZnO substrates instead of
sapphire [369]. They could achieve good InGaN material with In mole fractions as
1.5 Ternary and Quaternary Alloys
large as 23%. Nakamura and Mukai [394] discovered that the film quality of
InxGa1xN could be significantly improved if these films are grown on high-quality
GaN films. Thus, from the reports cited above it may be concluded that the major
challenge for obtaining high-mobility InGaN is to find a compromise in the growth
temperature, because InN is unstable at typical GaN deposition temperatures. This
growth temperature would undoubtedly be a function of the dopant atoms, as well
as the method (MBE, OMVPE, etc.) used for the growth. This is evident from a
study by Nakamura et al., who have since expanded the study of InGaN employing
Si [395] and Cd [396] as dopants. A review of various transport properties of GaInN
and AlInN by Bryden and Kistenmacher [296] is available but predates the bandgap
reconsideration of InN; the growth and mobility of p-GaInN is discussed by
Yamasaki et al. [344].
1.5.3
InAlN Alloy
In1xAlxN is an important compound that can provide a lattice-matched barrier to
GaN, low fraction AlGaN, and InGaN, and consequently lattice-matched AlInN/
AlGaN or AlInN/InGaN heterostructures. Although there was some discrepancy as
to which composition really lattice-matched GaN, continued improvement in layer
quality and persistence narrowed the In composition for matching. Compositions
In0.29Al0.71N and In0.17Al0.83N have been reported as matching, but the value around
the latter composition is gaining more acceptance [397]. The growth and electrical
properties of this semiconductor have not yet been as extensively studied compared to
the other two ternaries, particularly AlGaN, as the growth of this ternary is also
challenging because of diverse thermal stability, lattice constant, and cohesive energy
of AlN and InN. Moreover, thermal instability resulting from, for example, the
spinodal phase separation phenomenon, which is more of an issue in Al1xInxN than
in InyGa1yN [398], must be considered. Despite the above-mentioned difficulties,
lattice matching and the lack of crack formation when AlGaN is replaced with InAlN
in distributed Bragg reflectors (DBRs) and other structures requiring relatively
thicker layers are more than enough to pursue this material. In fact, light emitters,
field effect transistors, and DBRs, as mentioned, using InAlN barriers as opposed to
AlGaN are gaining considerable momentum.
As in the case of AlGaN and InGaN, the calculated lattice parameter of this alloy
follows Vegards law [347] (also reviewed in Ref. [17]) as
aAlx In1 x N ¼ 3:58480 4753x Å
and c Alx In1 x N ¼ 5:8002 0:8063x Å:
ð1:29Þ
By utilizing various tools such as HRXRD, the experimental data for various AlInN
layers support the applicability of Vegards law in that the experimental data
aAlx In1 x N ¼ ð3:560 0:019Þ ð0:449 0:019Þx Å and c ¼ ð5:713 0:014Þ ð0:745 0:024Þx Å are within about 2% of that predicted by linear interpolation,
the Vegard law. As in the case of AlGaN and InGaN, the bond lengths exhibit a
j97
j 1 General Properties of Nitrides
98
nonlinear behavior, deviating from the virtual crystal approximation. Essentially, the
nearest neighbor bond lengths are not as dependent on composition as might be
expected from the virtual crystal approximation.
Early experimental results [288] for the bandgap of In0.29Al0.71N, which was
thought lattice matched to GaN, indicate that this alloy has an energy gap of 3.34 eV
at low temperatures (the room-temperature value is actually closer to 4.5 eV) that is
even below that for GaN. The estimations by Wright and Nelson [399] that followed
pointed to a bandgap of about 5 eV for the zinc blende variety. The accompanying
bowing parameter reported by Wright and Nelson is 2.53 eV at the time when the
larger InN bandgap was accepted. Naturally, when the bandgap is in question the
bowing parameter is even more in question. As in the case of AlGaN and InGaN,
the compositional dependence of the bandgap of AlInN can be expressed with the
following empirical expression using a bowing parameter, bAlInN, as
g
g
g
E Alx InN ¼ xE AlN þ ð1 xÞE InN bAlInN xð1 xÞ
¼ 6:1x þ 0:7ð1 xÞ bAlInN xð1 xÞ eV:
ð1:30Þ
In addition to the aforementioned calculations, experimental data for the bowing
parameter, b, exist. Using a bandgap of 6.2 eV for AlN (the new figure is closer to
6 eV), the values that have been reported encompass b ¼ 3.1 eV deduced by fitting the
bandgap of this alloy determined by PL [400], b ¼ 2.384 eV by absorption measurements but by using 2.0 eV for the bandgap of InN and 5.9 eV for AlN [401], and
b ¼ 5.4 eV in a review where 1.95 eV was used for InN bandgap [17]. Scaling the
bandgap of AlN to about 6.0 eV would reduce the bowing parameter a little. Despite
the scattered data, reasonably useful bandgap variation of AlInN with composition
can be obtained as shown in Figure 1.40.
Kim et al. [404] deposited thin AlInN films with X-ray rocking curve FWHM values
between 10 and 20 arcmin. They observed an increase of In content in AlInN of up to
8% by lowering the substrate temperature to 600 C. A further reduction of substrate
temperature during OMVPE is not useful because of the needed efficient pyrolysis of
ammonia. Yamaguchi et al. [405] also reported on OMVPE growth of AlInN on GaN
templates that were in turn deposited on low-temperature AlN nucleation layers on cplane sapphire. In macroscopic sense, the alloys grown were not phase separated and
the bandgap variation followed the compositional variations in the InN composition
range of 19–44%. From the square of the absorption coefficient versus E–Eg, the
bandgap of the alloy was determined.
Starosta [406] and later Kubota et al. [407] grew InAlN alloy by radio frequency (RF)
sputtering. Kistenmacher et al. [408], however, used the RF magnetron sputtering
(RF MS) from a composite metal target to grow InAlN at 300 C. It was observed that
the energy bandgap E of this semiconductor varies between 2.0 eV (this is supposed
to represent the InN binary end point, which assumes the old and incorrect value)
and 6.20 eV (this too represents the old value for the bandgap of AlN with the new
figure being approximately 6 eV) for x between 0 and 1 [407]. The carrier concentration and the mobility of In1xAlxN for x ¼ 0.04 were 2 · 1020 cm3 and 35 cm2
V1 s1, respectively, and for x ¼ 0.25 were 8 · 1019 cm3 and 2 cm2 V1 s1,
1.5 Ternary and Quaternary Alloys
Lattice constant (Å)
7
a
b
c
d
e
AlN
6
Energy gap (eV)
5
f
g
h
i
j
k
l
4
3
2
InN
1
Eg=6(1–x)+0.7x–3.1x(1–x)
0
0.0
0.2
0.4
0.6
0.8
1.0
Composition (x)
Figure 1.40 Dependence of bandgap of the
InAlN alloy on composition. Unless otherwise
stated, the measurement temperature is room
temperature. The solid line between the 0.8 eV
gap of InN and 6 eV of AlN is deemed as being
reasonably accurate. (a) Absorption; (b) RT PL;
(c) RT absorption [404]; (d) Ref. [401]; (e)
absorption, poly [416]; (f) absorption [402]; (g) a
theory [403]; (h) RT PL and CL [400]; (i) 8 K optical
reflection [400]; (j) RT absorption [405]; (k) RT
PL [405]; (l) fit to Eg ¼ 6(1 x) þ 0.7 x 3.1 x
(1 x). In part courtesy of Wladek Walukiewicz.
respectively [296]. Thus, the mobility decreases substantially with an increase in the
Al mole fraction because the structure of the InAlN approaches the structure of the
insulating AlN.
1.5.4
InAlGaN Quaternary Alloy
By alloying InN together with GaN and AlN, the bandgap of the resulting alloy(s) can
be increased from 1.9 eV (or near 0.7 eV if we use the updated InN bandgap) to a value
of 6.2 eV (or 6 eV if we use the updated value), which is critical for making highefficiency visible light sources and detectors. In addition, the bandgap of this
quaternary can be changed while keeping the lattice constant matched to
GaN [409,410]. In quaternary alloys of nitrides, the N atoms constitute anion
sublattice, whereas group III elements (In, Ga, Al) constitute the cation sublattice.
Use of this quaternary material allows almost independent control of the bandgap
and thus the band offset in AlInGaN-based heterostructures. However, among other
difficulties brought about by the four-component system, the optimal growth
temperature is important to optimize and control, as aluminum-based compounds
generally require higher growth temperatures and In-based ones require lower
j99
j 1 General Properties of Nitrides
100
temperatures. Higher temperatures are also desirable for reducing the O incorporation in the growing film as oxides of Ga and In desorb from the surface. The growth
temperature will therefore govern the limits of In and Al incorporation into the
AlGaInN quaternary alloy [409]. The quaternary alloy (Ga1xAlx)In1yN is expected to
exist in the entire composition range 0 < x < 1 and 0 < y < 1. Unfortunately, as in the
case of the InGaN alloy, incorporation of indium in these quaternary alloys is not easy.
To prevent InN dissociation, InGaN crystals were originally grown at low temperatures (about 500 C) [411], which also applies to InGaAlN. The use of a high nitrogen
flux rate allowed the high-temperature (800 C) growth of high-quality InGaN and
InGaAlN films on (0 0 0 1) sapphire substrates. Note that the incorporation of indium
into InGaN film is strongly dependent on the flow rate, N/III ratio, and growth
temperature in an OMVPE environment. The incorporation efficiency of indium
decreases with increasing growth temperatures. Observations made in the case of
InGaN should be applicable to In incorporation in quaternary nitrides.
Ryu et al. [412] reported on the optical emission in this quaternary system and
AlInGaN/AlInGaN multiple quantum wells grown by pulsed metalorganic chemical
vapor deposition. Strong blue shift with excitation intensity was observed in both the
quaternary layers and quantum wells that was attributed to localization. This would
imply the inhomogeneous nature of the structures and/or presence of band tail states
indicative of early stages of material development and/or serious technological
problems involved.
The relationships between composition and bandgap (or lattice constant) can be
predicted by the equation below, which was originally developed for the InGaAsP
system [413].
Qðx; y; zÞ ¼
xyT 12 ðð1 x þ yÞ=2Þ þ yzT 23 ðð1 y þ zÞ=2Þ þ zxT 31 ðð1 z þ xÞ=2Þ
;
xy þ yz þ zx
T ij ðaÞ ¼ aBj þ ð1 aÞBi þ að1 aÞbij :
The parameters x, y, and z represent the composition of GaN, InN, and AlN. If GaN,
InN, and AlN are represented by 1, 2, and 3, T12 would represent GaxInyN. Further, the
term T12 can be expressed as T 12 ðaÞ ¼ aB2 þ ð1 aÞB1 þ að1 aÞb12 , where b12 is
the bowing parameter for the GaxInyN alloy and a ¼ ð1 x þ yÞ=2 or
ð1 x þ yÞ=2 or ð1 z þ xÞ=2 is the effective molar fraction for GaInN, InAlN, and
AlGaN, respectively, B2 the bandgap of InN, and B1 is the bandgap of GaN. Similar
expressions can be constructed for T23 and T31 by appropriate permutations. An
alternative approach is discussed in conjunction with Equation 1.31.
The results of these calculations for the bandgap and lattice constant dependence
on composition are shown in the three-dimensional diagrams of Figures 1.41–1.43.
An empirical expression similar to that used for the ternaries can also be
constructed for the quaternary as
g
g
g
g
E Alx Iny Ga1 x y N ¼ xE AlN þ yE InN þ ð1 x yÞE GaN
bAlGaN xð1 xÞ bInGaN yð1 yÞ;
ð1:31Þ
1.5 Ternary and Quaternary Alloys
Figure 1.41 Bandgap versus composition for quaternary
AlxInyGa1xyN (assumed InN bandgap ¼ 0.8 eV). (Please find a
color version of this figure on the color tables.)
Figure 1.42 Bandgap versus composition for quaternary
AlxInyGa1xyN (assumed InN bandgap ¼ 1.9 eV). (Please find a
color version of this figure on the color tables.)
j101
j 1 General Properties of Nitrides
102
Figure 1.43 Lattice constant a versus composition for quaternary AlxInyGa1xyN.
where the first three parameters on the right-hand side of the equation are contributions by the binaries to the extent of their presence in the lattice, the third term
represents the bowing contribution related to Al, and the last term depicts the bowing
contribution due to In. The bowing parameters, bAlGaN and bInGaN, indicated in
Equation 1.31 are the same as those discussed in conjunction with InGaN and AlInN.
As such, the values are the same. The parameters x, y, and z represent the molar
fraction of binaries in the quaternary.
After discussing all three ternary alloys of the nitride semiconductor family, the
bandgap (both in terms of energy and also corresponding air wavelength) versus the
lattice parameter is shown in Figure 1.44 for convenience.
The discussion of alloys individually up to this point paves the way to a collective
discussion of alloys in terms of structural parameters for a rapid observation of
trends. This discussion would be of special value particularly for the least discussed of
alloys, InAlN. Let us first discuss the structural properties such as the lattice constants
and bond lengths, and angles of nitride semiconductor alloys, following the discussion in Sections 1.5.1–1.5.3 and that surrounding Figure 1.8, Equation 1.3, and
Table 1.2. Following Ref. [17], the lattice parameter calculated using Equation 1.25
(for AlGaN), Equation 1.27 (for InGaN), and Equation 1.29 (for InAlN) can be used to
calculate the lattice constants for the three ternaries for all compositions and
compared with experiments for AlGaN [414], InGaN [415], and InAlN [416], as
shown in Figure 1.45.
1.5 Ternary and Quaternary Alloys
Figure 1.44 The bandgap versus the lattice parameter for AlGaN,
InGaN, and InAlN using bowing parameters in the same order, 1,
1.43, and 3.1 eV, and bandgap values of 6 eV for AlN, 3.4 eV
for GaN, and 0.8 eV for InN. The lattice constants used for the
binary AlN, GaN, and InN are 3.11, 3.199, and 3.585 Å,
respectively.
Following the case for the binaries tabulated in Table 1.2 and discussed from a
theoretical point of view in Refs [347,417], and the experimental points of view in
Refs [414] (for AlGaN), [415] (for InGaN), and [416] (for AlxIn1xN), the cell
parameter, u, has been calculated for randomly distributed A0.5B0.5N (here A and
B represent the metal components forming the alloy) alloys by the theoretical
approach of Ref. [418], the pertinent parts of which are succinctly discussed in
Section 1.1. The internal cell parameter can be approximately expressed by the
quadratic equation
uAx B1 x N ¼ xuAN þ ð1 xÞuBN bAB xð1 xÞ;
ð1:32Þ
where bAB is the bowing parameter defined as
bAB ¼ 2Y AN þ 2Y BN 4Y A0:5 B0:5 N :
ð1:33Þ
j103
j 1 General Properties of Nitrides
104
3.8
(a)
Lattice constant, a (x) (Å)
3.6
Alx In1–xN
Inx Ga1–xN
3.4
Alx Ga1–x N
3.2
Theory (T = 0 K)
Experiment (T = 300 K)
3.0
0
0.2
0.6
0.4
Molar fraction, x
0.8
6.0
1.0
(b)
5.8
Alx In1–xN
Lattice constant c(x) (Å)
Inx Ga1–xN
5.6
5.4
5.2
Alx Ga1–x N
5.0
Theory (T = 0 K)
Experiment (T = 300 K)
4.8
0
0.2
0.4
0.6
0.8
1.0
Molar fraction, x
Figure 1.45 (a) The a(x) lattice parameter and
(b) the c(x) lattice parameter for random ternary
alloys of AlxGa1xN, InxGa1xN, and AlxIn1xN as
measured by HRXRD at room temperature (solid
lines) and the calculated values using
Equation 1.25 (for AlGaN) and Equation 1.27 (for
InGaN) for T ¼ 0 K (dashed lines). The
agreement between calculations and measured
lattice constants is better than 2% over the entire
range of compositions, compiled in Ref. [17]
utilizing Refs [414,415]. Courtesy of O.
Ambacher.
1.5 Ternary and Quaternary Alloys
The internal cell parameters for each of the three alloys then are
uAlx Ga1 x N ¼ 0:3819x þ 0:3772ð1 xÞ 0:0032xð1 xÞ;
uInx Ga1 x N ¼ 0:3793x þ 0:3772ð1 xÞ 0:0057xð1 xÞ;
uAlx In1 x N ¼ 0:3819x þ 0:3793ð1 xÞ 0:0086xð1 xÞ:
ð1:34Þ
The structural and other polarization related parameters of ternaries do not follow a
linear relationship of the composition, as discussed in detail in Section 2.7. The
nonlinearity in question for an alloy, AxB1xN, where A and B represent the metal
components, is approximated by quadratic equations of the form [418]
Y Ax B1 x N ¼ xY AN þ ð1 xÞY BN bAB xð1 xÞ;
ð1:35Þ
where Y represents any parameter, namely the lattice constant, the u parameter or
polarization, and the bowing parameter is defined in Equation 1.33.
As in the case of binaries discussed in Section 1.1, the cell parameter, u, and the c/a
ratio do not follow the ideal crystal values for the three ternaries of nitride semiconductors. They are shown for the three ternaries for varying composition in
Figure 1.46. Similar to the binaries, tabulated in Table 1.2 in conjunction with
Figure 1.8, the aforementioned two parameters, the nearest and the second neighbor
bond lengths, as well as the bond angles have been calculated for the three ternaries
and those associated with 50% alloys are tabulated in Table 1.34.
As displayed in Figure 1.46, the cell internal parameter increases as one goes from
GaN to InN and, more significantly, to AlN. The nonlinear dependencies on the alloy
composition are described by a bowing parameter, bAB, whose values are 0.0032,
0.0057, and 0.0086 for AlxGa1xN, InxGa1xN, and AlxIn1xN, respectively. The
bowing parameter increases from AlxGa1xN to InxGa1xN, and continues on to
AlxIn1xN. It is worth noting that the bowing parameter is negative for all the three
ternaries, the average cell internal parameter of the same alloys is always above the
ideal value of 0.375. If the lattice constants scale linearly with the alloy composition
but the internal parameter does not, the bond angles and/or the bond lengths of the
real and the virtual crystal must depend nonlinearly on the alloy composition. The
average nearest neighbor bond lengths (b and b1, see Figure 1.8 for a graphical
description) and bond angles (see Figure 1.8 for a graphical description) calculated by
using Equations 1.2–1.4 are shown in Figure 1.47a and b and listed in Tables 1.2
and 1.34. The average cation–anion distances to the nearest and second nearest
neighbors scale nearly linearly with alloy composition for AlxGa1xN, InxGa1xN,
and AlxIn1xN. The average bond length along the c-axis is 0.7–0.9% longer than the
nearest neighbor bonds in the direction of the basal plane (Figure 1.47a).
1.5.5
Dilute GaAs(N)
When small amounts of N and As are incorporated into GaAs and GaN lattices,
respectively, a large negative bandgap bowing parameter results. Consequently, with
j105
j 1 General Properties of Nitrides
106
very small amounts of N in the GaAs lattice, its bandgap can be made very small, to a
point where 1.3 mm lasers and 1.5 mm lasers if In and Sb are also added to the lattice
can all be built with GaAs technology. Anomalously large bandgap bowing parameters exhibited by GaAsN and GaNAs are caused by large chemical and size
1.64
(a)
Ideal
1.63
c(x)/a(x) ratio
1.62
Inx Ga1–xN
Alx Ga1–xN
1.61
Alx In1–xN
1.60
Theory (T = 0 K)
Experiment (T = 300 K)
1.59
0
0.2
0.4
1.0
0.8
0.6
molar fraction, x
0.383
(b)
cell-internal parameter u(x)
0.381
Alx Ga1–x N
b = –0.0032
0.379
Alx In1–xN
b = 0.0086
Inx Ga1–xN
0.377
b = –0.0057
Ideal
0.375
0
0.2
0.6
0.4
molar fraction, x
0.8
1.0
1.5 Ternary and Quaternary Alloys
Table 1.34 Calculated cell internal parameter, a lattice parameter,
c/a ratio, cation–anion distance between the nearest and second
nearest neighbors, and bond angles (given in degrees) for the
three ternary random alloys in the virtual crystal limit with a
composition of 50%.
u
a (Å)
c/a
b (Å)
b1 (Å)
0
b1 (Å)
0
b2 (Å)
0
b3 (Å)
a
b
Al0.5Ga0.5N
In0.5Ga0.5N
In0.5Al0.5N
0.379
3.154
1.620
1.935
1.924
3.175
3.701
3.694
108.80
110.14
0.377
3.392
1.625
2.078
2.073
3.436
3.977
3.975
109.13
109.81
0.378
3.347
1.612
2.042
2.041
3.354
3.921
3.920
108.76
110.18
The distance is in Å and the angles are in degrees [17].
~
differences between As and N [419–422]. Dependence of the bandgap energy in
GaAsN and InPN on nitrogen content is shown in Figure 1.48. To a first extent, the
dashed lines originating from both GaN end (in which case small amounts of As are
added to GaN) and GaAs end (in which case small amounts of N are added to GaAs)
represent the bandgap dependence of GaNAs. However, one must keep in mind that
for both GaAsN and InPN the simple treatment behind the aforementioned
statement fails and that the arrows shown in the figure indicate the boundaries of
the regions where the gap dependence on composition may be predicted with any
accuracy. Also shown is the bandgap variation with composition for other commonly
used ternaries. The thicker vertical line through GaAs represents the bandgap
attainable with GaInAsN, at least in theory, while maintaining lattice matching to
GaAs. The decrease in the lattice constant caused by N can be compensated with In
added to the lattice. The potential of covering a large range of bandgap energies on
GaAs substrates has attracted a great deal of interest in this material system. In fact,
the first laser containing N was an InGaAs(N) active layer one. Owing in part to
Figure 1.46 (a) The c/a ratio for the three
random ternary alloys determined by HRXRD at
room temperature (solid lines) and calculated
using Equation 1.24 for T ¼ 0 K (dashed lines).
The measured and calculated data confirm that
the c/a ratios of Wz InGaN, AlGaN, and AlInN
crystals are always less than the value of 1.633 for
ideal hexagonal crystal. (b) The cell internal
parameter, u, for three random AlGaN, InGaN,
and AlInN alloys calculated using the quadratic
Equation 1.35. The nonlinearity of the internal
cell internal parameter in its compositional
dependence can be described by a negative
bowing parameter b. This bowing parameter is
0.0032, 0.0057, and 0.0086 for AlxGa1xN,
InxGa1xN, and AlxIn1xN, respectively, as
indicated in the figure as well. The u parameter of
the ternaries is always larger than 0.375 that is the
value for an ideal hexagonal crystal [17]. Courtesy
of O. Ambacher.
j107
j 1 General Properties of Nitrides
108
(a)
M-Nc1
InN
2.2
InN
Metal-N average bond length (A)
M-Nb1
2.1
Inx Ga1–xN
Alx In1–xN
2.0
Alx Ga1–xN
M-Nc1
GaN
b=M-Nc1
b1= M-Nb1
1.9
0
0.2
0.4
M-Nb1
AlN
0.6
0.8
1.0
Molar fraction, x
111
(b)
T=0K
AlN
Bond angle of virtual lattice (deg)
InN
Alx In1-x N
110
InN
Alx Ga1-x N
GaN
Inx Ga1-x N
ideal: α = β =109.47 ο
Inx Ga1-x N
109
β
α
Alx Ga1-x N
Alx In1-x N
AlN
108
0
0.2
0.8
0.6
0.4
Molar fraction, x
Figure 1.47 (a) The compositional dependence
of the average nearest neighbor bond lengths, b
and b1 (see Figure 1.8 for a graphical description)
in the virtual crystal limit for the metal–nitrogen
bonds along the c-axis (solid line) and off c-axis
(dashed line). (b) The compositional
dependence of the average bond angles a
(dashed lines) and b (solid lines) of random
1.0
AlxGa1xN, InxGa1xN, and AlxIn1xN alloys (see
Figure 1.8 for a graphical description). Clearly,
the average bond angles deviate noticeably
from the ideal hexagonal crystal for which
a ¼ b ¼ 109.47. Moreover, the deviation
increases from GaN to InN and continues onto
AlN in a nonlinear fashion [17]. Courtesy of O.
Ambacher.
1.5 Ternary and Quaternary Alloys
Γ valley energy gap (eV)
6
AlN
zinc blende
T=0K
5
4
AlP
AlAs
3
GaP
GaN
2
InN
1
AlSb
GaAs
GaAsN
InPN
0
4.5
5.0
5.5
InP
GaSb
InSb
InAs
6.0
6.5
Lattice constant (A)
Figure 1.48 Direct G valley energy gap as a
function of lattice constant for the zinc blende
form of 12 III–V binary compound
semiconductors (filled circles) and some of their
random ternary alloys (lines connecting the solid
circles) at zero temperature. The energy gaps for
certain ternaries such as AlAsP, InAsN, GaAsN,
InPN, and GaPN are extended into regions where
no experimental data have been reported. For
GaAsN and InPN, the arrows indicate the
boundaries of the regions where the gap
dependence on composition may be predicted
with any accuracy, patterned after Ref. [423] with
necessary changes, particularly the one reflecting
the small bandgap of InN.
extreme nonequilibrium conditions employed for growth, MBE is the dominant
growth approach for dilute arsenides with nitrogen. The critical issues are compositional control, incorporation of more than a small percentage of N, doping inefficiency, and layer quality. The situation is exacerbated on all fronts when the N
concentration is increased for achieving 1.5 mm wavelength of emission. Postgrowth
annealing is often employed to improve the crystal quality and/or to increase Si
dopant incorporation, however, at the expense of blue shift in the bandgap. While
GaAsN is chosen here for the present discussion, there are many other dilute nitride
semiconductors as discussed in Section 2.11 in conjunction with band parameters.
As alluded to earlier, the chemical and size differences between the N and As atoms
are the challenges facing experimentalists. In addition, the generation of atomic
nitrogen, although not that different from the technology required for hexagonal GaN
growth [424], deserves some attention. While basic mismatch between N and As can
be dealt with by growing the layers under nonequilibrium conditions, the issue of
atomic nitrogen can be handled by compact RF sources that have seen a good degree
of improvement lately. By adjusting the RF power and pressure in the cell, one can
tailor the source to produce mostly the atomic species by optimizing the emission at
745 nm of wavelength. Note that the substrate and most of the structure are zinc
blende and, consequently, the dilute material assimilates and assumes the same
crystalline structure. The desired nitrogen concentrations are in the range of 1–10%
for red shifting the transitions out to as long as 1.55 mm. Larger growth rates lead to a
reduced incorporation of N in the lattice. Similarly, higher growth temperatures lead
to the same. Consequently, when 1.55 mm wavelength material is desired, lower
j109
j 1 General Properties of Nitrides
110
growth rates must be employed as well as lower growth temperatures. At substrate
temperatures of 500 C or below, if very large As overpressure is employed,
incorporation of N is limited because the flux of atomic nitrogen is small. However,
atomic nitrogen is very reactive and, therefore, compositional control should be
much better as compared to quaternaries relying on P and As (InGaAsP).
As expected, owing to dissimilarities of N and As, the luminescence properties of
GaInNAs degrade rapidly with increasing nitrogen concentration. Employing remedies such as postgrowth annealing enhances the luminescence efficiency of GaInNAs. However, this enhancement is accompanied by a blue shift in the transition in
bulk and quantum well materials. Nitrogen and possibly In diffusion out of GaInAsN
are responsible for the observed luminescence shift to shorter wavelengths.
For completeness, a one-paragraph discussion of device issues will be made in
conjunction with the edge emitting and vertical cavity lasers operating at 1.3 and
1.5 mm portion of the optical spectrum, although other applications such as heterojunction bipolar transistors are possible. Several groups have reported lasers operating at 1.3 mm region [425–442], where the silica-based fiber dispersion is zero, and
1.5 mm region [443–447] (albeit with addition of Sb to the lattice as the quality
required for laser operation for InGaAsN layers cannot be obtained), where the loss is
low, again for the silica-based fibers. Both are intended for telecommunication
purposes. Even 8 W [448] and 12 W [449] CW operation has been reported. Highspeed testing of these lasers has also been performed [450] with data transmission
rates as high as Gbit s1 having been achieved already [451]. For interconnects and
high-speed data links, vertical cavity surface emitting lasers (VCSELs) have received a
great deal of attention. Now that dilute nitrides are becoming potential candidates for
long-wavelength lasers, efforts are under way to explore VCSELs in this material
system as well [452,453].
References
1 Lei, T., Fanciulli, M., Molnar, R.J.,
Moustakas, T.D., Graham, R.J. and
Scanlon, J. (1991) Applied Physics Letters,
59, 944.
2 Paisley, M.J., Sitar, Z., Posthill, J.B. and
Davis, R.F. (1989) Journal of Vacuum
Science & Technology, 7, 701.
3 Powell, R.C., Lee, N.E., Kim, Y.W. and
Greene, J.E. (1993) Journal of Applied
Physics, 73, 189.
4 Mizita, M., Fujieda, S., Matsumoto, Y. and
Kawamura, T. (1986) Japanese Journal of
Applied Physics, 25, L945.
5 Xia, Q., Xia, H. and Ruoff, A.L. (1993)
Journal of Applied Physics, 73, 8198.
6 Perlin, P., Jauberthie-Carillon, C., Itie,
J.P., San Miguel, A., Grzegory, I. and
Polian, A. (1992) Physical Review B:
Condensed Matter, 45, 83.
7 Ueno, M., Yoshida, M., Onodera, A.,
Shimommura, O. and Takemura, K.
(1994) Physical Review B: Condensed
Matter, 49, 14.
8 Pirouz, P. and Yang, J.W. (1993)
Ultramicroscopy, 51, 189.
9 Ruterana, P., Sanchez, A.M. and Nouet,
G. (2003) Extended defects in wurtzite
GaN layers: atomic structure, formation
and interaction mechanisms, in Nitride
Semiconductors – Handbook on Materials
References
10
11
12
13
14
15
16
17
18
and Devices (eds P. Ruterana, M. Albrecht
and J. Neugebauer), Wiley-VCH Verlag
GmbH, Weinheim, Germany.
Harris, W.A. (1980) Electronic Structure
and Properties of Solids, Dover, NY, pp.
174–179.
Yeh, C.-Y., Lu, Z.W., Froyen, S. and
Zunger, A. (1992) Physical Review B:
Condensed Matter, 46, 10086.
Bernardini, F., Fiorentini, V. and
Vanderbilt, D. (2001) Accurate calculation
of polarization-related quantities in
semiconductors. Physical Review B:
Condensed Matter, 63, 193–201.
Bechstedt, F., Großner, U. and
Furthm€
uller, J. (2000) Physical Review B:
Condensed Matter, 62, 8003.
Wei, S.-H. and Zunger, A. (1996) Applied
Physics Letters, 69, 2719.
Leszczynski, M., Teisseyre, H., Suski, T.,
Grzegory, I., Bockowski, M., Jun, J.,
Porowski, S., Pakula, K., Baranowski,
J.M., Foxon, C.T. and Cheng, T.S. (1996)
Lattice parameters of gallium nitride.
Applied Physics Letters, 69, 73.
Leszczynski, M. (1999) Common crystal
structure of the group III-nitrides, in
Properties, Processing and Applications of
Gallium Nitride and Related
Semiconductors (eds J.H. Edgar, S. Strite, I.
Akasaki, H. Amano and C. Wetzel), EMIS
Data Review Series, No. 23, INSPEC, The
Institution of Electrical Engineers,
Stevenage, UK, pp. 3–5.
Ambacher, O., Majewski, J., Miskys, C.,
Link, A., Hermann, M., Eickhoff, M.,
Stutzmann, M., Bernardini, F.,
Fiorentini, V., Tilak, V., Schaff, B. and
Eastman, L.F. (2002) Pyroelectric
properties of Al(In)GaN/GaN heteroand quantum well structures. Journal of
Physics: Condensed Matter, 14,
3399–3434.
Leszczynski, M., Suski, T., Perlin, P.,
Teisseyre, H., Grzegory, I., Bockowski,
M., Jun, J., Porowski, S., Pakula, K.,
Baranowski, J.M., Foxon, C.T. and Cheng,
T.S. (1996) Applied Physics Letters,
69, 73.
19 Jeffery, G.A., Parry, G.S. and Mozzi, R.L.
(1956) Journal of Chemical Physics, 25,
1024.
20 Schulz, H. and Theimann, K.H. (1977)
Solid State Communications, 23, 815.
21 Tanaka, M., Nakahata, S., Sogabe, K.,
Nakata, H. and Tabioka, M. (1997)
Japanese Journal of Applied Physics, 36,
L1062.
22 Angerer, H., Brunner, D., Freudenberg,
F., Ambacher, O., Stutzmann, M., H€opler,
R., Metzger, T., Born, E., Dollinger, G.,
Bergmaier, A., Karsch, S. and K€orner,
H.-J. (1997) Applied Physics Letters, 71,
1504.
23 Domagala, J., Leszczynski, M., Prystawko,
P., Suski, T., Langer, R., Barski, A. and
Bremser, M. (1999) Journal of Alloys and
Compounds, 286, 284.
24 Kim, K., Lambrecht, W.R.L. and Segall, B.
(1996) Physical Review B: Condensed
Matter, 53, 16310.
25 Wright, A.F. and Nelson, J.S. (1995)
Physical Review B: Condensed Matter, 51,
7866.
26 Detchprohm, T., Hiramatsu, K., Itoh, K.
and Akasaki, I. (1992) Japanese Journal of
Applied Physics, 31, L1454.
27 Leszczynski, M., Teisseyre, H., Suski, T.,
Grzegory, I., Bockowski, M., Jun, J.,
Porowski, S. and Major, J. (1995) Journal of
Physics D: Applied Physics, 69, A149.
28 Deguchi, T., Ichiryu, D., Toshikawa, K.,
Sekiguchi, K., Sota, T., Matsuo, R.,
Azuhata, T., Yamaguchi, M., Yagi, T.,
Chichibu, S. and Nakamura, S. (1999)
Journal of Applied Physics, 86, 1860.
29 Paszkowicz, W. (1999) Powder Diffraction,
14, 258.
30 Wu, J., Walukiewicz, W., Yu, K.M., Ager,
J.W., III, Haller, E.E., Lu, H., Schaff, W.J.,
Saito, Y. and Nanishi, Y. (2003) Applied
Physics Letters, 80, 3967.
31 Phillips, J.C. (1973) Bonds and Bands in
Semiconductors, Academic Press, New
York, p. 31.
32 Xu, Y.-N. and Ching, W.Y. (1993)
Electronic, optical, and structural
properties of some wurtzite crystals.
j111
j 1 General Properties of Nitrides
112
33
34
35
36
37
38
39
40
41
42
Physical Review B: Condensed Matter, 48,
4335–4350.
Morkoc, H., Strite, S., Gao, G.B., Lin,
M.E., Sverdlov, B. and Burns, M. (1994)
Large-band-gap SiC, III–V nitride, and
II–VI ZnSe-based semiconductor device
technologies. Journal of Applied Physics, 76
(3), 1363–1398.
Akasaki, I. and Amano, H. (1994)
Properties of Group III Nitrides (ed. J.H.
Edgar), EMIS Data Review Series, No. 11,
INSPEC, The Institution of Electrical
Engineers, Stevenage, UK, pp. 30–34.
Christensen, N.E. and Gorczyca, I. (1994)
Optical and structural properties of III–V
nitrides under pressure. Physical Review
B: Condensed Matter, 50, 4397–4415.
Levinshtein, M., Rumyantsev, S. and
Shur, M. (eds) (1996/1999) Handbook
Series on Semiconductor Parameters, vols
1 and 2, World Scientific, London.
Bougrov, V., Levinshtein, M.E.,
Rumyantsev, S.L. and Zubrilov, A. (2001)
Properties of Advanced Semiconductor
Materials GaN, AlN, InN, BN, SiC, SiGe
(eds M.E. Levinshtein, S.L. Rumyantsev
and M.S. Shur), John Wiley & Sons, Inc.,
New York, pp. 1–30.
Polian, A., Grimsditch, M. and Grzegory,
I. (1996) Journal of Applied Physics, 79,
3343–3344.
Kisielowski, C., Kr€
uger, J., Ruvimov, S.,
Suski, T., Ager, J.W., III, Jones, E.,
Liliental-Weber, Z., Rubin, M., Weber,
E.R., Bremser, M.D. and Davis, R.F.
(1996) Physical Review B: Condensed
Matter, 54, 17745.
Nikolaev, V., Shpeizman, V. and Smirnov,
B. (1998) The Second Russian Workshop
on GaN, InN, and AIN-Structures and
Devices, June 2, St Petersburg Technical
University, St Petersburg, Russia.
Drory, M.D., Ager, J.W., Suski, T.,
Grzegory, I. and Porowski, S. (1996)
Hardness and fracture toughness of bulk
single crystal gallium nitride. Applied
Physics Letters, 69 (26), 4044–4046.
Wright, A.F. (1997) Elastic properties of
zinc-blende and wurtzite AlN, GaN, and
43
44
45
46
47
48
49
50
51
52
53
InN. Journal of Applied Physics, 82 (6),
2833–2839.
Madelung, O. (ed.) (1991) Semiconductor:
Group IV Elements and III–V Compound,
Data in Science and Technology (series ed.
R. Poerschke), Springer, Berlin.
Truell, R., Elbaum, C. and Chick, B.B.
(1969) Ultrasonic Methods in Solid State
Physics, Academic Press, New York.
Kim, K., Lambrecht, W.R.L. and Segall, B.
(1994) Physical Review B: Condensed
Matter, 50, 1502.
Ueno, M., Yoshida, M., Onodera, A.,
Shimomura, O. and Takemura, K. (1994)
Physical Review B: Condensed Matter,
49, 14.
Leszczynski, M., Podlasin, S. and Suski, T.
(1993) Journal of Applied Crystallography,
32, 1528.
Qian, W., Skowronski, M. and Rohrer,
G.R. (1996) Structural defects and their
relationship to nucleation of GaN thin
films, in III-Nitride, SiC, and Diamond
Materials for Electronic Devices,
Materials Research Society Symposium
Proceedings, vol. 423 (eds D.K. Gaskill,
C.D. Brandt and R.J. Nemanich),
Pittsburgh, PA, pp. 475–486.
Sheleg, A.U. and Savastenko, V.A. (1977)
Vestsi Akademii Nauk, Seria. Fizika i
Matematika Nauk, 126.
Porowski, S. (1997) Materials Science &
Engineering B: Solid State Materials for
Advanced Technology, 44, 407–413.
Barin, I., Knacke, O. and Kubaschewski,
O. (1977) Thermochemical Properties of
Inorganic Substances, Springer, Berlin.
Fritsch, D., Schmidt, H. and Grundmann,
M. (2003) Band-structure pseudopotential
calculation of zinc-blende and wurtzite
AlN, GaN, and InN. Physical Review B:
Condensed Matter, 67, 235205.
Chow, T.P. and Ghezzo, M.J. (1996) SiC
power devices, in III-Nitride, SiC, and
Diamond Materials for Electronic
Devices, Materials Research Society
Symposium Proceedings, vol. 423 (eds
D.K. Gaskill, C.D. Brandt and R.J.
Nemanich), Pittsburgh, PA, pp. 69–73.
References
54 Ejder, E. (1971) Physica Status Solidi a:
Applied Research, 6, 445–448.
55 Barker, A.S., Jr and Ilegems, M. (1973)
Infrared lattice vibrations and freeelectron dispersion in GaN. Physical
Review B: Condensed Matter, 7, 743.
56 Manchon, D.D., Jr, Barker, A.S., Jr, Dean,
P.J. and Zetterstrom, R.B. (1970) Solid
State Communications, 8, 1227.
57 Siegle, H., Kaczmarczyk, G., Filippidis, L.,
Litvinchuk, L., Hoffmann, A. and
Thornsen, C. (1997) Zone-boundary
phonons in hexagonal and cubic GaN.
Physical Review B: Condensed Matter, 55
(11), 7000–7004.
58 Karch, K., Wagner, J.-M. and Bechstedt, F.
(1998) Ab initio study of structural,
dielectric, and dynamical properties of
GaN. Physical Review B: Condensed Matter,
57, 7043–7049.
59 Zi, J., Wan, X., Wei, G., Zhang, K. and Xie,
X. (1996) Journal of Physics: Condensed
Matter, 8, 6323–6328.
60 Lemos, V., Arguello, C.A. and Leite,
R.C.C. (1972) Solid State Communications,
11, 1351.
61 Dingle, R. and Ilegems, M. (1971) Solid
State Communications, 9, 175.
62 Rheinlander, A. and Neumann, H. (1974)
Physica Status Solidi b: Basic Research, 64,
K123.
63 Bloom, S., Harbeke, G., Meier, E. and
Ortenburger, I.B. (1974) Physica Status
Solidi b: Basic Research, 66, 161–168.
64 Pankove, J.I., Bloom, S. and Harbeke, G.
(1975) RCA Review, 36, 163.
65 Look, D.C. and Sizelove, J.R. (2001)
Predicted mobility in bulk GaN. Applied
Physics Letters, 79 (8), 1133–1135.
66 Stroscio, M.A. and Dutta, M. (2001)
Phonons in Nanostructures, Cambridge
University Press.
67 Duboz, J.Y. (2002) Hot photoluminescence
in GaN: carrier energy relaxation and
hot phonon effects. Journal of Applied
Physics, 92 (8), 4312–4319.
68 Lee, B.C., Kim, K.W., Dutta, M. and
Stroscio, M.A. (1997) Physical Review B:
Condensed Matter, 56, 997.
69 Ramirez-Flores, G., Navarro-Contreras,
H., Lastras-Martinez, A., Powell, R.C. and
Greene, J.E. (1994) Physical Review B:
Condensed Matter, 50, 8433.
70 Fan, J.W., Li, M.F., Chong, T.C. and Xia,
J.B. (1996) Electronic properties of zincblende GaN, AlN, and their alloys
Ga1 xAlxN. Journal of Applied Physics, 79
(1), 188–194.
71 Chang, W.Y. and Harmon, B.N. (1986)
Physical Review B: Condensed Matter, 34,
5305.
72 Hejda, B. and Hauptmanova, K. (1969)
Physica Status Solidi, 36, K95.
73 Blum, S. (1971) Journal of Physics and
Chemistry of Solids, 32, 2027.
74 Jones, D. and Lettington, A.H. (1972)
Solid State Communications, 11, 701.
75 Kobayasi, A., Sankey, O.F., Volz, S.M. and
Dow, J.D. (1983) Physical Review B:
Condensed Matter, 28, 935.
76 Huang, M.Z. and Ching, W.Y. (1985)
Journal of Physics and Chemistry of Solids,
46, 977.
77 Slack, G.A. (1973) Journal of Physics and
Chemistry of Solids, 34, 321–335.
78 Goldberg, Y. (2001) Properties of Advanced
Semiconductor Materials GaN, AlN, InN,
BN, SiC, SiGe (eds M.E. Levinshtein, S.L.
Rumyantsev and M.S. Shur), John Wiley
& Sons, Inc., New York, pp. 31–47.
79 Gerlich, D., Dole, S.L. and Slack, G.A.
(1986) Journal of Physics and Chemistry of
Solids, 47, 437.
80 Thokala, R. and Chaudhuri, J. (1995) Thin
Solid Films, 266 (2), 189–191.
81 Kawabi, K., Tredgold, R.H. and Inuishi, Y.
(1967) Electrical Engineering in Japan,
87, 62.
82 Yonenaga, I., Shima, T. and Sluiter,
M.H.F. (2002) Japanese Journal of Applied
Physics, 41, 4620.
83 McNeil, E., Grimsditch, M. and French,
R.H. (1993) Journal of the American
Ceramic Society, 76 (5), 1132–1136.
84 Vurgaftman, I. and Meyer, J.R. (2003)
Band parameters for nitrogen-containing
semiconductors. Journal of Applied
Physics, 94 (6), 3675–3696.
j113
j 1 General Properties of Nitrides
114
85 Slack, G.A. and Bartram, S.F. (1975)
Journal of Applied Physics, 46 (1), 89–89.
86 Touloukian, Y.S., Kirby, R.K., Taylor, R.E.
and Lee, T.Y.R. (eds) (1977)
Thermophysical Properties of Matter, vol. 13,
Plenum Press, New York.
87 Meng, W.J. (1994) Properties of Group III
Nitrides (ed. J.H. Edgar), IEE EMIS Data
Review Series, No. 11, INSPEC, The
Institution of Electrical Engineers,
Stevenage, UK, pp. 22–29.
88 Krukowski, S., Leszczynski, M. and
Porowski, S. (1999) Thermal properties of
the group III nitrides, in Properties,
Processing and Applications of Gallium
Nitride and Related Semiconductors (eds
J.H. Edgar, S. Strite, I. Akasaki, H. Amano
and C. Wetzel), EMIS Data Review Series,
No. 23, INSPEC, The Institution of
Electrical Engineers, Stevenage, UK, pp.
21–28.
89 Sirota, N.N. and Golodushko, V.Z. (1974)
Tezisy Dokl., Vses Konf. Khi., Svyazi
Poluprovdn. Polumetallakh 5th, p. 98.
90 Slack, G.A., Tanzilli, R.A., Pohl, R.O. and
Vandersande, J.W. (1987) Journal of
Physics and Chemistry of Solids, 48 (7),
641–647.
91 Slack, G., Schowalter, L., Rojo, J.,
Morelli, D. and Freitas, J. (2002)
Proceedings of the International
Workshop on Bulk Nitrides, May 2002,
Amazonas, Brazil, Journal of Crystal
Growth, 246 (3–4), 287–298; For an
application of the method to synthetic
diamond, see Morelli, D.T., Beetz, C.P.
and Perry, T.A. (1988) Thermal
conductivity of synthetic diamond
films. Journal of Applied Physics, 64 (6),
3063–3066.
92 MacChesney, J.B., Bridenbaugh, P.M.
and OConnor, P.B. (1970) Materials
Research Bulletin, 5, 783.
93 Koshchenko, V.I., Grinberg, Ya.Kh. and
Demidienko, A.F. (1984) Inorganic
Materials, 20 (11), 1550–1553.
94 Ribeiro, C.T.M., Alvarez, F. and Zanatta,
A.R. (2002) Applied Physics Letters,
81, 1005.
95 See, for example, Colthup, N.B., Daly,
L.H. and Wiberley, S.E. (1990) Infrared
and Raman Spectroscopy, Academic Press,
San Diego, FL.
96 Ren, Z.M., Lu, Y.F., Ni, H.Q., Liew, T.Y.F.,
Cheong, B.A., Chow, S.K., Ng, M.L. and
Wang, J.P. (2000) Journal of Applied
Physics, 88, 7346.
97 Chang, W.Y. and Harmon, B.N. (1986)
Physical Review B: Condensed Matter, 34,
5305.
98 Hejda, B. and Hauptmanova, K. (1969)
Physica Status Solidi, 36, K95.
99 Blum, S. (1971) Journal of Physics and
Chemistry of Solids, 32, 2027.
100 Jones, D. and Lettington, A.H. (1972)
Solid State Communications, 11, 701.
101 Kobayashi, A., Sankey, O.F., Volz, S.M.
and Dow, J.D. (1983) Physical Review B:
Condensed Matter, 28, 935.
102 Huang, M.Z. and Ching, W.Y. (1985)
Journal of Physics and Chemistry of Solids,
46, 977.
103 Guo, Q. and Yoshida, A. (1994) Japanese
Journal of Applied Physics, Part 1: Regular
Papers, Short Notes & Review Papers, 33
(5A), 2453–2456.
104 Teisseyre, H., Perlin, P., Suski, T.,
Grzegory, I., Porowski, S., Jun, J.,
Pietraszko, A. and Moustakas, T.D. (1994)
Temperature dependence of the energy
gap in GaN bulk single crystals and
epitaxial layer. Journal of Applied Physics,
76 (4), 2429–2434.
105 Yamashita, H., Fukui, K., Misawa, S. and
Yoshida, S. (1979) Journal of Applied
Physics, 50, 896.
106 Roskovcova, L. and Pastrnak, J. (1980)
Czechoslovak Journal of Physics B, 30, 586.
107 Silveira, E., Freitas, J.A., Jr, Kneissl, M.,
Treat, D.W., Johnson, N.M., Slack, G.A.
and Schowalter, L.J. (2004) Nearbandedge cathodoluminescence of an
AlN homoepitaxial film. Applied Physics
Letters, 84 (18), 3501–3503.
108 Silveira, E., Freitas, J.A., Jr, Glembocki,
O.J., Slack, G.A. and Schowalter, L.J.
(2005) Excitonic structure of bulk AlN
from optical reflectivity and
References
109
110
111
112
113
114
115
116
117
118
119
120
121
cathodoluminescence measurements.
Physical Review B: Condensed Matter, 71,
041201(R)-1–041201(R)-4.
Gorczyca, I. and Christensen, N.E. (1993)
Physica B, 185, 410–414.
Gorczyca, I., Svane, A. and Christensen,
N.E. (1997) Internet Journal of Nitride
Semiconductor Research, 2, article 18.
Collins, A.T., Lightowlers, E.C. and Dean,
P.J. (1967) Physical Review, 158 (3),
833–838.
Chu, T.L. and Keln, R.W., Jr (1975) Journal
of the Electrochemical Society, 122, 995.
Moore, W.J., Freitas, J.A., Jr, Holm, R.T.,
Kovalenkov, O. and Dmitriev, V. (2005)
Infrared dielectric function of wurtzite
aluminum nitride. Applied Physics Letters,
86, 141912-1–141912-3.
Suzuki, M. and Uenoyama, T. (1995)
Physical Review B: Condensed Matter, 52,
8132.
Kim, K., Lambrecht, W.R.L. and Segall, B.
(1997) Physical Review B: Condensed
Matter, 56, 7363.
Suzuki, M. and Uenoyama, T. (1996)
Strain effect on electronic and optical
properties of GaN/AlGaN quantum-well
lasers. Journal of Applied Physics, 80 (12),
6868–6874.
Sanjurjo, J.A., Lopez-Cruz, E., Vogi, P.
and Cardona, M. (1983) Dependence on
volume of the phonon frequencies and
the IR effective charges of several III–V
semiconductors. Physical Review B:
Condensed Matter, 28, 4579.
Carlone, C., Lakin, K.M. and Shanks, H.R.
(1984) Journal of Applied Physics, 55, 4010.
MacMillan, M.F., Devaty, R.P. and
Choyke, W.J. (1993) Infrared reflectance
of thin aluminum nitride films on various
substrates. Applied Physics Letters, 62 (7),
750–752.
Perlin, P., Polian, A. and Suski, T. (1993)
Raman-scattering studies of aluminum
nitride at high pressure. Physical Review B:
Condensed Matter, 47 (5), 2874–2877.
Van Camp, P.E., Van Doren, V.E. and
Devreese, J.T. (1991) High-pressure
properties of wurtzite- and rocksalt-type
122
123
124
125
126
127
128
129
130
131
aluminum nitride. Physical Review B:
Condensed Matter, 44 (16), 9056–9059.
Pearson, W.B. (1967) A Handbook of
Lattice Spacings and Structures of Metals
and Alloys, Pergamon Press, Oxford.
Pichugin, I.G. and Tiachala, M. (1978)
Izvestia Akademii Nauk SSSR,
Neorganicheskie Materialy, 14, 175.
Zubrilov, A. (2001) Properties of Advanced
Semiconductor Materials GaN, AlN, InN,
BN, SiC, SiGe (eds M.E. Levinshtein, S.L.
Rumyantsev and M.S. Shur), John Wiley
& Sons, Inc., New York, pp. 49–66.
Edgar, J.H., Wei, C.H., Smith, D.T.,
Kistenmacher, T.J. and Bryden, W.A.
(1997) Journal of Materials Science, 8, 307.
Perlin, P., Iota, V., Weinstein, B.A.,
Wisniewski, P., Suski, T., Eliseev, P.G. and
Osinski, M. (1997) Influence of pressure
on photoluminescence and
electroluminescence in GaN/InGaN/
AlGaN quantum wells. Applied Physics
Letters, 70, 2993–2995.
Krukowski, S., Witek, A., Adamczyk, J.,
Jun, J., Bockowski, M., Grzegory, I.,
Lucznik, B., Nowak, G., Wroblewski, M.,
Presz, A., Gierlotka, S., Stelmach, S.,
Palosz, B., Porowski, S. and Zinn, P.
(1998) Thermal properties of indium
nitride. Journal of Physics and Chemistry of
Solids, 59, 289–295.
Davydov, V.Yu., Emtsev, V.V., Goncharuk,
A.N., Smirnov, A.N., Petrikov, V.D.,
Mamutin, V.V., Vekshin, V.A., Ivanov,
S.V., Smirnov, M.B. and Inushima, T.
(1999) Experimental and theoretical
studies of phonons in hexagonal InN.
Applied Physics Letters, 75, 3297–3299.
Tansley, T.L. (1994) Properties of Group III
Nitrides (ed. J.H. Edgar), INSPEC,
London, p. 39.
Sobolev, V.V. and Zlobina, M.A. (1999)
Semiconductors, 33, 395.
Tyagai, V.A., Evstigneev, A.M., Krasiko,
A.N., Andreeva, A.F. and Malakhov,
V.Ya. (1977) Soviet Physics: Semiconductors,
11, 1257–1259; Fizika i Tekhnika
Poluprovodnikov, 1977, 11, 2142 (in
Russian).
j115
j 1 General Properties of Nitrides
116
132 Inushima, T., Shiraishi, T. and Davydov,
V.Yu. (1999) Phonon structure of InN
grown by atomic layer epitaxy. Solid State
Communications, 110 (9), 491–495.
133 Lambrecht, W.R. and Segall, B. (1993)
Anomalous band-gap behavior and phase
stability of c-BN-diamond alloys. Physical
Review B: Condensed Matter, 47,
9289–9296.
134 Yeo, Y.C., Chong, T.C. and Li, M.F. (1998)
Electronic band structures and effectivemass parameters of wurtzite GaN and
InN. Journal of Applied Physics, 83,
1429–1436.
135 Pugh, S.K., Dugdale, D.J., Brand, S. and
Abram, R.A. (1999) Semiconductor Science
and Technology, 14, 23–31.
136 Foley, C.P. and Tansley, T.L. (1986)
Pseudopotential band structure of indium
nitride. Physical Review B: Condensed
Matter, 33, 1430.
137 Yeh, C.-Y., Lu, Z.W., Froyen, S. and
Zunger, A. (1992) Zinc-blende-wurtzite
polytypism in semiconductors. Physical
Review B: Condensed Matter, 46,
10086–10097.
138 Bechstedt, F. and Furthm€
uller, J. (2002)
Do we know the fundamental energy gap
of InN? Journal of Crystal Growth, 246
(3–4), 315–319.
139 Johnson, W.C., Parson, J.B. and Crew,
M.C. (1932) The Journal of Physical
Chemistry, 36, 2561.
140 Maruska, H.P. and Tietjen, J.J. (1969)
Applied Physics Letters, 15, 327.
141 Pankove, J.I. (1972) Journal of the
Electrochemical Society, 119, 1110.
142 Chu, T.L. (1971) Journal of the
Electrochemical Society, 118, 1200.
143 Lakshmi, E. (1981) Thin Solid Films, 83,
L137.
144 Morimoto, Y. (1974) Journal of the
Electrochemical Society, 121, 1383.
145 Shintani, A. and Minagawa, S. (1976)
Journal of the Electrochemical Society, 123,
706.
146 Itoh, K., Amano, H., Hiramatsu, K. and
Akasaki, I. (1991) Japanese Journal of
Applied Physics, 30, 1604.
147 Ito, K., Hiramatsu, K., Amano, H. and
Akasaki, I. (1990) Journal of Crystal
Growth, 104, 533.
148 Visconti, P., Reshchikov, M.A., Jones,
K.M., Wang, D.F., Cingolani, R., Morkoc,
H., Molnar, R.J. and Smith, D.J. (2001)
Highly selective photoelectro-chemical
etching of nitride materials for defect
investigation and device fabrication.
Journal of Vacuum Science & Technology B:
Microelectronics and Nanometer Structures,
19 (4), 1328–1333.
149 Mohammad, S.N., Salvador, A. and
Morkoc, H. (1995) Proceedings of the IEEE,
83, 1306.
150 Pearton, S.J., Zolper, J.C., Shul, R.J. and
Ren, F. (1999) GaN: processing, defects,
and devices. Journal of Applied Physics,
86, 1.
151 Maruska, H.P., Anderson, L.J. and
Stevenson, D.A. (1974) Journal of the
Electrochemical Society, 121, 1202.
152 Lagerstedt, O. and Monemar, B. (1979)
Physical Review B: Condensed Matter, 19,
3064.
153 Maruska, H.P., Anderson, L.J. and
Stevenson, D.A. (1974) Journal of the
Electrochemical Society, 121, 1202.
154 Kr€
uger, J., Sudhir, G.S., Corlatan, D.,
Cho, Y., Kim, Y., Klockenbrink, R.,
Ruvimov, S., Liliental-Weber, Z.,
Kisielowski, C., Rubin, M., Weber, E.R.,
McDermott, B., Pittman, R. and Gertner,
E.R. (1997) Proceedings of the Fall 97
Meeting of the Materials Research
Society, Boston.
155 Leszczynski, M., Suski, T., Domagala, J.
and Prystawko, P. (1999) Lattice
parameters of the group III-nitrides, in
Properties, Processing and Applications of
Gallium Nitride and Related
Semiconductors (eds J.H. Edgar, S. Strite,
I. Akasaki, H. Amano and C. Wetzel),
EMIS Data Review Series, No. 23,
INSPEC, The Institution of Electrical
Engineers, Stevenage, UK, pp. 6–10.
156 Liu, C., Bensching, B., Volz, K. and
Rauschenbach, B. (1997) Applied Physics
Letters, 71, 2313.
References
157 Akasaki, I. and Amano, H. (1994)
Properties of Group III Nitrides (ed. J.H.
Edgar), IEE EMIS Data Review Series,
INSPEC, The Institution of Electrical
Engineers, Stevenage, UK, p. 222.
158 Van Camp, P.E., Van Doren, V.E. and
Devreese, J.T. (1992) Solid State
Communications, 81, 23.
159 Murnaghan, F.D. (1944) Proceedings of the
National Academy of Sciences of the United
States of America, 30, 244.
160 Polian, A. (1999) Mechanical properties of
the group III nitrides, in Properties,
Processing and Applications of Gallium
Nitride and Related Semiconductors (eds
J.H. Edgar, S. Strite, I. Akasaki, H. Amano
and C. Wetzel), EMIS Data Review Series,
No. 23, INSPEC, The Institution of
Electrical Engineers, Stevenage, UK, pp.
11–20.
161 Miwa, K. and Fukumoto, A. (1993)
Physical Review B: Condensed Matter, 48,
7897.
162 Savastenko, V.A. and Sheleg, A.U. (1978)
Physica Status Solidi a: Applied Research,
48, K135.
163 Chetverikova, I.F., Chukichev, M.V. and
Rastorguev, L.N. (1986) Inorganic
Materials, 22, 53.
164 Sherwin, M.E. and Drummond, T.J.
(1991) Journal of Applied Physics, 69, 8423.
165 Deger, C., Born, E., Angerer, H.,
Ambacher, O., Stutzmann, M.,
Hornsteiner, J., Riha, E. and Fischerauer,
G. (1998) Applied Physics Letters, 72, 2400.
166 Yamaguchi, M., Yagi, T., Azuhata, T., Sota,
T., Suzuki, K., Chichibu, S. and
Nakamura, S. (1997) Journal of Physics:
Condensed Matter, 9, 241.
167 Kim, K.W., Lambert, W.R.L. and Segall, B.
(1996) Physical Review B: Condensed
Matter, 53, 16310.
168 Kim, K.W., Lambert, W.R.L. and Segall, B.
(1994) Physical Review B: Condensed
Matter, 50, 1502.
169 Davydov, V.Yu., Kitaev, Yu.E., Goncharuk,
I.N., Smirnov, A.N., Graul, J.,
Semchinova, O., Uffmann, D., Smirnov,
M.B., Mirgorodsky, A.P. and Evarestov,
170
171
172
173
174
175
176
177
178
179
180
R.A. (1998) Physical Review B: Condensed
Matter, 58, 12899.
Yamaguchi, M., Yagi, T., Sota, T., Deguchi,
T., Shimada, K. and Nakamura, S. (1999)
Journal of Applied Physics, 85, 8502.
Schwarz, R.B., Khachaturyan, K. and
Weber, E.R. (1997) Applied Physics Letters,
70, 1122.
Bergman, L., Dutta, M. and Nemanich,
R.J. (2000) Raman scattering spectroscopy
and analyses of III–V nitride based
materials, in Raman Scattering in Material
Science, Springer, Berlin, Chapter 7.
Cardona, M. (1982) Light Scattering in
Solids II, Springer Topics in Applied Physics,
vol. 50 (eds M. Cardona and G.
G€
untherodt), Springer, Berlin, pp.
19–178.
Harima, H. (2002) Properties of GaN
and related compounds studied by
means of Raman scattering. Journal
of Physics: Condensed Matter, 14,
R967–R993.
Arguello, C.A., Rousseau, D.L. and Porto,
S.P.S. (1969) Physical Review B: Condensed
Matter, 181, 1351.
Cingolani, A., Ferrara, M., Lugara, M. and
Scamarcio, G. (1986) Solid State
Communications, 58, 823.
Huang, D., Yun, F., Visconti, P.,
Reshchikov, M.A., Wang, D., Morkoc, H.,
Rode, D.L., Farina, L.A., Kurdak,C., Tsen,
K.T., Park, S.S. and Lee, K.Y. (2001)
Hall mobility and carrier concentration in
GaN free-standing templates grown by
hydride vapor phase epitaxy with high
quality. Solid State Electronics, 45 (5),
711–715.
Wagner, J.M. and Bechstedt, F. (2002)
Physical Review B: Condensed Matter, 66,
115202.
Miyoshi, S., Onabe, K., Ohkouchi, N.,
Yaguchi, H., Ito, R., Fukutsu, S. and
Shraki, Y. (1992) Journal of Crystal Growth,
124, 439.
Miller, S.C. and Love, W.F. (1967) Tables of
Irreducible Representations of Space Groups
and Corepresentations of Magnetic Space
Groups, Pruett, Boulder, CO.
j117
j 1 General Properties of Nitrides
118
181 Davydov, V.Yu., Averkiev, N.S.,
Goncharuk, I.N., Nelson, D.K., Nikitina,
I.P., Polkovnikov, A.S., Smirnov, A.N.,
Jacobson, M.A. and Semchinova, O.K.
(1997) Journal of Applied Physics,
82, 5097.
182 Leszczynski, M. and Walker, J.F. (1993)
Applied Physics Letters, 62, 1484–1487.
183 Sime, J.R. and Margrave, J.L. (1956) The
Journal of Physical Chemistry, 60, 810.
184 Morimoto, Y. (1974) Journal of the
Electrochemical Society, 121, 1383.
185 Furtado, M. and Jacob, G. (1983) Journal of
Crystal Growth, 64, 257.
186 Gordienko, S.P., Samsonov, G.V. and
Fesenko, V.V. (1964) Soviet Journal of
Physical Chemistry, 38, 1620.
187 Munir, Z.A. and Searcy, A.W. (1965)
Journal of Chemical Physics, 42, 4223.
188 Groh, R., Gerey, G., Bartha, L. and
Pankove, J.I. (1974) Physica Status Solidi a:
Applied Research, 26, 353.
189 Munir, Z.A. and Searcy, A.W. (1965)
Journal of Chemical Physics, 42, 4223.
190 Thurmond, C.D. and Logan, R.A. (1972)
Journal of the Electrochemical Society, 119,
622.
191 Karpinski, J., Jun, J. and Porowski, S.
(1984) Journal of Crystal Growth, 66, 1.
192 Grzegory, I., Krukowski, S., Leszczynski,
M., Perlin, P., Suski, T. and Porowski, S.
High pressure crystallization of GaN, in
Nitride Semiconductors – Handbook on
Materials and Devices (eds P. Ruterana, M.
Albrecht and J. Neugebauer), Wiley-VCH
Verlag GmbH, Weinheim, Germany.
193 Sasaki, T. and Matsuoka, T. (1995) Journal
of Applied Physics, 77, 192.
194 Madar, R., Jacob, G., Hallais, J. and
Fruchart, R. (1975) Journal of Crystal
Growth, 31, 197.
195 Slack, G.A. and McNelly, T.F. (1976)
Journal of Crystal Growth, 34, 263.
196 Ambacher, O. (1998) Growth and
applications of group III-nitrides. Journal
of Physics D: Applied Physics, 31, 2653.
197 Matsuoka, T. (2004) Progress in nitride
semiconductors from GaN to InN –
MOVPE growth and characteristics.
198
199
200
201
202
203
204
205
206
207
208
209
Superlattices and Microstructures, 37 (1),
19–32.
Popovici, G. and Morkoc, H. (1999)
Growth and doping of and defects in IIInitrides, in GaN and Related Materials II:
Optoelectronic Properties of Semiconductors
and Superlattices (ed. S.J. Pearton, series
ed. M.O. Manaresh), vol. 7, Gordon and
Breach, Amsterdam, pp. 93–172.
Ambacher, O., Brandt, M.S., Dimitrov, R.,
Metzger, T., Stutzmann, M., Fischer, R.A.,
Miehr, A., Bergmaier, A. and Dollinger, G.
(1996) Journal of Vacuum Science &
Technology B: Microelectronics and
Nanometer Structures, 14, 3532.
Redhead, P.A. (1962) Vacuum, 12, 203;
Redhead, P.A. (1962) Journal of Crystal
Growth, 9, 158.
Madar, R., Jacob, G., Hallais, J. and
Fruchart, R. (1975) Journal of Crystal
Growth, 31, 197.
Karpinski, J. and Porowski, S. (1984)
Journal of Crystal Growth, 66, 11.
Glushko, W.P. (ed.) (1979)
Termodinamiczeskije swojstwa
indiwidualnych weszczestw, Nauka,
Moscow (in Russian).
Zinowiev, W.E. (1989) Teplofiziczeskije
swojstwa metallow pri wysokich
temperaturach, Metallurgia, Moscow (in
Russian).
Bhandari, C.M. and Rowe, D.M. (1988)
Thermal Conduction in Semiconductors,
John Wiley & Sons, Inc., New York.
Kittel, C. (1986) Introduction to Solid State
Physics, 6th edn, John Wiley & Sons, Inc.,
New York, p. 150.
Sichel, E.K. and Pankove, J.I. (1977)
Journal of Physics and Chemistry of Solids,
38, 330.
Witek, A. (1998) Some aspects of
thermal conductivity of isotopically
pure diamond – a comparison with
nitrides. Diamond & Related Materials, 7
(7), 962–964.
Florescu, D.I., Asnin, V.M., Pollak, F.H.,
Jones, A.M., Ramer, J.C., Schurman, M.J.
and Ferguson, I. (2000) Applied Physics
Letters, 77, 1464.
References
210 Kotchetkov, D., Zouand, J., Balandin, A.,
Florescu, D.I. and Pollak, F.H. (2001)
Effect of dislocations on thermal
conductivity of GaN. Applied Physics
Letters, 79 (26), 4316–4318.
211 Florescu, D.I., Asnin, V.M., Pollak, F.H.
and Molnar, R.J. (2000) Materials Research
Society Symposium Proceedings, 595,
3.89.1.
212 Florescu, D.I., Asnin, V.M., Pollak, F.H.,
Molnar, R.J. and Wood, C.E.C. (2000)
Journal of Applied Physics, 88, 3295.
213 Florescu, D.I., Pollak, F.H., Lanford,
W.B., Khan, F., Adesida, I. and Molnar,
R.J. (2000) Plasma-induced effects on the
thermal conductivity of hydride vapor
phase epitaxy grown n-GaN/sapphire
(0001). Materials Research Society
Symposium Proceedings, 639, G11.57.
214 Kittel, C. Introduction to Solid State Physics,
4th edn, John Wiley & Sons, Inc., New
York, p. 215.
215 Koshchenko, V.I., Demidienko, A.F.,
Sabanova, L.D., Yachmenev, V.E., Gran,
V.E. and Radchenko, A.E. (1979) Inorganic
Materials, 15, 1329–1330 (translation of
Izv. Akad. Nauk SSSR, Neorg. Mater.,
1979, 15, 1686–1687).
216 Demidienko, A.F., Koshchenko, V.I.,
Sabanova, L.D. and Gran, V.E. (1975)
Russian Journal of Physical Chemistry, 49,
1585.
217 Elwell, D. and Elwell, M.M. (1988) Progress
in Crystal Growth and Characterization of
Materials, 17, 53.
218 Porowski, S. and Grzegory, I. (1994)
Properties of Group III Nitrides (ed. J.H.
Edgar), IEE EMIS Data Review Series,
No. 11, INSPEC, The Institution of
Electrical Engineers, Stevenage, UK,
pp. 71, 76, 83.
219 Oliner, A.A. (ed.) (1978) Acoustic Surface
Waves, Topics in Applied Physics, vol. 24,
Springer, Berlin.
220 Petrov, I., Mojab, E., Powell, R., Greene, J.,
Hultman, L. and Sundgren, J.E. (1992)
Applied Physics Letters, 60, 2491.
221 Vollstadt, H., Ito, E., Akaishi, M.,
Akimoto, S. and Fukunaga, O. (1990)
222
223
224
225
226
227
228
229
230
231
232
233
234
235
Proceedings of the Japan Academy, Series B,
66, 7.
Ruiz, E., Alvarez, S. and Alemany, P.
(1994) Physical Review B: Condensed
Matter, 49, 7115.
Tsubouchi, K. and Mikoshiba, N. (1985)
Zero-temperature coefficient SAW
devices on AlN epitaxial films. IEEE
Transactions on Sonics and Ultrasonics, 32,
634–644.
Tsubouchi, T., Sugai, K. and Mikoshiba,
N. (1981) Ultrosonic Symposium
Proceedings, IEEE, New York, p. 375.
Taylor, K.M. and Len, C. (1960) Journal of
the Electrochemical Society, 107, 308.
Cline, C.F. and Kalm, J.S. (1963) Journal of
the Electrochemical Society, 110, 773.
Gleize, J., Renucci, M.A., Frandon, J.,
Bellett-Amalric, E. and Daudin, B. (2003)
Journal of Applied Physics, 93, 2065.
Tischler, J.G. and Freitas, J.A., Jr (2004)
Anharmonic decay of phonons in strain
free wurtzite AlN. Applied Physics Letters,
85, 1943.
Bergman, L., Alexson, D., Murphy, P.L.,
Nemanich, R.J., Dutta, M., Stroscio, M.A.,
Balkas, C., Shin, H. and Davis, R.F. (1999)
Physical Review B: Condensed Matter, 59,
12977.
Ribeiro, C.T.M., Alvarez, F. and Zanatta,
A.R. (2002) Applied Physics Letters, 81,
1005.
See, for example, Colthup, N.B., Daly,
L.H. and Wiberley, S.E. (1990) Infrared
and Raman Spectroscopy, Academic Press,
San Diego, FL.
Ren, Z.M., Lu, Y.F., Ni, H.Q., Liew, T.Y.F.,
Cheong, B.A., Chow, S.K., Ng, M.L. and
Wang, J.P. (2000) Journal of Applied
Physics, 88, 7346.
Karch, K. and Bechstedt, F. (1997) Physical
Review B: Condensed Matter, 56, 7404.
T€
ut€
unc€
u, H.M. and Srivastava, G.P.
(2000) Phonons in zinc-blende and
wurtzite phases of GaN, AlN, and BN with
the adiabatic bond-charge model. Physical
Review B: Condensed Matter, 62 (8),
5028–5235.
Class, W. (1968) NASA Report CR-1171.
j119
j 1 General Properties of Nitrides
120
236 Van Vechten, J.A. (1973) Physical Review
B: Condensed Matter, 7, 1479.
237 Yim, W.M. and Paff, R.J. (1974) Journal of
Applied Physics, 45, 1456.
238 Mah, A.D., King, E.G., Weller, W.W. and
Christensen, A.U. (1961) Bureau of Mines
Report of Investigations, 5716, 18.
239 Glushko, V.P., Gurevich, L.V., Bergman,
G.A., Weitz, I.V., Medvedev, V.A.,
Chachkurov, G.A. and Yungman, V.S.
(1979) Thermodinamicheskiie swoistwa
indiwidualnych weshchestw, vol. 1, Nauka,
Moscow, pp. 164–165.
240 Slack, G.A., Tanzilli, R.A., Pohl, R.O.
and Vandersande, J.W. (1987) Journal of
Physics and Chemistry of Solids,
48, 641.
241 Slack, G.A. and McNelly, T.F. (1977)
Journal of Crystal Growth, 42, 560 (AlN
from pellet drop, sublimation, and final
growth).
242 Florescu, D.I., Asnin, V.M. and Pollak, F.H.
(2001) Thermal conductivity of GaN and
AlN. Compound Semiconductor, 7 (2), 62.
243 Nikolaev, A., Nikitina, I., Zubrilov, A.,
Mynbaeva, M., Melnik, Y. and Dmitriev, V.
(2000) Materials Research Society
Symposium Proceedings, 595, 6.5.1.
244 Edwards, J., Kawabe, K., Stevens, G. and
Tredgold, R.H. (1965) Solid State
Communications, 3, 99.
245 Cox, G.A., Cummins, D.O., Kawabe, K.
and Tredgold, R.H. (1967) Journal of
Physics and Chemistry of Solids,
28, 543.
246 Yim, W.M., Stotko, E.J., Zanzucchi, P.J.,
Pankove, J.I., Ettenberg, M. and Gilbert,
S.L. (1973) Journal of Applied Physics,
44, 292.
247 Yoshida, S., Misawa, S., Fujii, Y., Takada,
S., Hayakawa, H., Gonda, S. and Itoh, A.
(1979) Journal of Vacuum Science &
Technology, 16, 990.
248 Chu, T.L., Ing, D.W. and Noreika, A.J.
(1967) Solid State Electronics, 10, 1023.
249 Rutz, R.F. (1976) Applied Physics Letters,
28, 379.
250 Rutz, R.F., Harrison, E.P. and Cuome, J.J.
(1973) IBM Journal of Research, 17, 61.
251 Edwards, J., Kawabe, K., Stevens, G. and
Tredgold, R.H. (1965) Solid State
Communications, 3, 99.
252 Kawabe, K., Tredgold, R.H. and Inuishi, Y.
(1967) Electrical Engineering in Japan, 87,
62.
253 Harris, J.H. and Youngman, R.A. (1994)
Properties of Group III Nitrides (ed. J.H.
Edgar), IEE EMIS Data Review Series, No.
11, INSPEC, The Institution of Electrical
Engineers, Stevenage, UK, p. 203.
254 Pacesova, S. and Jastrabik, L. (1979)
Czechoslovak Journal of Physics B, 29, 913.
255 Youngman, R.A. and Harris, J.H. (1990)
Journal of the American Ceramic Society, 73,
3238.
256 Harris, J.H., Youngman, R.A. and Teller,
R.G. (1990) Journal of Materials Research,
5, 1763.
257 Shishkin, Y., Devaty, R.P., Choyke, W.J.,
Feng Yun, King, T. and Morkoc, H., (2001)
Near bandedge cathodoluminescence
studies of AlN films: dependence on MBE
growth conditions. International
Conference on Nitride Semiconductors,
Denver, CO, July; Physica Status Solidi a:
Applied Research, 188 (2), 591–594.
258 Perry, P.B. and Rutz, R.F. (1978) Applied
Physics Letters, 33, 319.
259 Pastrnak, J. and Roskovcova, L. (1968)
Physica Status Solidi, 26, 591.
260 Pastrnak, J. and Souckova, L. (1963)
Physica Status Solidi, 9, K71.
261 Pastrnak, J. and Roskovcova, L. (1965)
Physica Status Solidi, 11, K73.
262 Karel, F., Pastrnak, J., Hejduk, J. and
Losik, V. (1966) Physica Status Solidi, 15,
693.
263 Karel, F. and Pastrnak, J. (1969)
Czechoslovak Journal of Physics B, 19, 78.
264 Karel, F. and Pastrnak, J. (1970)
Czechoslovak Journal of Physics B, 20, 46.
265 Karel, F. and Mares, J. (1972) Czechoslovak
Journal of Physics B, 22, 847.
266 Karel, F. and Mares, J. (1973) Czechoslovak
Journal of Physics B, 23, 652.
267 Juza, R. and Hahn, H. (1938) Zeitschrift f€
ur
Anorganishce und Allgemeine Chemie,
239, 282.
References
268 Matsuoka, T. (2004) MOVPE growth and
characteristics of nitride semiconductors
from GaN to InN, in Advanced Materials in
Electronics (ed. Q. Guo), Research
Signpost, pp. 46–83.
269 Osamura, K., Naka, S. and Murakami,
Y. (1975) Journal of Applied Physics, 46,
3432.
270 Puychevrier, N. and Menoret, M. (1976)
Thin Solid Films, 36, 141.
271 Tansley, T.L. and Foley, C.P. (1986) Journal
of Applied Physics, 59, 3241.
272 Westra, K.L., Lawson, R.P.W. and Brett,
M.J. (1988) Journal of Vacuum Science &
Technology A: Vacuum Surfaces and Films,
6, 1730.
273 Bhuiyan, A.G., Hashimoto, A. and
Yamamoto, A. (2003) Indium nitride
(InN): a review on growth,
characterization, and properties. Journal
of Applied Physics, 94 (5), 2779–2808.
274 Davydov, V.Yu., Klochikhin, A.A., Emtsev,
V.V., Ivanov, S.V., Vekshin, V.V.,
Bechstedt, F., Furthm€
uller, J., Harima,
H., Mudryi, A.V., Hashimoto, A.,
Yamamoto, A., Aderhold, J., Graul, J. and
Haller, E.E. (2002) Physica Status Solidi b:
Basic Research, 230, R4.
275 Araki, T., Saito, Y., Yamaguchi, T.,
Kurouchi, M., Nanishi, Y. and Naoi, H.
(2004) Radio frequency-molecular beam
epitaxial growth of InN epitaxial films on
(0 0 0 1) sapphire and their properties.
Journal of Vacuum Science & Technology B:
Microelectronics and Nanometer Structures,
22 (4), 2139–2143.
276 Fritsch, D., Schmidt, H. and Grundmann,
M. (2004) Band dispersion relations of
zinc-blende and wurtzite InN. Physical
Review B: Condensed Matter, 69, 165204.
277 Wu, J. and Walukiewicz, W. (2004) Band
gaps of InN and group III-nitride alloys.
Superlattices and Microstructures. 34, 63–75.
278 Butcher, K.S.A. (2004) InN, a historic
review – from obscurity to controversy, in
Advanced Materials in Electronics (ed. Q.
Guo), Research Signpost.
279 Butcher, K.S.A. and Tansley, T.L. (2005)
InN, latest development and a review of
280
281
282
283
284
285
286
287
288
the band-gap controversy. Superlattices
and Microstructures, 38 (1), 1–37.
Shubina, T.V., Ivanov, S.V., Jmerik, V.N.,
Solnyshkov, D.D., Vekshin, V.A., Kopev,
P.S., Vasson, A., Leymarie, J., Kavokin, A.,
Amano, H., Shimono, K., Kasic, A. and
Monemar, B. (2004) Mie resonances,
infrared emission, and the band gap of
InN. Physical Review Letters, 92 (11),
117407–117410.
Butcher, K.S.A., Wintrebert-Fouquet, M.,
Chen, P.P., Timmers, H. and Sherestha,
S.K. (2003) Detailed analysis of the
absorption data for InN. Presented at the
International Symposium on Point
Defects and Nonstoichiometry, March
2003, Sendai, Japan; Material Science in
Semiconductor Processing, 2003, 6,
351–354.
Wolfe, C.M. and Stillman, G.E. (1975)
Apparent mobility enhancement in
inhomogeneous crystals, in
Semiconductors and Semimetals, vol. 10
(eds R.K. Willardson and A.C. Beer),
Academic Press, New York,
pp. 175–220.
Yamaguchi, S., Kariya, M., Nitta, S.,
Takeuchi, T., Wetzel, W., Amano, H. and
Akasaki, I., (1999) Structural properties of
InN on GaN grown by metalorganic
vapor-phase epitaxy. Journal of Applied
Physics, 85 (11), 7682–7688.
Strite, S., Chandrasekhar, D., Smith, D.J.,
Sariel, J., Chen, H., Teraguchi, N. and
Morkoc, H. (1993) Journal of Crystal
Growth, 127, 204.
Hahn, H. and Juza, R. (1940) Zeitschrift f€
ur
Anorganishce und Allgemeine Chemie, 244,
111.
Tansley, T.L. (1994) Properties of Group III
Nitrides, (ed. J.H. Edgar), IEE EMIS Data
Review Series, No. 11, INSPEC, The
Institution of Electrical Engineers,
Stevenage, UK, p. 35.
van Camp, P.E., van Doren, V.E. and
Devreese, J.T. (1990) Physical Review B:
Condensed Matter, 41, 1598.
Kubota, K., Kobayashi, Y. and Fujimoto, K.
(1989) Journal of Applied Physics, 66, 2984.
j121
j 1 General Properties of Nitrides
122
289 Inushima, T., Yaguchi, T., Nagase, A., Iso,
A., Shiraishi, T. and Ooya, S. (1995)
Optical and electrical properties of InN
grown by the atomic layer epitaxy. 7th
International Conference on Indium
Phosphide and Related Materials (Cat.
No. 95CH35720), IEEE, New York, pp.
187–190.
290 Inushima, T., Yaguchi, T., Nagase, A., Iso,
A. and Shiraishi, T. (1996) Investigation of
optical properties of InN grown by atomic
layer epitaxy. Presented at the
International Meeting on Silicon Carbide
and Related Materials, 1995, Kyoto,
Japan, IOP Publishing, Bristol, UK,
pp. 971–974.
291 Osamura, K., Naka, S. and Murakami, Y.
(1975) Journal of Applied Physics, 46, 3432.
292 Sheleg, A.V. and Savastenko, V.A. (1976)
Vestsi Akademii Nauk USSR, Seria Fiziki i
Matematika Nauk, 3, 126.
293 Krukowski, S., Witek, A., Adamczyk, J.,
Jun, J., Bockowski, M., Grzegory, I.,
Lucznik, B., Nowak, G., Wroblewski, M.,
Presz, A., Gierlotka, S., Stelmach, S.,
Palosz, B., Porowski, S. and Zinn, P.
(1998) Journal of Physics and Chemistry of
Solids, 59, 289–295.
294 Tansley, T.L. and Foley, C.P. (1985)
Proceedings of the 3rd International
Conference on Semiinsulating III–V
Materials, Warm Springs, OR, 1984 (ed.
J.S. Blakemore), Shiva, London.
295 Bryden, W.R., Ecelberger, S.A., Hawley,
M.E. and Kistenmacher, T.J. (1994)
Diamond SiC, Nitride Wide Bandgap
Semiconductors (eds C.H. Carter Jr, G.
Gildenblat, S. Nakamura and R.J.
Nemanich), Materials Research Society,
Pittsburgh, PA; Materials Research Society
Symposium Proceedings, 339, 497.
296 Bryden, W.R. and Kistenmacher, T.J.
(1994) Properties of Group X Nitrides (ed.
J.H. Edgar), IEE EMIS Data Review
Series, No. 11, INSPEC, The Institution of
Electrical Engineers, Stevenage, UK, pp.
117–118.
297 Abernathy, C.R., Pearton, S.J., Ren, F. and
Wisk, P.W. (1993) Journal of Vacuum
298
299
300
301
302
303
304
305
306
307
308
309
Science & Technology B: Microelectronics
and Nanometer Structures, 11, 179.
Maruyama, T. and Morishita, T. (1994)
Journal of Applied Physics, 76, 5809.
Yamaguchi, S., Kariya, M., Nitta, S.,
Takeuchi, T., Wetzel, C., Amano, H. and
Akasaki, I. (1999) Journal of Applied
Physics, 85, 7682.
Lu, H., Schaff, W.J., Hwang, J., Wu, H.,
Yeo, W., Pharkya, A. and Eastman, L.F.
(2000) Applied Physics Letters, 77, 2548.
Aderhold, J., Davydov, V.Yu., Fedler, F.,
Klausing, H., Mistele, D., Rotter, T.,
Semchinova, O., Stemmer, J. and Graul, J.
(2001) Journal of Crystal Growth, 222, 701.
Lu, H., Schaff, W.J., Hwang, J., Wu, H.,
Koley, G. and Eastman, L.F. (2001) Applied
Physics Letters, 79, 1489.
Motlan, Goldys, E.M. and Tansley, T.L.
(2002) Journal of Crystal Growth, 241, 165.
Saito, Y., Yamaguchi, T., Kanazawa, H.,
Kano, K., Araki, T., Nanishi, Y., Teraguchi,
N. and Suzuki, A. (2002) Journal of Crystal
Growth, 237–239, 1017.
Lu, H., Schaff, W.J., Eastman, L.F., Wu, J.,
Walukiewicz, W., Yu, K.M., Auger, J.W.,
III, Haller, E.E. and Ambacher, O. (2002)
Conference Digest of the 44th Electronic
Materials Conference, Santa Barbara, p. 2;
Schaff, W.J., Lu, H., Eastman, L.F.,
Walukiewicz, W., Yu, K.M., Keller, S.,
Kurtz, S., Keyes, B. and Gevilas, L. (2004)
Electrical properties of InN grown by
molecular beam epitaxy. Fall 2004 ECS
Meeting. State-of-the-Art Program on
Compound Semiconductors XLI and Nitride
and Wide Bandgap Semiconductors for
Sensors, Photonics, and Electronics, vol.
2004-06 (eds V.-H. Ng and A.G. Baca).
Higashiwaki, M. and Matsui, T. (2003)
Journal of Crystal Growth, 252, 128.
Look, D.C., Lu, H., Schaff, W.J., Jasinski, J.
and Liliental-Weber, Z. (2002) Applied
Physics Letters, 80, 258.
Tansley, T.L. and Foley, C.P. (1984)
Electronics Letters, 20, 1066.
OLeary, S.K., Foutz, B.E., Shur, M.S.,
Bhapkar, U.V. and Eastman, L.F. (1998)
Journal of Applied Physics, 83, 826.
References
310 Bellotti, E., Doshi, B.K., Brennan, K.F.,
Albrecht, J.D. and Paul Ruden, P. (1999)
Ensemble Monte Carlo study of electron
transport in wurtzite InN. Journal of
Applied Physics, 85, 916.
311 Foutz, B.E., Eastman, L.F., Bhapkar, U.V.
and Shur, M.S. (1997) Comparison of
high field electron transport in GaN and
GaAs. Applied Physics Letters, 70,
2849–2851.
312 Foutz, B.E., OLeary, S.K., Shur, M.S. and
Eastman, L.F. (1999) Transient electron
transport in wurtzite GaN, InN, and AlN.
Journal of Applied Physics, 85, 7727–7734.
313 Wakahara, A., Tsuchiya, T. and Yoshida, A.
(1990) Journal of Crystal Growth, 99, 385.
314 Kubota, K., Kobayashi, Y. and Fujimoto, K.
(1989) Journal of Applied Physics, 66,
2984–2988.
315 Guo, Q., Kato, O., Fujisawa, M. and
Yoshida, A. (1992) Optical constants of
indium nitride. Solid State
Communications, 83 (9), 721–723.
316 Guo, Q., Ogawa, H., and Yoshida, A.,
(1996) Optical properties of indium
nitride in vacuum ultraviolet region.
Journal of Electron Spectroscopy & Related
Phenomena, 79, 9.
317 Yodo, T., Yona, H., Ando, H., Nosei, D.
and Harada, Y. (2002) Applied Physics
Letters, 80, 968.
318 Inushima, T., Mamutin, V.V., Vekshin,
V.A., Ivanov, S.V., Sakon, T., Motokawa,
M. and Ohoya, S. (2001) Journal of Crystal
Growth, 481, 227–228.
319 Davydov, V.Yu., Klochikhin, A.A., Seisyan,
R.P., Emtsev, V.V., Ivanov, S.V., Bechstedt,
F., Furthmuller, J., Harima, H., Mudryi,
A.V., Aderhold, J., Semchinova, O. and
Graul, J. (2002) Physica Status Solidi b:
Basic Research, 229, R1.
320 Hamberg, I. and Granqvist, C.G. (1986)
Journal of Applied Physics Reviews, 60,
R123.
321 Alexandrov, D., Butcher, K.S.A. and
Wintrebert-Fouquet, M. (2004)
Absorption and photoluminescence
features caused by defects in InN. Journal
of Crystal Growth, 269 (1), 77–86.
322 Alexandrov, D., Butcher, K.S.A. and
Wintrebert-Fouquet, M. (2004) Energy
band gaps of InN containing oxygen and
of the InxAl1xN interface layer formed
during InN film growth. Journal of
Vacuum Science & Technology A: Vacuum
Surfaces and Films, 22, 954–961.
323 Trainor, J.W. and Rose, K. (1974) Journal of
Electronic Materials, 3, 821.
324 Haddad, D.B., Takur, J.S., Naik, V.M.,
Auner, G.W., Naik, R. and Wenger, L.E.
(2003) Materials Research Society
Symposium Proceedings, 743, 701.
325 Hovel, H.J. and Cuomo, J.I. (1972) Applied
Physics Letters, 20, 71.
326 Nataraian, B.R., Eltoukhy, A.H., Greene,
J.E. and Barr, T.L. (1980) Thin Solid Films,
69, 201.
327 Sullivan, B.T., Parsons, R.R., Westra, K.L.
and Brett, M.J. (1988) Journal of Applied
Physics, 64, 4144.
328 Wintrebert-Fouquet, M., Butcher, K.S.A.
and Chen, P.P.-T. (2003) InN grown by
remote plasma enhanced chemical
vapour deposition. Presented at the First
International InN Workshop, November
16–20, Fremantle, Australia.
329 Butcher, K.S.A., Wintrebert-Fouquet, M.,
Chen, P.P.-T., Tansley, T.L. and Srikeaw, S.
(2002) Materials Research Society
Symposium Proceedings, 693, 341.
330 Demangeot, F., Frandon, J., Pinquier, C.,
Caumont, M., Briot, O., Maleyre, B., ClurRuffenach, S. and Gil, B. (2003) Raman
scattering in large single indium nitride
dots: correlation between morphology
and strain. Physical Review B: Condensed
Matter, 68, 245308.
331 Briot, O., Maleyre, B., Ruffenach, S.,
Pinquier, C., Demangeot, F., Frandon, J.
and Gil, B. (2003) Absorption and Raman
scattering processes in InN films and
dots. Presented at the 1st International
InN Workshop, November 16–20,
Fremantle, Australia.
332 Tsen, K.T., Liang, W., Ferry, D.K., Hai Lu,
Schaff, W.J., Özg€
ur, Ü., Fu, Y., Moon, Y.T.,
Yun, F., Morkoc, H. and Everitt, H.O.
(2005) Optical studies of carrier dynamics
j123
j 1 General Properties of Nitrides
124
333
334
335
336
337
338
339
340
341
and non-equilibrium optical phonons in
nitride-based wide bandgap
semiconductors. Superlattices and
Microstructures, 38 (2), 77–114.
Shan, W., Song, J.J., Feng, Z.C.,
Schurman, M. and Stall, R.A. (1997)
Applied Physics Letters, 71, 2433.
Shan, W., Ager, J.W., III, Yu, K.M.,
Walukiewicz, W., Haller, E.E., Martin,
M.C., McKinney, W.R. and Yang, W.
(1999) Journal of Applied Physics, 85, 8505.
Wolford, D.J. and Bradley, J.A. (1985)
Solid State Communications, 53, 1069.
Perlin, P., Gorczyca, I., Suski, T.,
Wisniewski, P., Lepkowski, S.,
Christansen, N.E., Svane, A., Hansen, M.,
DenBaars, S.P., Damilano, D.,
Grandjean, N. and Massies, S.P. (2001)
Physical Review B: Condensed Matter, 64,
115319.
Tyagai, V.A., Snitko, O.V., Evstigneev,
A.M. and Krasiko, A.N. (1981) Physica
Status Solidi b: Basic Research, 103, 589.
Davydov, V., Klochikhin, A., Ivanov, S.,
Aderhold, J. and Yamamoto, A. (2003)
Growth and properties of InN, in Nitride
Semiconductors – Handbook on Materials
and Devices, (eds. P. Ruterana, M.
Albrecht and J. Neugebauer), Wiley-VCH
Verlag GmbH, Weinheim.
Kasic, A., Schebert, M., Saito, Y., Nanishi,
Y. and Wagner, G. (2002) Effective
electron mass and phonon modes in
n-type hexagonal InN. Physical Review B:
Condensed Matter, 65, 115206.
Perlin, P., Litwin-Staszewska, E.,
Suchanek, B., Knap, W., Camassel, J.,
Suski, T., Piotrzkowski, R., Grzegory, I.,
Porowski, S., Kaminska, E. and Chervin,
J.C. (1996) Determination of the effective
mass of GaN from infrared reflectivity and
Hall effect. Applied Physics Letters, 68 (8),
1114–1116.
Wu, J., Walukiewicz, W., Shall, W., Yu,
K.M., Ager, J.W., III, Haller, E.E., Lu, H.
and Schaff, W.J. (2002) Effects of the
narrow band gap on the properties of InN.
Physical Review B: Condensed Matter, 66,
201403.
342 Osamura, K., Naka, S. and Murakami, Y.
(1975) Journal of Applied Physics, 46, 3432.
343 Matsuoka, T. (1994) Properties of Group III
Nitrides (ed. J.H. Edgar), IEE EMIS Data
Review Series, No. 11, INSPEC, The
Institution of Electrical Engineers,
Stevenage, UK, pp. 231–238.
344 Yamasaki, S., Asami, S., Shibata, N.,
Koike, M., Manabe, K., Tanaka, T.,
Amano, H. and Akasaki, I. (1995) Applied
Physics Letters, 66, 1112.
345 Yoshida, S., Misawa, S. and Gonda, S.
(1982) Journal of Applied Physics, 53, 6844.
346 Khan, M.R.H., Koide, Y., Itoh, H., Sawaki,
N. and Akasaki, I. (1986) Solid State
Communications, 60, 509.
347 Zoroddu, A., Bernardini, F., Ruggerone,
P. and Fiorentini, V. (2001) Firstprinciples prediction of structure,
energetics, formation enthalpy, elastic
constants, polarization, and piezoelectric
constants of AlN, GaN, and InN:
comparison of local and gradientcorrected density-functional theory.
Physical Review B: Condensed Matter, 64
(45), 208.
348 Amano, H., Takeuchi, T., Sota, S., Sakai,
H. and Akasaki, I. (1997) III–V Nitrides
(eds. F.A. Ponce, T.D. Moustakas, I.
Akasaki and B. Monemar), Materials
Research Society Symposium Proceedings,
449, 1143–1150.
349 Yoshida, S., Misawa, S. and Gonda, S.
(1983) Journal of Vacuum Science &
Technology B: Microelectronics and
Nanometer Structures, 1, 250.
350 Wickenden, D.K., Bargeron, C.B.,
Bryden, W.A., Miragliova, J. and
Kistenmacher, T.J. (1994) Applied Physics
Letters, 65, 2024.
351 Koide, Y., Rob, H., Khan, M.R.H.,
Hiramatsu, K., Sawaki, N. and Akasaki, I.
(1987) Journal of Applied Physics,
61, 4540.
352 Khan, M.A., Skogman, R.A., Schulze,
R.G. and Gershenzon, M. (1983) Applied
Physics Letters, 43, 492.
353 Korkotashvili, G.A., Pikhtin, A.N.,
Pichugin, I.G. and Tsaregorodtsev, A.M.
References
354
355
356
357
358
359
360
361
362
363
364
365
(1984) Soviet Physics: Semiconductors, 18,
913.
Wethkamp, T., Wilmers, K., Esser, N.,
Richter, W., Ambacher, O., Angerer, H.,
Jungk, G., Johnson, R.L. and Cardona, M.
(1998) Thin Solid Films, 313, 745.
Paduano, Q.S., Weyburne, D.W.,
Bouthillette, L.O. and Alexander, M.N.
(2002) 7th International Workshop on
Wide Bandgap Nitrides, Abstract Book,
Richmond, VA, 10–14 March, p. 21.
Nikishin, S.A., Faleev, N.N., Zubrilov,
A.S., Antipov, V.G. and Temkin, H. (2000)
Applied Physics Letters, 76, 3028.
Shan, W., Ager, J.W., III, Yu, K.M.,
Walukiewicz, W., Haller, E.E., Martin,
M.C., McKinney, W.R. and Yang, W.
(1999) Journal of Applied Physics,
85, 8505.
Özg€
ur, Ü., Webb-Wood, G., Everitt, H.O.,
Yun, F. and Morkoc, H. (2001) Applied
Physics Letters, 79, 4103.
Wagner, J., Obloh, H., Kunzer, M., Maier,
M., Kohler, K. and Johs, B. (2000) Journal
of Applied Physics, 89, 2779.
Jiang, H., Zhao, G.Y., Ishikawa, H.,
Egawa, T., Jimbo, T. and Umeno, M.
(2001) Journal of Applied Physics, 89, 1046.
Ochalski, T.J., Gil, B., Lefebvre, P.,
Grandjean, M., Leroux, M., Massies, J.,
Makamura, S. and Morkoc, H. (1999)
Applied Physics Letters, 74, 3353.
Lee, S.R., Lee, S.R., Wright, A.F.,
Crawford, M.H., Petersen, G.A., Han, J.
and Biefeld, R.M. (1999) Applied Physics
Letters, 74, 3344.
Yun, F., Reshchikov, M.A., He, L., King,
T., Morkoc, H., Novak, S.W. and Wei, L.,
(2002) Energy band bowing parameter in
AlxGa1xN alloys. Journal of Applied
Physics: Rapid Communications, 92,
4837–4839.
Khan, M.A., Van Hove, J.M., Kuznia, J.N.
and Olson, D.T. (1991) Applied Physics
Letters, 58, 2408.
Tanaka, T., Watanabe, A., Amano, H.,
Kobayashi, Y., Akasaki, I., Yamazaki, S.
and Koike, M. (1994) Applied Physics
Letters, 65, 593.
366 Nagatomo, T., Kuboyama, T., Minamino,
H. and Omoto, O. (1989) Japanese Journal
of Applied Physics, 28, L1334.
367 Matsuoka, T., Tanaka, H., Sasaki, T. and
Katsui, K. (1990) Proceedings of the 16th
International Symposium on GaAs and
Related Compounds, 1989, Karuizawa,
Institute of Physics Conference Series,
vol. 106, Institute of Physics, Bristol,
p. 141.
368 Yoshimoto, N., Matsuoka, T., Sasaki, T.
and Katsui, A. (1991) Applied Physics
Letters, 59, 2251.
369 Matsuoka, T., Yoshimoto, N., Sasaki, T.
and Katsui, A. (1992) Journal of Electronic
Materials, 21, 157.
370 Shimizu, M., Hiramatsu, K. and Sawaki,
N. (1994) Journal of Crystal Growth, 145,
209.
371 Ho, I.-H. and Stringfellow, G.B. (1996)
Applied Physics Letters, 69, 2701.
372 Osamura, K., Naka, S. and Murakami, Y.
(1975) Journal of Applied Physics, 46,
3432.
373 Koukitu, A., Takahashi, N., Taki, T. and
Seki, H. (1996) Japanese Journal of Applied
Physics, 35, L673.
374 Kawaguchi, Y., Shimizu, M., Hiramatsu,
K. and Sawaki, N. (1997) Materials
Research Society Symposium Proceedings,
449, 89.
375 Nakamura, S. and Mukai, T. (1992)
Japanese Journal of Applied Physics, 31,
L1457.
376 Piner, E.L., Behbehani, M.K., El-Mastry,
N.A., McIntosh, F.G., Roberts, J.C.,
Boutros, K.S. and Bedair, S.M. (1997)
Applied Physics Letters, 70, 461.
377 Wright, A.F., Leung, K. and van
Schilfgaarde, M. (2001) Effects of biaxial
strain and chemical ordering on the band
gap of InGaN. Applied Physics Letters, 78
(2), 189–191.
378 Ferhat, M. and Bechstedt, F. (2002) Firstprinciples calculations of gap bowing in
InxGa1xN and InxAl1xN alloys: relation
to structural and thermodynamic
properties. Physical Review B: Condensed
Matter, 65, 0752131–0752137.
j125
j 1 General Properties of Nitrides
126
379 S€
okeland, F., Rohlfing, M., Kr€
uger, P. and
Pollmann, J. (2003)Density functional and
quasiparticle band-structure calculations
for GaxAl1xN and GaxIn1xN alloys.
Physical Review B: Condensed Matter, 68,
075203-1–075203-11.
380 Pugh, S.K., Dugdale, D.J., Brand, S. and
Abram, R.A. (1999) Journal of Applied
Physics, 86, 3768.
381 Teles, L.K., Furthm€
uller, J., Scolfaro,
L.M.R., Leite, J.R. and Bechstedt, F. (2000)
Physical Review B: Condensed Matter, 62,
2475.
382 Teles, L.K., Furthm€
uller, J., Scolfaro,
L.M.R., Leite, J.R. and Bechstedt, F. (2001)
Influence of composition fluctuations and
strain on gap bowing in InxGa1xN.
Physical Review B: Condensed Matter, 63,
085204.
383 Nakamura, S. and Mukai, J. (1995) Journal
of Vacuum Science & Technology A: Vacuum
Surfaces and Films, 13, 6844.
384 ODonnell, K.P., Martin, R.W., TragerCowan, C., White, M.E., Esona, K.,
Deatcher, C., Middleton, P.G., Jacobs, K.,
van der Stricht, W., Merlet, C., Gil, B.,
Vantomme, A. and Mosselmans, J.F.W.
(2001) Materials Science & Engineering B:
Solid State Materials for Advanced
Technology, 82, 194.
385 Monroy, E., Gogneau, N., Enjalbert, F.,
Fossard, F., Jalabert, D., Bellet-Amalric,
E., Dang, L.S. and Daudin, B. (2003)
Molecular-beam epitaxial growth and
characterization of quaternary III-nitride
compounds. Journal of Applied Physics, 94
(5), 3121–3127.
386 Aumer, M.E., LeBoeuf, S.F., McIntosh,
F.G. and Bedair, S.M. (1999) Applied
Physics Letters, 75, 3315.
387 Dimakis, E., Georgakilas, A.,
Androulidaki, M., Tsagaraki, K., Kittler,
G., Kalaitzakis, F., Cengher, D., BelletAmalric, E., Jalabert, D. and Pelekanos,
N.T. (2003) Plasma-assisted MBE growth
of quaternary InAlGaN quantum well
heterostructures with room temperature
luminescence. Journal of Crystal Growth,
251 (1–4), 476–480.
388 Pereira, S., Correia, M.R., Monteiro, T.,
Pereira, E., Alves, E., Sequeira, A.D. and
Franco, N. (2001) Compositional
dependence of the strain-free optical band
gap in InxGa1xN layers. Applied Physics
Letters, 78 (15), 2137–2139.
389 McCluskey, M.D., Van der Walle, C.G.,
Romano, L.T., Krusor, B.S. and Johnson,
N.M. (2003) Effect of composition on the
band gap of strained InxGa1xN alloys.
Journal of Applied Physics, 93, 4340–4342.
390 Wu, J., Walukiewicz, W., Yu, K.M., Ager,
J.W., III, Haller, E.E., Hai Lu and Schaff,
W.J. (2002) Small bandgap bowing in
In1 xGaxN alloys. Applied Physics Letters,
80, 4741.
391 Shan, W., Walukiewicz, W., Haller, E.E.,
Little, B.D., Song, J.J., McCluskey, M.D.,
Johnson, N.M., Feng, Z.C., Schurman, M.
and Stall, R.A. (1998) Journal of Applied
Physics, 84, 4452.
392 Pereira, S., Correia, M.R., Monteiro, T.,
Pereira, E., Alves, E., Sequeira, A.D. and
Franco, N. (2001) Applied Physics Letters,
78, 2137.
393 ODonnell, K.P., Martin, R.W., TragerCowan, C., White, M.E., Esona, K.,
Deatcher, C., Middleton, P.G., Jacobs, K.,
van der Stricht, W., Merlet, C., Gil, B.,
Vantomme, A. and Mosselmans, J.F.W.
(2001) Materials Science & Engineering B:
Solid State Materials for Advanced
Technology, 82, 194.
394 Nakamura, S. and Mukai, T. (1992)
Japanese Journal of Applied Physics, 3 (1),
L1457.
395 Nakamura, S., Mukai, T. and Seno, M.
(1993) Japanese Journal of Applied Physics,
31, L16.
396 Nakamura, S., Iwasa, N. and Nagahama,
S. (1993) Japanese Journal of Applied
Physics, 32, L338–341.
397 See, for example, Butte, R., Carlin, J.-F.,
Feltin, E., Gonschorek, M., Nicolay, S.,
Christmann, G., Simeonov, D., Castiglia,
A., Dorsaz, J., Buehlmann, H.J.,
Christopoulos, S., Baldassarri, G., von
Hogersthal, H., Grundy, A.J.D., Mosca,
M., Pinquier, C., Py, M.A., Demangeot, F.,
References
398
399
400
401
402
403
404
405
406
Frandon, J., Lagoudakis, P.G., Baumberg,
J.J. and Grandjean, N. (2007) Current
status of AlInN layers lattice-matched to
GaN for photonics and Electronics.
Journal of Physics D: Applied Physics, 40,
1–17.
Nakamura, S., Senoh, M., Nagahata, S.,
Iwasa, N., Yamada, T., Matsushita, T.,
Hiyoku, H. and Sugimoto, Y. (1996)
Japanese Journal of Applied Physics, Part 2:
Letters, 35 L74.
Wright, A.F. and Nelson, J.S. (1995) Firstprinciples calculations for zinc-blende
AlInN alloys. Applied Physics Letters, 66
(25), 3465–3467.
Onuma, T., Chichibu, S.F., Uchinuma, Y.,
Sota, T., Yamaguchi, S., Kamiyama, S.,
Amano, H. and Akasaki, I. (2003)
Recombination dynamics of localized
excitons in Al1 xInxN epitaxial films on
GaN templates grown by metalorganic
vapor phase epitaxy. Journal of Applied
Physics, 94 (4), 2449–2453.
Lukitsch, M.J., Danylyuk, Y.V., Naik, V.M.,
Huang, C., Auner, G.W., Rimai, L. and
Naik, R. (2001) Optical and electrical
properties of Al1 xInxN films grown by
plasma source molecular-beam epitaxy.
Applied Physics Letters, 79 (5), 632–634.
Guo, Q., Ogawa, H. and Yoshida, A.
(1995) Journal of Crystal Growth, 146, 462.
Goano, M., Bellotti, E., Ghillino, E.,
Garetto, C., Ghione, G. and Brennen, K.F.
(2000) Journal of Applied Physics, 88, 6476.
Kim, K.S., Saxler, A., Kung, P., Razeghi, R.
and Lim, K.Y. (1997) Determination of the
band-gap energy of Al1 xInxN grown by
metal–organic chemical-vapor
deposition. Applied Physics Letters, 71 (6),
800–802.
Yamaguchi, S., Kariya, M., Nitta, S.,
Takeuchi, T., Wetzel, C., Amano, H. and
Akasaki, I. (1998) Observation of
photoluminescence from Al1 xInxN
heteroepitaxial films grown by
metalorganic vapor phase epitaxy. Applied
Physics Letters, 73, 830.
Starosta, K. (1981) Physica Status Solidi a:
Applied Research, 68, K55–K57.
407 Kubota, K., Kobayashi, Y. and Fujimoto, K.
(1989) Journal of Applied Physics, 66, 2984.
408 Kistenmacher, T.J., Ecelberger, S.A. and
Bryden, W.A. (1993) Journal of Applied
Physics, 74, 1684.
409 Bedair, S.M., McIntosh, F.G., Roberts,
J.C., Piner, E.L., Boutros, K.S. and
El-Masry, N.A. (1997) Journal of Crystal
Growth, 178, 32.
410 Mohammad, S.N., Salvador, A. and
Morkoc, H. (1995) Emerging GaN based
devices. Proceedings of the IEEE, 83,
1306–1355.
411 Nagamoto, T., Kuboyama, T., Minamino,
H. and Omoto, O. (1989) Japanese Journal
of Applied Physics, 28, L1334.
412 Ryu, M-.Y., Chen, C.Q., Kuokstis, E., Yang,
J.W., Simin, G. and Asif Khan, M. (2002)
Luminescence mechanisms in
quaternary AlxInyGa1xyN materials.
Applied Physics Letters, 80 (20), 3730–3733.
413 Williams, C.K., Glisson, T.H., Hauser, J.R.
and Littlejohn, M.A. (1978) Journal of
Electronic Materials, 7, 639.
414 Angerer, H., Brunner, D., Freudenberg,
F., Ambacher, O., Stutzmann, M., H€opler,
R., Metzger, T., Born, E., Dollinger, G.,
Bergmaier, A., Karsch, S. and K€orner,
H.-J. (1997) Determination of the Al mole
fraction and the band gap bowing of
epitaxial AlxGa1xN films. Applied Physics
Letters, 71 (11), 1504–1506.
415 G€orgens, L., Ambacher, O., Stutzmann,
M., Miskys, C., Scholz, F. and Off, J.
(2000) Characterization of InGaN thin
films using high-resolution X-ray
diffraction. Applied Physics Letters, 76 (5),
577–579.
416 Peng, T., Piprek, J., Qiu, G., Olowolafe,
J.O., Unruh, K.M., Swann, C.P. and
Schubert, E.F. (1997) Band gap bowing
and refractive index spectra of
polycrystalline AlxIn1xN films deposited
by sputtering. Applied Physics Letters, 71
(17), 2439–2441.
417 Bellaiche, L., Wei, S.-H. and Zunger, A.
(1997) Bond-length distribution in
tetrahedral versus octahedral
semiconductor alloys: the case of
j127
j 1 General Properties of Nitrides
128
418
419
420
421
422
423
424
425
426
427
Ga1 xInxN. Physical Review B: Condensed
Matter, 56 (21), 13872–13877.
Bernardini, F. and Fiorentini, V. (2001)
Nonlinear macroscopic polarization in
III–V nitride alloys. Physical Review B:
Condensed Matter, 64, 085207.
Bellaiche, L. (1999) Band gaps of latticematched (Ga,In)(As,N) alloys. Applied
Physics Letters, 75 (17), 2578–2580.
Bellaiche, L., Wei, S.-H. and Zunger, A.
(1997) Band gaps of GaPN and GaAsN
alloys. Applied Physics Letters, 70, 3558.
Wang, L.-W. (2001) Large-scale localdensity-approximation band gapcorrected GaAsN calculations. Applied
Physics Letters, 78 (11), 1565–1567.
Kent, P.R.C. and Zunger, A. (2001)
Evolution of III–V nitride alloy electronic
structure: the localized to delocalized
transition. Physical Review Letters, 86 (12),
2613–2616.
Vurgaftman, I., Meyer, J.R. and RamMohan, L.-R. (2001) Band parameters for
III–V compound semiconductors and
their alloys. Journal of Applied Physics, 89
(11), 5815–5875.
Morkoc, H. (2001) III-nitride
semiconductor growth by MBE: recent
issues. Journal of Materials Science, 12,
677–695.
Kondow, M., Kitatani, T., Nakatsuka, S.,
Larson, M.C., Nakahara, K., Yazawa, Y.,
Okai, M. and Uomi, K. (1997) GaInNAs: a
novel material for long wavelength
semiconductor lasers. IEEE Journal of
Selected Topics in Quantum Electronics, 3,
719–730.
Harris, J.S., Jr (2000) Tunable longwavelength vertical-cavity lasers: the
engine of next generation optical
networks? IEEE Journal of Selected Topics
in Quantum Electronics, 6 (6), 1145–1160.
Kitatani, T., Nakahara, K., Kondow, M.,
Uomi, K. and Tanaka, T. (2000) A 1.3-mm
GaInNAs/GaAs single-quantum-well
laser diode with a high characteristic
temperature over 200 K. Japanese Journal
of Applied Physics, Part 2: Letters, 39 (2A),
L86–L87.
428 Yang, K., Hains, C.P. and Cheng, J.L.
(2000) Efficient continuous-wave lasing
operation of a narrow-stripe oxideconfined GaInNAs–GaAs multiquantumwell laser grown by MOCVD. IEEE
Photonics Technology Letters, 12 (1), 7–9.
429 Kondow, M., Kitatani, T., Nakahara, K. and
Tanaka, T. (1999) A 1.3-mm GaInNAs laser
diode with a lifetime of over 1000 hours.
Japanese Journal of Applied Physics, Part 2:
Letters, 38 (12A), L1355–L1356.
430 Li, N.Y., Hains, C.P.,Yang, K., Lu, J., Cheng,
J. and Li, P.W. (1999) Organometallic vapor
phase epitaxy growth and optical
characteristics of almost 1.2 mm GaInNAs
three-quantum-well laser diodes. Applied
Physics Letters, 75 (8), 1051–1053.
431 Sato, S. and Satoh, S. (1998) Roomtemperature pulsed operation of strained
GaInNAs/GaAs double quantum well
laser diode grown by metal organic
chemical vapour deposition. Electronics
Letters, 34 (15), 1495–1497.
432 Kitatani, T., Kondow, M., Nakahara, K.,
Larson, M.C. and Uomi, K. (1998)
Temperature dependence of the threshold
current and the lasing wavelength in
1.3-mm GaInNAs/GaAs single quantum
well laser diode. Optical Review, 5 (2),
69–71.
433 Nakatsuka, S., Kondow, M., Kitatani, T.,
Yazawa, Y. and Okai, M. (1998) Index-guide
GaInNAs laser diode for optical
communications. Japanese Journal of
Applied Physics, Part 1: Regular Papers, Short
Notes & Review Papers, 37 (3B), 1380–1382.
434 Ougazzaden, A., Bouchoule, S., Mereuta,
A., Rao, E.V.K. and Decobert, J. (1999)
Room temperature laser operation of bulk
InGaAsN/GaAs structures grown by APMOVPE using N2 as carrier gas.
Electronics Letters, 35 (6), 474–475.
435 Fischer, M., Reinhardt, M. and Forchel, A.
(2000) High temperature operation of
GaInAsN laser diodes in the 1.3 mm
regime. 58th Device Research
Conference, IEEE, pp. 119–120.
436 Shimizu, H., Kumada, K., Uchiyama, S.
and Kasukawa, A. (2000) High
References
437
438
439
440
441
442
443
444
performance CW 1.26 mm GaInNAsSbSQW and 1.2 mm GaInAsSb-SQW ridge
lasers. Electronics Letters, 36 (20),
1701–1703.
Setiagung, C., Shimizu, H., Ikenaga, Y.,
Kumada, K. and Kasukawa, A. (2003)
Very low threshold current density of 1.3mm-range GaInNAsSb–GaNAs 3 and 5
QWs lasers. IEEE Journal of Selected
Topics in Quantum Electronics, 9 (5),
1209–1213.
Bank, S.R., Yuen, H.B., Bae, H., Wistey,
M.A. and Harris, J.S. (2006)
Overannealing effects in GaInNAs(Sb)
alloys and their importance to laser
applications. Applied Physics Letters, 88,
article 221115.
Gollub, D., Moses, S., Fischer, M. and
Forchel, A. (2003) 1.42 mm continuouswave operation of GaInNAs laser diodes.
Electronics Letters, 39 (10), 777–778.
Ikenaga, Y., Miyamoto, T., Makino, S.,
Kageyama, T., Arai, M., Koyama, F. and
Iga, K. (2002) 1.4 mm GaInNAs/GaAs
quantum well laser grown by chemical
beam epitaxy. Japanese Journal of Applied
Physics, 41, 664–665.
Tansu, N. and Mawst, L.J. (2002) Lowthreshold strain-compensated InGaAs(N)
(l ¼ 1.19–1.31 mm) quantum well lasers.
IEEE Photonics Technology Letters, 14 (4),
444–446.
Peng, C.S., Jouhti, T., Laukkanen, P.,
Pavelescu, E.-M., Konttinen, J., Li, W. and
Pessa, M. (2002) 1.32-mm GaInNAs-GaAs
laser with a low threshold current density.
IEEE Photonics Technology Letters, 14 (3),
275–277.
Yang, X., Jurkovic, M.J., Heroux, J.B. and
Wang, W.I. (1999) Molecular beam
epitaxial growth of InGaAsN:Sb/GaAs
quantum wells for long-wavelength
semiconductor lasers. Applied Physics
Letters, 75 (2), 178–180.
Yang, X., Heroux, J.B., Mei, L.F. and
Wang, W.I. (2001) InGaAsNSb–GaAs
quantum wells for 1.55 mm lasers grown
by molecular-beam epitaxy. Applied Physics
Letters, 78 (26), 4068–4070.
445 Vurgaftman, I., Meyer, J.R., Tansu, N. and
Mawst, L.J. (2003) (In)GaAsN–GaAsSb
type-II W quantum-well lasers for
emission at l ¼ 1.55 mm. Applied Physics
Letters, 83 (14), 2742–2744.
446 Bank, S.R., Wistey, M.A., Goddard, L.L.,
Yuen, H.B., Lordi, V. and Harris, J.S., Jr
(2004) Low-threshold continuous-wave
1.5-mm GaInNAsSb lasers grown on
GaAs. IEEE Journal of Quantum
Electronics, 40 (6), 656–664.
447 Fischer, M., Reinhardt, M. and Forchel, A.
(2000) Room-temperature operation of
GaInAsN/GaAs laser diodes in the 1.5 mm
range. Conference Digest, 2000 IEEE 17th
International Semiconductor Laser
Conference (Cat. No. 00CH37092), IEEE,
Piscataway, NJ, pp. 115–116.
448 Livshits, D.A., Egorov, Yu.A. and Riechert,
H. (2000) 8 W continuous wave operation
of InGaAsN lasers at 1.3 mm. Electronics
Letters, 36 (16), 1381–1382.
449 Bugge, F., Erbert, G., Fricke, J., Gramlich,
S., Staske, R., Wenzel, H., Zeimer, U. and
Weyers, M. (2001) 12 W continuous-wave
diode lasers at 1120 nm with InGaAs
quantum wells. Applied Physics Letters, 79
(13), 1965–1967.
450 Reinhardt, M., Fischer, M., Kamp, M. and
Forchel, A. (2000) 7.8 GHz small-signal
modulation bandwidth of 1.3 mm DQW
GaInAsN/GaAs laser diodes. Electronics
Letters, 36 (12), 1025–1026.
451 Steinle, G., Mederer, F., Kicherer, M.,
Michalzik, R., Kristen, G., Egorov, A.Y.,
Riechert, H., Wolf, H.D. and Ebeling, K.J.
(2001) Data transmission up to 10 Gbit/s
with1.3 mmwavelengthInGaAsNVCSELs.
Electronics Letters, 37 (10), 632–634.
452 Fischer, M., Reinhardt, M. and Forchel, A.
(2000) A monolithic GaInAsN verticalcavity surface-emitting laser for the
1.3-mm regime. IEEE Photonics Technology
Letters, 12 (10), 1313–1315.
453 Schneider, H.C., Fischer, A.J., Chow, W.W.
and Klem, J.F. (2001) Temperature dependence of laser threshold in an In GaAsN
vertical-cavity surface-emitting laser.
Applied Physics Letters, 78 (22), 3391–3393.
j129
j131
2
Electronic Band Structure and Polarization Effects
Introduction
The band structure of a given semiconductor is pivotal in determining its potential
utility. Consequently, an accurate knowledge of the band structure is critical if the
semiconductor in question is to be incorporated in the family of materials considered
for serious investigations and device applications. The group III–V nitrides are no
exception, and it is their direct bandgap nature and the size of the energy gap that
spurred the interest in nitride semiconductors. Nitride semiconductors can be
classified into two groups. One group pertains to stoichiometric systems where N
represents 50% of the constituents while the other half is made of metal constituents.
These stoichiometric nitrides come in wurtzitic and zinc blende (ZB) forms. The
other class of nitrides is the dilute compound semiconductors, wherein very small
amounts of N are added to the lattice for remarkably large negative bowing of the
bandgap, making these dilute nitride systems compete for longer wavelength
applications. For example, the bandgap of GaAs can be extended to 1.3 mm applications. Likewise, the bandgap of InGaAs coherently grown on GaAs can be extended
with dilute amounts of N in the lattice to be a contender for 1.5 mm applications,
which has been the domain of In0.53Ga0.47As lattice matched to InP. The impact of
dilute nitrides is that, in at least the aforementioned example, what used to be the
domain of InP-based technology can be met by GaAs technology with untold
consequences in terms of not only technology but also the cost of that technology.
A number of researchers have published band structure calculations for both
wurtzite (Wz) and zinc blende GaN, AlN, and InN. To make matters more interesting,
the bandgap of InN transmogrified from 1.9 eV downward to about 0.7 eV between
the first edition and the current one. It is argued that the first set of bandgap
measurements might have been conducted in films containing a large amount of O,
which could have caused an upward shift in the measured data. The initial estimate of
the 1.9 eV bandgap of InN, in addition to creating confusion concerning the nature
and applications of InN, caused uncertainties in the bandgap of the InGaN ternary as
well. The situation is exacerbated by inhomogeneities in composition and strain as
well as poor sample quality. The situation in fact transformed into one in which
Handbook of Nitride Semiconductors and Devices. Vol. 1. Hadis Morkoç
Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 978-3-527-40837-5
j 2 Electronic Band Structure and Polarization Effects
132
multiple bowing parameters, depending on the InN composition in the alloy, were
proposed. It can now be said that, with the small bandgap of about 0.8 eV for InN as
agreed on, only a single bowing parameter can indeed account for the data dating
back to 1980s where incompatibilities with the 1.9 eV InN bandgap were reported [1].
The first Wz GaN band structure found through a pseudopotential method led to a
3.5 eV direct bandgap. The band structure for ZB GaN has been obtained by a firstprinciples technique within the local density functional framework with a direct
bandgap of 3.40 eV and a lattice constant of 4.50 Å. A treatise of the band structure in
bulk and quantum wells (QWs) with and without strain for conventional nitrides,
GaN, InN, and AlN, along with their alloys in the wurtzite and zinc blende form, and
dilute nitrides (all of which are zinc blende ternaries and quaternaries, for example,
GaAsN and GaInAsN) is discussed. Complete and consistent parameter sets are
provided with tabulations of the direct and indirect energy gaps and spin–orbit and
crystal field splitting. The alloy bowing parameters, electron and hole effective
masses, deformation potentials, elastic constants, and piezoelectric (PE) and spontaneous polarization coefficients are given in many tables in Chapter 1. However, the
basis of spontaneous and piezoelectric polarization effects and their practical impact
on single- and dual-interface heterostructures are discussed in this chapter following
the band structure discussion. The heterostructure band offsets are discussed in
Volume 2, Chapter 4. The temperature dependence of the bandgap parameter of
nitrides inclusive of the case of band tailing, primarily GaN, is discussed in this
chapter as well as in Volume 2, Chapter 5 in conjunction with its optical properties.
2.1
Band Structure Calculations
A glossary of band structure calculations will be given before delving into the specific
calculations employed to determine the band structure of nitride semiconductors.
This is not an all-inclusive treatment of the field, but it is a compact treatment of the
salient features of methods used to illustrate and/or calculate the band structure. The
first exposure of many students to band structure calculations is that of a free-electron
or nearly free electron approach in a periodic lattice [2–4]. In this one-electron model,
the periodic potential can be thought of as arising from the periodic charge
distribution associated with ion cores that are situated at the lattice sites. To expand
the picture to include many electrons, an average constant potential contribution is
added to account for all the other electrons in the system. This problem was first
considered by Felix Bloch [5]. In its simplest form, the wave function representing the
electrons in a periodic potential would be composed of the product of a plane wave
function representing an electron in free space and a function representing the
periodicity of the crystal.
In the nearly free electron approach, the effect of the crystalline potential on the
electronic structure is considered to be weak, and the energy levels of the electrons
have little resemblance with those of the atoms. The allowed energies occur in bands
of allowed states separated by forbidden energy regions (gaps). Within an allowed and
2.1 Band Structure Calculations
mainly occupied energy band, the electron motion is in many ways similar to that of
free particles with an appropriate charge and effective mass. Whether the crystal is an
insulator or conductor depends on whether the states within a band or set of bands
are completely filled or partially empty. The crystal is considered a semiconductor if
the gap between a filled band and the empty band is small, the exact value of which has
changed over the years. For example, a gap of 3 eV was taken to be associated with
insulators, which is smaller than the gap of GaN. The one-electron picture is an
approximation and does not take into consideration processes such as electron–
electron interaction, which is neglected other than what is convoluted in the average
potential. To understand the basic aspects of band structure, it is instructive to
consider an infinite periodic one-dimensional square well potential, which forms the
basis for the Kronig–Penney approach [6]. This approximation leads to an exact
solution of the Schr€odinger equation. Even though the square well potential
approximation is very crude, it serves to illustrate explicitly many important characteristic features of the electron behavior in periodic lattices.
In contrast to the free-electron approximation in which the potential energy of the
electron is assumed to be small in comparison to its total energy, the opposite is
assumed in another commonly practiced approximation, called the tight binding
(TB) approximation. Specifically, it is assumed that the potential energy of the
electrons accounts nearly for all of the total energy in the case of which the allowed
energy bands are narrow compared to the forbidden ones. Unlike the free-electron
model, the electronic wave functions are more or less localized around the atoms.
Thus, the interaction between neighboring atoms is relatively weak, and the wave
functions and the allowed energy levels of the crystal as a whole resemble the wave
function and energy levels of isolated atoms. In a sense, an electron associated with
an atom is assumed to remain in an orbit associated with that atom, and these orbitals
are combined linearly in a form to be consistent with the Bloch–Floquent theorem to
represent a state running throughout the crystal. Again, each orbital is localized on a
particular atom. As such the results are very sensitive to overlap integrals, which in
turn are sensitive to the details of the orbitals outside the cores and to the lattice
spacing. Naturally, in this approach, electrons are not affected by atoms more than a
single atomic spacing away. The choice of whether the free electron or the tight
binding method is good depends on the particular crystal. In fact, in some crystals
neither of these is good. The tight binding approach was assumed by Bloch in his
original discussion of energy bands. If there is an appreciable interatomic interaction, the tight binding approach must use a linear combination of atomic orbitals
(LCAO), in which a quantum mechanical variational procedure is employed to find
the combination of s, p, and d orbitals that correspond to the lowest energy in the
system. Not surprisingly, the tight binding methods are successful when the effect of
the periodic core potential is quite large. This is true, for example, when the band is
derived from the 3d states in the first series of transition elements. In these elements,
the 3d states are partially filled and one or two electrons are present in 4s subshells.
When atoms of these elements are brought together to make a solid, the interaction
between the 4s states is very strong while the overlap between the partially filled inner
shells is rather weak.
j133
j 2 Electronic Band Structure and Polarization Effects
134
There are other band structure methods that provide much improvement over the
nearly free electron model and the tight binding model. These methods rely on
choosing an appropriate basis for the electrons that represent electron behavior both
inside and outside the atomic sphere. For example, while a single plane wave may be
adequate to represent electron wave function in the interstitial space, to account for
the rapid change in the function near the core region, a combination of a large
number of plane waves would be necessary.
Because the core functions are described in terms of radial functions and spherical
harmonics, all unknown wave functions of crystals can be expanded in a set of known
functions such as plane waves, radial functions, and spherical harmonics. The
various methods for band structure calculations, therefore, differ in the initial choice
of boundary conditions that the wave functions must satisfy. A brief qualitative
discussion of these methods is given in the following sections.
2.1.1
Plane Wave Expansion Method
Here, the Bloch wave is expanded as a linear combination of plane waves (LCOPW),
namely,
! ! !
X
!
Þ:r
ð
Ck ðKÞei k þ k :
ð2:1Þ
Fk ð r Þ ¼
!
k
The combination coefficients Ck(K) are determined by solving the determinantal
equation
"
#
X
!
!
h2 ðk þ KÞ2
0
E K jCk ðKÞj2 dK;K 0 þ
Ck ðKÞCk ðK ÞVð k k 0 Þ ¼ 0;
2m
0
K;K
!
!
0
ð2:2Þ
where EK is the energy eigenvalue and Vð k k Þ is the crystalline potential.
Although this is a simple method, its practical implementation is difficult, as it
requires a large number of plane waves to represent the behavior of electrons near a
core region. Consequently, the convergence in the eigenvalues is poor and it requires
solving a large determinantal equation.
2.1.2
Orthogonalized Plane Wave (OPW) Method
The method originally proposed by Herring and Hill [7] and later discussed by
Woodruff [8] considers the Bloch function to be a linear combination of OPW basis.
The OPW basis consists of a plane wave orthogonalized to the atomic core functions,
such that the electron behaves like a core electron while inside the core and like a
plane wave while in the interstitial region. The OPW basis can be written as
! !
X
1
!
!
mkj Fkj ð r Þ;
ð2:3Þ
X k ð r Þ ¼ pffiffiffiffiffiffiffiffi ei k : r NW
j
2.1 Band Structure Calculations
where Fkj are Ðthe core wave functions for constant j. The term mkj is evaluated by
!
!
requiring that Fkj ð r ÞX k ð r Þd3 r ¼ 0mX , where W is the volume of the Wigner–Seitz
cell, and N is the number of atoms.
The Bloch function is then expressed as a linear combination of the OPWs, and the
energy eigenvalues are computed by solving the appropriate determinantal equation.
This method has been applied to band structure calculation of metallic and nonmetallic solids.
2.1.3
Pseudopotential Method
Phillips and Kleinman [9] later demonstrated that it is possible to rewrite the crystal
potential that includes contributions from the core and valence electrons in such a
way that the Bloch function can be written in terms of a linear combination of planes
as in the plane wave expansion method without compromising the convergence
advantages of the OPW method [10,11]. The crystal potential thus obtained is called
the pseudopotential.
As mentioned earlier, the orbitals between the cores (outer regions) are smooth
where the wave functions are somewhat like plane waves. Near the core regions,
however, the wave functions are complicated by the strong and rapidly varying
potential. The orthogonality requirement causes nodes (zeros) in the wave function
in the core region. The weak potential experienced by the electrons in the outer region
can be treated as a perturbation that mixes the plane wave components (strongly only
at the Brillouin zone (BZ) boundaries). While the orbitals are not like plane waves
near the cores and potentials vary rapidly, it is argued that what goes on near the core
is irrelevant to the dependence of the energy on the wave vector. The energy wave
vector dependence can be calculated by applying the Hamiltonian operator to an
orbital at any point in space, which when applied to the outer regions, the picture
would be that of nearly free electron energy. The actual potential energy in the core
region can be represented by an effective potential energy called the pseudopotential
that gives the same wave functions outside the core regions as the actual potential.
Surprisingly, the pseudopotential is nearly zero in the core region, which is arrived at
by experience with such potentials as well as theoretical considerations. Although use
of these pseudopotentials may lead to incorrect wave functions, doing so can, with
acceptable accuracy, indicate how the energy varies.
2.1.4
Augmented Plane Wave (APW) Method
This method proposed by Slater [12] also makes use of the fact that the wave function
inside the core behaves like atomic functions and outside the core like plane waves as
in the OPW method. The difference lies in how one applies the boundary conditions.
Unlike the OPW method where the wave function outside and inside the core are
matched by the Schmidt orthogonalization condition, in the APW scheme one
expands the wave functions outside the core region (r ri) by a set of plane waves and
j135
j 2 Electronic Band Structure and Polarization Effects
136
inside the core region (r ri) by a sum of spherical waves, namely,
!!
X
!
alm Qðr i rÞY lm ðq; fÞRl ðE; rÞ;
Fk ð r Þ ¼ a0 Qðr r i Þei k : r þ
l;m
where Y is the step function with Y(x) ¼ 1 for x 0 and Y(x) ¼ 0 for x < 0. The
!
coefficients alm are chosen in such a way that the wave function Fk ð r Þ is continuous
across the sphere of radius ri. The term Rl(E, r) represents the solution of the radial
equation.
Slater [12] proposed a reasonable approximation to the potential with a constant
potential outside the cores and an ordinary atomic potential inside a sphere
surrounding each ion core. The Schr€odinger equation is then solved separately for
each of the two regions and solutions are matched over the spherical boundaries
between them, and within each core the wave function can be expanded in spherical
harmonics. The solution outside the cores is a superposition of plane waves. The
combination gives this method its name, the augmented plane waves.
2.1.5
Other Methods and a Review Pertinent to GaN
Another class of calculations relies on what is called the all-electron approach. Here,
one develops potentials within the sphere around the core region and uses their
energy derivatives to expand the wave functions in a manner to match those outside
the sphere. Among the methods relying on this premise are linear muffin-tin orbital
(LMTO) and linearized augmented plane wave (LAPW) methods. Essentially, in the
LMTO method, the crystal is divided into nonoverlapping muffin-tin spheres
surrounding the atomic sites and an interstitial region outside of spheres. Inside
the muffin-tin sphere, the potential is assumed spherically symmetric while in the
interstitial region the potential is assumed constant or slowly varying. The early
application of LMTO method made use of what is called the atomic sphere
approximation (ASA), in which the crystal potential is treated as a superposition
of weakly overlapping spherical potentials centered around lattice sites but in such a
way as to fill the space and the total volume of muffin-tin sphere is the same as the
atomic volume. The potential is also assumed to be spherically symmetric inside each
muffin-tin sphere. Additionally, the kinetic energy of the basis function in the
interstitial regions is restricted to be a constant and that constant is typically assumed
to be zero in the calculations. In open structures, it is customary to include spheres
centered on interstitial sites. It is worth noting that the potential so determined is very
close to the true full potential and provide a much better representation of the
potentials than the original muffin-tin potentials that are constant in the interstitial
regions and spherical within nonoverlapping spheres. The limited precision,
0.1 eV, prevents this method from being applied satisfactorily to the very intricate
portions of the band structures. For example, very small differences in the total
energy between the zinc blende and wurtzitic forms of GaN are a troublesome point
for these ASA-LMTO calculations. Likewise, the crystal field splitting at the valence
band maxima presents similar problems.
2.1 Band Structure Calculations
The LMTO method has advantages such as using a minimal basis for computational efficiency and thus allowing large unit cell calculations. It also treats all
elements in the same manner and is accurate due to an augmentation procedure,
which gives the wave function a reasonably correct shape near the nuclei. The
method also uses atom-centered basis functions of well-defined angular momentum.
The full potential LMTO (FP-LMTO) calculations are all fully relativistic electrons
with the shape approximation to the charge density or the potential. As in the LMTO,
the crystal is divided into nonoverlapping muffin-tin spheres and interstitial
regions outside the spheres. Then, the wave function is presented differently in
those two regions in that inside the muffin-tin spheres, the basis functions are as in
the LMTO-ASA method and are of Bloch sum of linear muffin-tin orbitals and the
kinetic energy is not restricted to zero in the interstitial regions. All of the other above
methods rely on our knowledge of the potential in which the electrons inside a crystal
move. This potential results from the interaction between electrons, electrons and
nuclei, and between nuclei. This presents a many body problem that cannot be
solved exactly. Thus, approximations are needed. One of these techniques developed
by Hohenberg and Kohn and Sham, known as the density functional theory
(DFT) [13,14], has shown a great promise in treating energy bands of solids. DFT
can be construed as a self-consistent field method for attaining the crystal potential.
Over the years, many band structure calculations evolved to include not only the
electronic properties but also binding energies, through which the lattice constant,
bulk modulus, elastic constants, and vibrational properties such as phonon frequencies can be computed. The DFT method is well suited for calculating the aforementioned parameters. The basic quantity is the electronic charge density, and the
total energy is expressed as functional of this density. The total energy expression
as a function of density includes exchange and correlation effects in an average
electron gas like manner [15], which is termed as the local density approximation
(LDA). Most common form expresses the charge density in terms of occupied oneelectron wave functions, the corresponding eigenvalues of which constitute the band
structure [16].
Calculations of the self-energies are best done by the DFT-LDA method within the
GW approximation. The nomenclature is adopted from the original paper of Lars
Hedin [17] for one-electron Greens function and Hedin and Lundqvist [18] for
screened Coulomb interaction. This approximation represents the first term in a
perturbation expansion of the self-energy, which can also be viewed as the screened
Hartree–Fock (H–F) theory. It should be pointed out that in semiconductors the GW
method differs from the band structure determined by LDA chiefly due to a shift of
the conduction band relative to the valence band. Consequently, the fundamental
bandgap is underestimated. The genesis for Kohn–Sham [15] local DFT lies in
lowering the conduction band in a manner that is k-value dependent, as has amply
been pointed out by Sham and Schl€
uter [19] and Perdew and Levy [20] in back-to-back
articles. It is for this reason that DFT-based calculations are supplemented by a shift
of the calculated fundamental gap inspired by a body of experimental data. Most band
structure calculations for GaN and related materials rely on the DFT-LDA approach.
Computational development of the GW approximation was later accomplished by a
j137
j 2 Electronic Band Structure and Polarization Effects
138
series of authors, namely, Hyberstsen and Louie [21], Godby, Schl€
uter, and Sham [22],
and even later on by Aryasetiawan and Gunnarsson [23,24].
Let us now discuss the band structure of some specific nitride semiconductors.
This semiconductor family can exist in wurtzite and zinc blende crystal polytypes
with the Wz phase being the stable and widely used form. While both the Wz and ZB
polytypes of GaN have been given in quite some detail, the ZB varieties of AlN and
InN are not thermodynamically stable and reports are very sketchy. In fact, there are
predictions that ZB AlN has an indirect bandgap [25].
A review of the band structure calculation and the methodologies used for both
polytypes of all group III nitrides, BN, AlN, GaN, and InN, has been given by Lambrecht
and Segall [16]. Calculations of electronic and optical properties of Wz GaN and related
structures have been undertaken over many years [16,26] with more of them emerging
continually, as nitride-based devices become more popular. Methods such as the ab
initio, tight binding [27], pseudopotential, ASA LMTO, a general treatment of which has
been treated by Andersen [28], LCAO, LAPW, full potential linearized augmented plane
wave (FP-LAPW) method such as that reported by Wimmer et al. [29] for calculating the
electronic band structure of Wz semiconductors within the LDA [15,16], DFT, and GW
methods have been employed to calculate the energy bands for both wurtzite and zinc
blende GaN, InN, and AlN bulk materials. Christensen and Gorczyca [30,31] utilized
the ASA-LMTO method for group III-nitrides mainly GaN and AlN, albeit in relation to
their behavior under pressure. The ASA-LMTO method was also applied by Lambrecht
and Segall [32] to contrast and compare the nature of the direct and indirect bandgap of
various ZB and Wz nitrides and, in particular, in terms of the directness or indirectness
of the bandgap.
These methods change in their capabilities to varying degrees. Early versions of
pseudopotential calculations did not include the contribution by 3d-electrons because of the difficulty of attaining convergence in-plane wave expansions due to the
deep N pseudopotential [33]. A mixed basis set was later used to overcome this
apparent shortcoming [34] but with the consequence that ZB GaN would be lower in
energy, meaning favored over the Wz phase that is inconsistent with experiments,
and also was not confirmed with well-converged plane wave calculations performed
for GaN by Yeh et al. [35] and InAlN [36]. Yeh et al. [35] focused on the issue of
polymorphism for a large number of semiconductors including AlN, GaN, InN, AlP,
AlAs, GaP, GaAs, ZnS, ZnSe, ZnTe, CdS, C, and Si, and using the local density
formalism (LDF), developed a simple scaling at T ¼ 0 that systematizes the energy
difference (DE LDF
W-ZB ) between the ZB and Wz forms. This energy difference was found
~
to be linearly dependent on the atomistic orbital radii coordinate RðA;
BÞ that
depends only on the properties of the free atoms A and B, making up the binary
compound AB. Of special interest for the topic under discussion is that Yeh et al. [35]
found that DE LDF
W-ZB ðABÞ for GaN is 9.9 meV/atom, which is within 0.7 meV/atom of
the calculations by Van Camp et al. [37] and within 0.3 meV/atom of the calculations
by Min et al. [34]. The energy difference, DE LDF
W-ZB ðABÞ, for all three nitride binaries are
negative when this quantity is scaled with differences in tetrahedral radii and
Paulings electronegativity, implying that the equilibrium state of all three nitride
binaries is the Wurtzitic form.
2.1 Band Structure Calculations
The issue of d-electrons is an important one, as in Ga 3d, in relation to pseudopotential and all-electron calculations, specifically, to know whether these calculations can handle these bands. Because both In and Ga are heavy and have d cores, Ga
3d and In 4d states overlap with the deep N 2s states with serious implications about
bonding and band structure. The way in which the d-electrons are treated as core
orbitals with or without nonlinear core corrections or as valence states in pseudopotential methods caused some confusion, which has been the topic of some
discussion [32,38]. In the case of Fiorentini et al. [38], the structural and electronic
properties, albeit cubic GaN, were studied within the local density approximation by
the full potential linear muffin-tin orbitals method, wherein the Ga 3d-electrons were
treated as band states with no shape approximation to the potential and charge
density. Owing to the resonance of Ga 3d-states with nitrogen 2s states, the cation
d bands were found not to be inert, and features unusual for a III–V compound were
found in the lower part of the valence band as well as in the valence charge density and
density of states. Additional full and frozen (T ¼ 0) overlapped core calculations
performed for GaN, ZnS, GaAs, and Si (all cubic) showed that an explicit description
of closed-shell interaction has a noticeable effect on the cohesive properties of GaN.
The resulting energy resonance causes the Ga 3d-electrons to strongly hybridize with
both the upper and lower valence band s and p levels. Such hybridization is predicted
to have a profound influence on the GaN properties, including quantities such as the
bandgap, the lattice constant, acceptor levels, and valence band heterojunction
offsets. Because Al has no 3d core states, there is no hybridization between the
cation d states and the N 2s states. In short, the band structure and cohesive
properties of GaN are very sensitive to the cation d bands. On the pseudopotential
side, Wright and Nelson [36] provided a framework in which accurate calculations
treating the Ga 3d- and In 4d-electrons explicitly as valence states were performed by
extending the plane wave cutoff to 240 Ry to ensure convergence. An interesting
observation is that while d-electrons are important in bonding, they appear as
separate states in considering quasi-particle excitations in photoemission
experiments [39].
Somewhat of a side note but with legitimate relevance, it has been predicted in the
cases of ZnS and ZnSe that potential acceptors, such as Cu, whose d-electrons are
resonant with the lower valence band, are repelled by the d-hybridized upper valence
band, resulting in a deep level. Impurities without d-electron resonance form shallow
acceptors. Mg has no d-electrons and turns out to be sufficiently shallow for roomtemperature p-type doping of GaN. On the contrary, Zn, Cd, and Hg, which all have
d-electrons, form deep levels in GaN [40]. Further insight is warranted before
conclusive statements can be made with certainty as, for example, photoemission
data show the N 2s to be well below the Ga 3d band.
Pseudopotential calculations can also be applied to defects [41] and surfaces [42], as
has been done for GaN. The calculations relating to surfaces are discussed in
Section 3.2.7.2 in reasonable detail. Likewise, the calculations in relation to defects
are treated in Section 4.3.1. Other applications of the calculations discussed above are
for the determination of dielectric properties and susceptibility [43] and vibrational
properties [44].
j139
j 2 Electronic Band Structure and Polarization Effects
140
Calculations in the Hartree–Fock approximation rather than the local density
approximation have also been performed for nitride semiconductors, notably
GaN [45] and AlN [46,47]. While LDA calculations underestimate the bandgap, the
opposite is the case for Hartree–Fock calculations. In the H–F method, exchange is
treated exactly but the correlations are fully ignored. Specific to the case of GaN, its
total energy as a function of unit cell volume has been calculated for the wurtzite, zinc
blende, and rock salt phases by the ab initio all-electron periodic Hartree–Fock
method by Pandey et al. [45]. In this case, the gallium 3d levels were treated as fully
relaxed band states, and the internal parameters c/a and u in the wurtzite phase were
optimized. The calculated transition pressure between the wurtzite and rock salt
phases were found to be about 52 GPa at the Hartree–Fock level and about 35 GPa at
the correlated level. The calculated electronic structure shows strong hybridization of
Ga 3d and N 2s states with the ordering as Ga 3d–N 2s–N 2p in all the phases. The
results indicate the bandgap to be direct at G in the wurtzite and zinc blende phases
and indirect in the high-pressure rock salt phase where the valence band maximum is
shifted away from the G point. The electronic structure of Wz AlN has been
investigated by means of periodic ab initio Hartree–Fock calculations for the purpose
of calculating the binding energy, lattice parameters (a, c), and the internal coordinate
or parameter (u) [47]. The values of the bulk modulus, its pressure derivative, the
optical phonon frequencies at the center of the Brillouin zone, and the full set of
elastic constants have been calculated and compared with experimental data.
When ab initio Hartree–Fock calculations were used to determine the electronic
structure of AlN in high pressure, the rock salt phase resulted [46]. In this phase, the
calculated lattice constant is 3.982 Å with the bulk modulus of 329 GPa. As in the
case of GaN, the rock salt phase is predicted to be indirect at the X point with a gap of
8.9 eV. Moreover, the bonding is essentially ionic between Al and N. The direct gap
shows a stronger linear dependence on pressure with a pressure derivative of
68 meV GPa1 compared to that of the indirect X-valley gap with a pressure
derivative of 31.7 meV GPa1. It should be emphasized that the rock salt phase
is favored to exist under high pressure, and as such throughout this book and
literature, nitrides are spoken of as if they are wurtzitic with GaN being cubic
also when grown away from thermodynamic equilibrium conditions on cubic
substrates along h0 0 1i directions.
As mentioned above, the electronic properties of nitride semiconductors can more
accurately be calculated using first-principles techniques like density functional
theory [25] within the Greens function theory with the characteristic GW approximation of the exchange correlation self-energy [17]. These calculations have been applied
to Wz and ZB GaN and AlN by Rubio et al. [25] and to ZB GaN by Palummo et al. [48].
The computational complexity of the full GW method is prohibitive for applications to
complex systems with large number of atoms, such as surfaces, interfaces, and
clusters. However, it should be mentioned that efficient simplified version of the GW
method has been reported to reduce the central processing unit (CPU) time by a factor
of 100 (in conjunction with semiconductors Si, GaAs, AlAs, and ZnSe) [49,50]. Unless
the simplified GW method is used [48,49], the full GW method is typically limited to
simple systems, for example, elemental or binary semiconductors.
2.1 Band Structure Calculations
The GW calculations are reasonably consistent with each other and also with
experiments in many cases. In the calculations of Rubio et al. [25], the ab initio
pseudopotential method within the local density approximation and the quasi-particle
approach have been employed to determine the electronic properties of both Wz and
ZB phases of AlN and GaN. The quasi-particle band structure energies were calculated
using a model dielectric matrix for the evaluation of the electron self-energy. In the zinc
blende structure, AlN was predicted to be indirect (G to X) with (4.9 eV) and that GaN to
be direct with 3.1 eV at the G point, the latter in good agreement with absorption
experiments on cubic GaN, showing the bandgap to be 3.2–3.3 eV. In the calculations
of Palummo et al. [48], models of diagonal and off-diagonal screening with LDA-RPA
full calculations in cubic GaN were considered. Simplified GW calculations relying on
these models were also compared with full GW calculations. At the time empirical
pseudopotential calculations were not available, necessitating ab initio RPA calculations to be done within the DFT-LDA approach. These calculations have already been
used for obtaining the full GW band structure of GaN [51].
It should be mentioned that with respect to pure LDA results, the valence band
shifts down and the conduction band shifts up, resulting in larger bandgap estimation. The amount of downward shift of the valence bands increases with the increase
in energy below the valence band maximum. The N2s states are about 1.5–2 eV more
than the valence band maximum. Moreover, the bottom of the N2p valence band, the
character of which is of a mixture of N2p cations, shifts by an amount of about 0.5 eV
more than the maximum. The absolute shift of the valence band maximum is a
problem in GW theories, which is also the case with this method for very established
materials such as Si. This seems to stem from the choice parameterization used for
the LDA starting point of the calculations. The GW method changes the gaps of GaN
and AlN by 1 and 2 eV, respectively [16]. The conduction band correction is on the
order of 0.1 eV across the k-points and specific states. This figure appears to increase
with the increase in energy dealing with higher conduction bands, awaiting further
refinement following any comparison with experiments when accurate measurements become available.
Unlike the full GW calculations, the TB approach provides an attractive possibility
for an extension of the system size accessible to electronic structure calculations
with atomic resolution. TB calculations have been applied to nitride-based
systems [52–54]. Moreover, the Slater–Koster parameters transferable between the
ZB and Wz crystal phases have been treated [27].
There has been a plethora of reports regarding band structure calculations in nitride
semiconductors. The calculated band structures of Wz GaN, AlN, and InN are
exhibited in Figure 2.1. For semblance of completeness, results from empirical
pseudopotential method are also included here. Fritsch et al. [55] investigated the
electronic band structure of both the wurtzite and zinc blende group III nitride
semiconductors GaN, AlN, and InN within the empirical pseudopotential approach.
Using ionic model potentials and a static dielectric screening function derived, the
cationic and anionic model potential parameters were obtained from the zinc blende
GaN, AlN, and InN experimental data. Using these model potentials, Fritsch et al. [55]
calculated the band structure of group III nitrides in both the wurtzite and zinc blende
j141
j 2 Electronic Band Structure and Polarization Effects
142
(a)
GaN
12
10
3
1
1
3
6
Energy ( eV )
6
1
8
3
1
2
4
3
2
1
1
6
5
0
–2
2
4
3
2
3
–4
3
–6
1
1
3
1
3
–8
–10
3
–12
3
–14
–16
3
1
3
3
1
3
A
S
H
R L
T
U
P
K
M
1
K
M
M U L
T
(b)
6
3
3
10
H
P K
4
1
3
8
3
3
1
1
6
S
A
R
AlN
12
3
2
1
4
Energy (eV)
3
4
3
2
1
5
4
3
1
3
3
1
6
0
3
–2
2
3
–4
1
–6
–8
A
S
Γ
R L
T
U
–10
3
–12
–14
M
K
T
3
1
1
3
M U
H
P
K
M
L
Figure 2.1 Calculated band structures of (a) wurtzite GaN,
(b) AlN, and (c) InN in the LDA within the FP-LMTO method at the
experimental lattice constant and optimized u-value. The first
Brillouin zone is also shown for convenience [16].
R
A
S
H P K
2.1 Band Structure Calculations
(c)
8
6
3
1
3
2
6
4
1
1
1
3
1
1
3
2
Energy (eV)
InN
4
3
1
2
10
1
6
0
2
–2
5
4
1
3
2
3
1
3
–4
–6
3
3
1
3
1
A
S
R L
T
U
–8
3
–10
3
1
3
–12
3
–14
M
K
1
3
T
H
P
K
M
3
1
M U
L
R
A
S
H P K
Figure 2.1 (Continued )
form, recognizing the necessity of including the anisotropy of wurtzite crystals in the
screening function. The band structures so calculated for wurtzitic GaN, AlN, and InN
are shown in Figure 2.2. The same for the zinc blende variety is shown in Figure 2.3.
It should be noted at the outset that all these binary materials, including alloy
compounds obtained by combinations of these binaries, are wide direct bandgap
semiconductors in both crystal phases, except zinc blende AlN that is expected to have
an indirect gap with the conduction band minimum being at the X valley. Due to the
lack of reliable experimental data, many details of these studies must be improved to
provide an accurate band description. Approaches such as the kp model [56,57]
including strain have been employed to calculate the valence band structure of Wz
GaN [56,58]. First-principles calculations of effective mass parameters and valence
band structures in bulk and confined systems with and without strain, utilizing the
FP-LAPW method [59–61,66] and envelope function formalism for valence bands in
wurtzite quantum wells, have been undertaken [62].
The wurtzite structure has a hexagonal unit cell and thus two lattice constants, c
and a. It contains six atoms of each type. The space group for the wurtzite structure is
P63mc (C46v ) [63]. The wurtzite structure consists of two interpenetrating hexagonal
close-packed (HCP) sublattices, each with one type of atoms, offset along the c-axis by
5/8 of the cell height (5c/8). The zinc blende structure has a cubic unit cell, containing
four group III elements and four nitrogen elements. The space group for the zinc
blende structure is T 2d : F 43m. The position of the atoms within the unit
cell is identical to the diamond crystal structure. Both structures consist of two
j143
j 2 Electronic Band Structure and Polarization Effects
144
interpenetrating face-centered cubic sublattices, offset by one quarter of the distance
along a body diagonal. Each atom in the structure may be viewed as positioned at the
center of a tetrahedron with its four nearest neighbors defining the four corners of the
tetrahedron. The zinc blende and wurtzite structures are similar. In both cases, each
Energy (eV)
10
5
0
–5
A
R
L U M
Σ
Γ Δ A
S
H P K
T
Γ
(a)
15
Energy (ev)
10
5
0
–5
A
R
L U M
Σ
Γ Δ A
S
(b)
Figure 2.2 Band structure of (a) wurtzitic GaN, (b) wurtzitic AlN,
and (c) wurtzitic InN along high-symmetry lines in the Brillouin
zone calculated within the empirical pseudopotential method
(EPM), using ionic model potentials obtained experimentally from
zinc blend varieties. Courtesy of Daniel Fritsch et al. [55].
H P K
T
Γ
2.1 Band Structure Calculations
j145
Energy (eV)
10
5
0
–5
A
R
L U M
Σ
Γ Δ A
S
H P K
Γ
T
(c)
Figure 2.2 (Continued )
Energy (eV)
15
L3
10
K1
X3
L1
5
X1
L3
0
X5
X3
L2
–5
L
Λ
Γ
Δ
X
(a)
Figure 2.3 Band structure of (a) zinc blende GaN, (b) zinc blende
AlN, and (c) zinc blende InN along high-symmetry lines in the
Brillouin zone calculated within the empirical pseudopotential
method (EPM), using ionic model potentials obtained
experimentally. Courtesy of Daniel Fritsch et al. [55].
K1
K2
K1
K1
UK
Σ
Γ
j 2 Electronic Band Structure and Polarization Effects
146
20
15
Energy (eV)
L3
10
X1
5
0
L3
–5
L2
10
K1
K2
X5
K1
X3
Λ
L
(b)
K1
X3
L1
Γ
Δ
K1
X
U,K
Σ
Γ
L3
Energy (eV)
K1
X3
5
X1
K1
L1
0
K2
L3
X5
K1
L2
–5
L
K1
X3
Λ
Γ
Δ
X
U, K
Σ
Γ
(c)
Figure 2.3 (Continued )
group III atom is coordinated by four nitrogen atoms. Conversely, each nitrogen atom
is coordinated by four group III atoms. The main difference between these two
structures lies in the stacking sequence of closest packed diatomic planes. For the
wurtzite structure, the stacking sequence of the (0 0 0 1) planes is ABABAB in the
h0 0 0 1idirection. For the zinc blende structure, the stacking sequence of the (1 1 1)
planes is ABCABC in the (1 1 1) direction.
2.1 Band Structure Calculations
The structure and the first Brillouin zone of a wurtzite and zinc blende crystal along
with the irreducible wedges, calculated using the LDA within the FP-LMTO method
at the experimental lattice constant and optimized u value, are displayed in
Figure 2.4a and b, respectively. In a crystal with Wz symmetry, the conduction band
kz
So
A
o
R
Δo
T
o
Γ
H
L
o
o
S’
oP
U
K
o
o
ky
T’
Σ
M
kx
(a)
kz
L
U
Q
S
Z
kx
K
X
ky
W
(b)
Figure 2.4 (a) Structure and the first Brillouin
zone of a wurtzite crystal. Schematics of the
irreducible wedges of Wz structure, indicating
the high-symmetry points and lines, are also
shown. The Umin point of the Wz phase is located
on the M–L line at two-thirds a distance away
from the M point. (b) Structure and the first
Brillouin zone of a zinc blende crystal.
Schematics of the irreducible wedges of ZB
structure indicating the high-symmetry points
and lines are also shown.
j147
j 2 Electronic Band Structure and Polarization Effects
148
Conduction band
E gA
E0
E0
C6v
or
5
9
A
6
7
7
7
Valence
band
9
, J = 3/2
Valence
band
B
cr
15
so
15
1
7
ZB
W
Crystal
field
cr =
so
W
Spin
orbit
7
C
W
8 , J = 1/2
Crystal
field
1
=
ZB
ZB
Spin
orbit
cr = 0
so
=
2
=
3
Figure 2.5 Schematic representation of the splitting of the valence
band in Wz crystals due to crystal field and spin–obit interaction.
From left to right, the crystal field splitting is considered first.
From right to left, the spin–orbit splitting is considered first.
Regardless of which is considered first, the end result is the same
in that there are three valence bands that are sufficiently close to
one another for band mixing to be nonnegligible.
wave functions are formed of the atomic s orbitals, which transform the G point
congruent with the G7 representation of the space group C 46v , The upper valence band
states are constructed out of appropriate linear combinations of products of p3-like
(px-, py-, and pz-like) orbitals with spin functions.
Under the influence of the crystal field and spin–orbit interactions, the hallmark of
the wurtzite structure, the sixfold degenerate G15 level associated with the cubic
system, splits into Gv9 , upper Gv7 and lower Gv7 levels (Figure 2.5). Figure 2.6 shows the
dispersion of the uppermost valence and conduction band structures in Wz GaN and
ZB GaN ((a) near the G band for Wz, (b) inclusive of M, L, and A minima in Wz, and (c)
inclusive of G, L, and X minima in ZB GaN). The influence of the crystal field
splitting, which is present only in the wurtzite structure, transforms the semiconductor from ZB to Wz, which is represented in the section on the left-hand side in
Figure 2.5. The crystal field splits the G15 band of the ZB structure into G5 and G1,
states of the wurtzite structure. These two states are further split into Gv9 , upper Gv7,
and lower Gv7 levels by spin–orbit interactions. Application of the spin–orbit splitting,
from right to left, splits the G15 band of the ZB crystal into G8 and G7 states while the
crystal possesses the zinc blende symmetry. Application of a crystal field further splits
these states into Gv9 , upper Gv7, and lower Gv7 levels, and the crystal now possesses the
wurtzite symmetry.
2.1 Band Structure Calculations
E(k)
c
Γ7
Γ9 :HH
Δ1
Γ7:LH
Γ7:ΧΗ
kz
(a)
Γ
Figure 2.6 (a) Schematic representation of the G
point valence and conduction bands in crystal
with wurtzite symmetry, such as GaN, where
the spin–orbit splitting leads to the bands
labeled as HH and LH. The one caused by
splitting due to crystal field is labeled as CH
[59,60]. (b) Schematic representation of the
band diagram for Wurtzite GaN showing the
separation between the G, A, and M–L band
symmetry points at 300 K. The values with
respect to the top of the valence band are
EG ¼ 3.4 eV, EM–L ¼ 4.5–5.3 eV, EA ¼ 4.7–5.5 eV,
kx , ky
Eso ¼ 0.008 eV, Ecr ¼ 0.04 eV [56]. The values of
EG ¼ 6 eV, EM–L ¼ 7 eV, and EA ¼ 8 eV are given by
Fritsch et al. [55]. (c) Schematic representation of
the band diagram for zinc blende GaN showing
the separation between the G, X, and L band
symmetry points at 300 K. The values with
respect to the top of the valence band are
EG ¼ 3.2 eV, EL ¼ 4.8–5.1 eV, Ex ¼ 4.6 eV, and
Eso ¼ 0.02 eV. Note that in the ZB structure, the
valence band is degenerate [56]. The values of
EG ¼ 3.2 eV, EL ¼ 5.1 eV, EX ¼ 4.3 eV are given by
Fritsch et al. [55].
j149
j 2 Electronic Band Structure and Polarization Effects
150
Energy
A valley
M and L valleys
valley
EA
EM–L
E
Ecr
HH band
LH band
kz
k x,y
Split–off band
(b)
Energy
L valleys
X valley
EL
valley
EX
E
<1 0 0>
Eso
HH band
<111>
LH band
Split–off band
(c)
Figure 2.6 (Continued )
Literature values of the calculated and experimental critical point transition
energies for wurtzitic GaN, AlN, and InN are tabulated in Tables 2.1–2.3.
Literature values of the calculated and experimental critical point transition
energies for zinc blende GaN, AlN, and InN are tabulated in Tables 2.4–2.6.
As shown in Figure 2.6a for wurtzitic GaN, the hole effective masses of the three
uppermost valence bands Gv9 , Gv7 , and Gv7 exhibit large k-dependence. The bands are
labeled as HH (heavy Gv9 ), LH (light, upper Gv7 spin–orbit split) and CH (Gv7 , crystal
field split). The mass of the Gv9 band is heavy in all k-directions, whereas that of
the upper Gv7 is relatively light in the x- and y-planes but heavy in the z-direction.
That of the lower Gv7 is light in the x- and y-planes, but it is heavy along the z-direction
2.1 Band Structure Calculations
Table 2.1 Literature values of calculated and experimental critical
point transition energies for wurtzitic GaN [55].
A (eV)
Parameter
Anisotropic
Isotropic
B (eV)
Mv2 Mc1
Mv4 Mc1
Mv4 Mc3
Gv6 Gc1
Gv3 Gv6
Gv1 Gv6
Gv5 Gv6
Gv5 Gc3
Gv5 Gc6
Hv3 Hc3
K v3 K c2
K v2 K c2
7.67
6.07
7.68
3.47
6.97
0.043
1.00
5.96
10.74
8.06
8.54
8.68
7.67
6.07
7.68
3.47
6.94
0.023
1.00
5.96
10.74
8.07
8.55
8.68
8.26
6.61
7.69
3.50
6.80
0.021
9.0
9.43
10.10
C (eV)
3.0
7.0
0.0
1.0
5.9
11.1
8.3
7.9
D (exp) (eV)
7.05
7.0a
7.05
3.6, 3.44a, 3.50a
7.0a
0.022a
5.3
9.4
7.9a
7.65, 9.0a
The term aniso represents the values derived using a band structure calculation with
anisotropically screened model potentials, whereas the term iso describes a comparative band
structure calculation on the basis of isotropically screened model potentials using an averaged e0
value by taking the spur of the dielectric tensor. A: empirical pseudopotential calculation from
Ref. [55]; B: ab initio pseudopotential calculation within local density approximation from
Ref. [25]; C: LCAO within local density approximation from Ref. [64]; D: unless stated otherwise,
the experimental values are taken from Ref. [65].
a
Experimental values taken from Ref. [66].
(c-direction). Two different definitions are prevalent in the literature. The G6, Gl
pair has been used in Refs [63,71] and the G5, G1 pair in Refs [62,72,73]; for
background information on group theory and symmetries in physics, see Refs [74,75].
We should mention that a carryover habit from the zinc blende nomenclature is
still used for wurtzite symmetry by referring to the crystal field split-off band with
the nomenclature SO as if it is the spin–orbit split-off band, because it happens to
be the farthest from the HH band. In the zinc blende symmetry, the crystal field
splitting is nonexistent, making the top of the valence band degenerate, and the
spin–orbit splitting is large. Portions of this book, unfortunately, participate in the
misuse of this nomenclature. Shown in Figure 2.6b are the most pertinent bands
near the zone center and A, M, and L valleys. The same for zinc blende GaN is shown
in Figure 2.6c.
Without the spin–orbit interaction, the valence band would consist of three
doubly degenerate bands: HH, LH, and CH bands. The spin–orbit interaction
removes this degeneracy and yields six bands. Some band calculations based on
the empirical pseudopotential method (EPM) [66] or the empirical tight binding
method (ETBM) [76] have shown this splitting to be about 10 meV near the G point,
which is comparable to the energy separation of the split-off band in GaN. In early
attempts, the general Hamiltonian in kp theory included the spin–orbit interaction,
j151
j 2 Electronic Band Structure and Polarization Effects
152
Table 2.2 Literature values of calculated and experimental critical
point transition energies for wurtzitic AlN [55].
A (eV)
Parameter
Anisotropic
Isotropic
B (eV)
Mv2 Mc1
Mv4 Mc1
Mv4 Mc3
Gv6 Gc1
Gv3 Gv6
Gv1 Gv6
Gv5 Gv6
Gv5 Gc3
Gv5 Gc6
Hv3 Hc3
K v3 K c2
K v2 K c2
9.56
7.88
8.81
6.11
6.44
0.13
1.04
8.95
12.99
10.10
9.43
9.59
9.54
7.87
8.83
6.11
6.41
0.16
1.03
8.94
12.97
10.91
9.43
9.57
10.0
8.3
8.5
6.0
6.7
0.2
0.9
9.4
14.0
10.5
9.6
9.7
C (exp) (eV)
6.29
8.02
14.00
10.39
The term aniso represents the values derived using a band structure calculation with
anisotropically screened model potentials, whereas the term iso describes a comparative band
structure calculation on the basis of isotropically screened model potentials using an averaged e0
value by taking the spur of the dielectric tensor. A: empirical pseudopotential calculation from
Ref. [55]; B: ab initio pseudopotential calculation within local density approximation from
Ref. [25]; C: experimental values taken from Ref. [67].
but it took a while for it to be applied to the calculations of the band structure of
wurtzite materials such as GaN.
Naturally, the band structures of wurtzitic and zinc blende polytypes are very
distinct due to the differences in underlying symmetries. For the zinc blende case, the
three Luttinger parameters and the spin–orbit splitting provide a minimal description of the valence band structure. Moreover, the energy gap and interband coupling
strength are also required for complete parameterization of both the conduction and
valence bands. The split-off hole mass can be treated as an independent parameter
within the commonly used eight-band kp model. The increase in the electron
effective mass due to interactions with higher conduction bands can be included
via the F parameter (see Section 2.2 for details) [72,77–79]. The set of band parameters
needed to describe the wurtzite lattice must be augmented due to its lower symmetry.
Neglecting the effect of valence band and upper conduction bands on the electron
effective mass allows one to omit the interband matrix element and the F parameter.
Owing to the reduced symmetry, the electron mass can display a rather weak
anisotropy. In contrast, a full description of the valence band in the wurtzite polytype
band structure requires both the spin–orbit splitting, Dso, and the crystal field
splitting, Dcr, along with the seven so-called A parameters. Analogous to the Luttinger
parameters in zinc blende materials, the latter parameterizes the hole masses along
the different directions. Figure 2.6a and b highlights the differences in wurtzite and
zinc blende varieties in terms of their band structure.
2.1 Band Structure Calculations
Table 2.3 Literature values of calculated and experimental critical
point transition energies for wurtzitic InN [55].
A (eV)
Parameter
Anisotropic
Isotropic
B (eV)
d (exp) (eV)
Mv2 Mc1
Mv4 Mc1
Mv4 Mc3
Gv6 Gc1
Gv3 Gv6
Gv1 Gv6
Gv5 Gv6
Gv5 Gc3
Gv5 Gc6
Hv3 Hc3
K v3 K c2
K v2 K c2
7.30
5.94
6.71
2.58
5.63
0.214
0.90
5.22
10.16
7.34
8.13
8.60
7.23
5.88
6.70
2.59
5.50
0.084
0.89
5.18
10.12
7.36
8.12
8.50
6.65
5.05
5.80
2.04
5.77
0.017
1.05
4.65
8.74
6.51
7.38
7.20
7.3, 4.95a
7.3
2.11, 2.0a;b
5.0, 5.5, 4.7a
8.8, 8.9a
5.4a
7.3, 7.2a
The term aniso represents the values derived using a band structure calculation with
anisotropically screened model potentials, whereas the term iso describes a comparative band
structure calculation on the basis of isotropically screened model potentials using an averaged e0
value by taking the spur of the dielectric tensor. A: empirical pseudopotential calculation from
Ref. [55]; B: empirical pseudopotential calculation from Ref. [66].
a
Experimental values taken from Ref. [68].
b
It should be pointed out that the values of bandgap values listed in this table compares with
1.8–2.1 eV values reported during the early stages of InN development and 0.7–0.8 eV reported
later on. For an in-depth discussion of this seemingly controversial bandgap determined
experimentally, the reader is referred to Section 2.9.1.
Table 2.4 High-symmetry point energies in zinc blende GaN in
reference to the top of the valence band for cases where spin–orbit
effects are neglected (included) [55].
Parameter
G15
c
Gc1
Gv15
X c3
X c1
X v5
X v3
Lc3
Lc1
Lv3
Lv2
(Gc7 )
(Gc6 )
(Gv8 )
(X c7 )
(X c6 )
(X v7 )
(X v6 )
(Lc4;5 )
(Lc6 )
(Lv4;5 )
(Lv6 )
A (eV)
B (eV)
C (eV)
10.098
3.308
0.000
6.010
4.428
2.459
6.294
10.416
5.149
0.834
6.812
10.300
3.383
0.000
6.805
4.571
2.693
6.149
9.916
5.636
0.931
6.743
10.248
3.213
0.000
6.265
4.585
2.086
5.923
10.606
5.510
0.772
6.644
A: empirical pseudopotential calculation from Ref. [55]; B: empirical pseudopotential calculation
from Ref. [69]; B: empirical pseudopotential calculation from Ref. [70].
j153
j 2 Electronic Band Structure and Polarization Effects
154
Table 2.5 High-symmetry point energies in zinc blende AlN in
reference to the top of the valence band for cases where spin–orbit
effects are neglected (included) [55].
Parameter
G15
c
Gc1
Gv15
X c3
X c1
X v5
X v3
Lc3
Lc1
Lv3
Lv2
(Gc7 )
(Gc6 )
(Gv8 )
(X c7 )
(X c6 )
(X v7 )
(X v6 )
(Lc4;5 )
(Lc6 )
(Lv4;5 )
(Lv6 )
A (eV)
B (eV)
12.579
5.840
0.000
8.794
5.346
2.315
5.388
12.202
8.264
0.718
6.251
13.406
5.936
0.000
10.661
5.102
2.337
5.262
12.014
9.423
0.728
6.179
A: empirical pseudopotential calculation from Ref. [55]; B: empirical pseudopotential calculation
from Ref. [69].
Table 2.6 High-symmetry point energies in zinc blende InN in
reference to the top of the valence band for cases where spin–orbit
effects are neglected (included) [55].
Parameter
G15
c
Gc1
Gv15
X c3
X c1
X v5
X v3
Lc3
Lc1
Lv3
Lv2
(Gc7 )
(Gc6 )
(Gv8 )
(X c7 )
(X c6 )
(X v7 )
(X v6 )
(Lc4;5 )
(Lc6 )
(Lv4;5 )
(Lv6 )
A (eV)
B (eV)
9.722
2.112
0.000
6.416
5.187
1.555
4.303
10.168
4.733
0.480
4.667
10.193
1.939
0.000
7.392
2.509
1.408
4.400
8.060
5.818
0.456
5.200
A: empirical pseudopotential calculation from Ref. [55]; B: empirical pseudopotential calculation
from Ref. [69].
2.2
General Strain Considerations
Strain–stress relationship or Hookes law can be used to describe the deformation of a
crystal ekl, due to external or internal forces or stresses sij,
X
sij ¼
Cijkl ekl ;
ð2:4Þ
k;l
2.2 General Strain Considerations
where Cijkl is the fourth ranked elastic tensor and represents the elastic stiffness
coefficients in different directions in the crystal, which due to the C6v symmetry can
be reduced to a 6 · 6 matrix using the Voigt notation: xx ! 1, yy ! 2, zz ! 3, yz,
zy ! 4, zx, xz ! 5, xy, yx ! 6. The elements of the elastic tensor can be rewritten as
Cijkl ¼ Cmn with i, j, k, l ¼ x, y, z and m, n ¼ 1, . . . , 6. With this notation, Hookes law
can be reduced to
si ¼
X
Cij ej :
ð2:5Þ
j
or as treated in Ref. [80] for C6v symmetry, we have
3 2
C11
sxx
6 syy 7 6 C12
7 6
6
6 szz 7 6 C13
7 6
6
6 sxy 7 ¼ 6 0
7 6
6
4 syz 5 4 0
0
szx
2
C12
C22
C13
0
0
0
C13
C13
C33
0
0
0
0
0
0
C44
0
0
0
0
0
0
C55
0
32
3
exx
0
7
6
0 7
76 eyy 7
7
6
0 76 ezz 7
7;
7
6
0 7
76 exy 7
0 54 eyz 5
C66
ezx
ð2:6Þ
with C66 ¼ C11 2 C12 . If the crystal is strained in the (0 0 0 1) plane, and allowed to
expand and constrict in the [0 0 0 1] direction, the szz ¼ sxy ¼ syz ¼ szx ¼ 0, sxx 6¼ 0,
and syy 6¼ 0, and the strain tensor has only three nonvanishing terms (with C11 ¼
C22), namely,
sxx ; syy
szz
¼
C11 þ C12
2C13
C 13
C 33
exx ; eyy
;
ezz
ð2:7Þ
with exx ¼ eyy ¼ a a0a0 and ezz ¼ c c0c0 ¼ CC1333 ðexx þ eyy Þ, the latter of which describes the Poisson effect, and a and a0 and c and c0 represent the in-plane and out-ofplane lattice constants of the epitaxial layer and the relaxed buffer (substrate),
respectively. The above assumes that the in-plane strain in x- and y-directions is
identical, namely, exx ¼ eyy. When the crystal is uniaxially strained in the (0 0 0 1)
c-plane and free to expand and constrict in all other directions, szz is the only
nonvanishing stress term, and the strain tensor is reduced to
1
eyy
C 12 C33 C213
¼ 2
exx :
ð2:8Þ
ezz
C13 C11 C33 C 11 C13 C12 C13
If the growth is performed on the ð1 1 0 0Þm-plane, meaning the growth plane is
the xz-plane with the growth direction being along the y-axis, the in-plane strain
anisotropy dictates that exx 6¼ eyy . The out-of-plane stress, syy ¼ 0, which when
utilized in the stress–strain relationship of Equation 2.6 leads to
eyy ¼ C12 exx þ C13 ezz
:
C11
ð2:9Þ
If the growth is performed on the ð1 1 2 0Þa-plane, meaning the growth plane is the
yz-plane with growth direction being along the x-axis, the in-plane strain anisotropy
j155
j 2 Electronic Band Structure and Polarization Effects
156
dictates that exx 6¼ eyy. The out-of-plane stress, sxx ¼ 0, which when utilized in the
stress–strain relationship of Equation 2.6 leads to
exx ¼ C12 eyy þ C13 ezz
:
C 11
ð2:10Þ
The values of the elastic stiffness coefficients for GaN have been measured
by Sheleg and Savastenko [81] and reproduced in Ref. [59]. These data are listed
in Table 1.24.
The inversion of the 6 · 6 elastic constant matrix in Equation 2.6 leads to the elastic
compliance constants as follows:
S11 ¼
C11 C 33 C213
;
ðC 11 C12 Þ½C33 ðC 11 þ C12 Þ 2C213 S12 ¼
C12 C 33 C213
;
ðC 11 C12 Þ½C33 ðC 11 þ C12 Þ 2C213 S13 ¼
C13
;
½C33 ðC11 þ C 12 Þ 2C 213 S33 ¼
C11 þ C12
;
½C33 ðC11 þ C 12 Þ 2C 213 S44 ¼
1
:
C44
ð2:11Þ
Through the use of the aforementioned compliance constants, a very useful figure of
merit can be determined, which in turn would lead to the directional hardness and
the reciprocal Youngs modulus as a function of orientation to the crystal axis. For
0
hexagonal symmetry, the reciprocal Youngs modulus S11 along an arbitrary direction
at an angle y with respect to the [0 0 0 1] axis is given by [82–84]
0
S11 ¼ S11 sin4 q þ S33 cos4 q þ ðS44 þ 2S13 Þsin2 qcos2 q:
ð2:12Þ
0
In Figure 2.7a and b, polar plots of S 11 as a function of the direction in reference to the
[0 0 0 1] axis and for directions along the basal plane are shown for InN, GaN, and AlN
binaries. Clearly, AlN and GaN are harder than InN by more than a factor of 2. The
hardness of AlN is almost isotropic, whereas that for GaN and InN exhibit some
preferential softness along the [0 0 0 1] and ½2 1
1 0 axes. Of paramount interest
here is that the hardness of all the wurtzitic binaries is isotropic in the basal plane.
This takes on a special meaning as the strain, with growth along the c-axis, caused in
epitaxial heterostructures by mismatch lattice and thermal mismatch is along the
basal plane. Lack of any force in the growth direction and the fact that the crystal can
relax freely in this direction leads to a biaxial strain e1 ¼ e2, which in turn causes
stresses
s1 ¼ s2
with s3 ¼ 0:
The internal strain is defined by the variation of the internal parameter under
strain, (u u0)/u0. In the limit of small deviations from the equilibrium, Hookes law
2.2 General Strain Considerations
[1 0 1 0]
8
S 11 [10 –12 m 2 /N–1]
(a)
6
In N
4
GaN
2
AlN
–8
–6
–4
–2
0
0
[1 2 1 0]
2
4
6
8
–2
–4
–6
- –8
[0 0 0 1]
8
S 11 [10 –12 m 2 / N–1]
(b)
6
InN
4
GaN
2
AlN
–8
–6
–4
–2
[2 1 1 0]
q
0
0
2
4
6
8
–2
–4
–6
- –8
Figure 2.7 (a) The reciprocal Youngs moduli,
S0 11, in the basal plane of InN, GaN, and AlN
indicating AlN and GaN are harder than InN to be
harder by more than a factor of 2. Of paramount
importance, the hardness of the wurtzitic crystals
is isotropic in the basal plane. (b) The reciprocal
Youngs modulus, S0 11, along an arbitrary
direction making an angle y with respect to the
c-axis in the [0 0 0 1] direction. In basal plane the
stiffness of AlN is isotropic whereas GaN and
InN show preferential softness along the
[0 0 0 1] and [2 1 1 0] directions [84].
gives the corresponding diagonal stress tensor s with the elements [85]
sxx ¼ syy ¼ ðC11 þ C12 Þexx þ C13 ezz ;
szz ¼ 2C13 exx þ C33 ezz :
ð2:13Þ
In Equation 2.13, four of the five independent stiffness constants Cij of the wurtzite
crystal are involved. The modifications of Equation 2.13 by the built-in electric field due
to the spontaneous and piezoelectric polarization are neglected, as the effect is small.
j157
j 2 Electronic Band Structure and Polarization Effects
158
In the case of uniaxial stress, for example, along the c-direction, there is an
elastic relaxation of the lattice in the c-plane. The ratio of the resulting in-plane
strain to the deformation along the stress direction is expressed by the Poissons
ratio, which in general can be anisotropic. For the wurtzite lattice subjected to a
uniaxial stress szz parallel to the c-axis, sxx ¼ syy ¼ 0 holds. Then Equation 2.13
gives the relation
exx ¼ ½C13 =ðC11 þ C12 Þezz ¼ nezz ;
ð2:14Þ
with n ¼ C13 =ðC11 þ C12 Þ being the Poissons ratio.
The uniaxial stress correlates with strain along the direction of the stress by the
Youngs modulus E as szz ¼ Eezz and thus
E ¼ C33 2C 213
:
C11 þ C 12
ð2:15Þ
A homogeneous biaxial stress in the basal plane is described by a constant force in
the plane with sxx ¼ syy and vanishing force in the c-direction szz ¼ 0. The Hookes
law of Equation 2.13 leads to a relationship between axial and basal plane strain
components as ezz ¼ RBexx, which reproduces Equation 2.7 with the biaxial
relaxation coefficient being
RB ¼
2C13
:
C33
ð2:16Þ
The biaxial relaxation coefficient is also referred to as simplylater in this chapter in
the polarization section. The in-plane stress–strain relationship using the biaxial
modulus is sxx ¼ Yexx, which leads to
Y ¼ C11 þ C12 2C 213
:
C33
ð2:17Þ
The strain–stress relationship along the c-axis is
sxx ¼ ðY=RB Þezz :
ð2:18Þ
We can then relate the Young modules E (is also commonly described by nomenclature as Yo) to biaxial modulus Y as
E¼
C33 Y
2n
or E ¼ B Y:
C11 þ C12
R
ð2:19Þ
Equation 2.13 can now be expressed as
sxx ¼ E
ezz :
2n
ð2:20Þ
In the case of hydrostatic pressure
sxx ¼ syy ¼ szz ;
ð2:21Þ
and from the Hookes law
ezz ¼ RH exx ;
ð2:22Þ
2.3 Effect of Strain on the Band Structure of GaN
with RH expressed as
RH ¼
C11 þ C12 2C13
:
C33 C 13
To calculate elastic stiffness constants, Wagner and Bechstedt [85] considered
C11 þ C12 as an independent quantity and made use of the relation between the
elastic constants and isothermal bulk modulus, B0 (nomenclature Bs is also used):
B0 ¼
ðC11 þ C12 ÞC13 2C213
:
C11 þ C12 þ 2C 33 4C13
ð2:23Þ
Equation 2.22 can be obtained from Equation 2.13 with the aid of linearized relation
DP/B0 ¼ DV/V0 ¼ 2(e11 þ e33) or Dp/B0 ¼ DV/V0 ¼ 2(e11 þ e33), where DV is
the variation of volume with pressure and V0 is the static volume. The values of the
bulk modulus B0 have been obtained by fitting the Vinet equation of state to the
calculated dependence of energy on volume [86,87], using
1
Y ¼ 2 þ 1=2
4 RB B 0 ;
ð2:24Þ
v
and
C13 ¼ Y=½ð1=vÞ RB :
ð2:25Þ
Wagner and Bechstedt [85] obtained the absolute values of the elastic stiffness
coefficients. The strain- and stress-related issues represent the cornerstone of the
discussion on piezoelectric polarization, and the above discussed section provides
sufficient material to embark on the discussion of polarization issues in nitride
semiconductor heterostructures.
2.3
Effect of Strain on the Band Structure of GaN
The strain in conventional group III–V semiconductors has been a much desired
feature for its beneficial effects [88]. In the world of GaN, however, it is not necessarily a
desirable commodity but could be construed as a nemesis brought upon by the lack of
lattice- and thermal-matched substrates and uncomfortably large lattice and thermal
mismatch with its ternaries. It is therefore imperative that strain effects be considered.
Figure 2.8 exhibits the valence band structure of GaN in the x- and y-planes under
biaxial compressive strain and uniaxial strain in the c-plane with the direction of strain
as in Figure 2.8c. There are no major changes in the HH, LH, and CH bands, other
than crystal splitting becoming larger, with the hole effective mass remaining heavy,
the density of states staying high, and the crystal symmetry remaining the same, C46v .
In contrast, the uniaxial strain in the c-plane causes an anisotropic energy splitting in
the x- and y-planes, which leads to a symmetry lowering from C 46v to C2v . When a
compressive uniaxial strain is induced along the y-direction, the HH band in the
x-direction and the LH band in the y-direction move to higher energies. This causes a
j159
j 2 Electronic Band Structure and Polarization Effects
160
(a)
(b)
E
E
(c)
Biaxial
eff
so
eff
cr
Uniaxial
z
kx
ky
kx
ky
y
x
Figure 2.8 The valence band structure of GaN under (a) biaxial
strain in the c-plane, (b) uniaxial strain in the c-plane, and
(c) schematic of the particulars of the strain [59,89].
reduction in the density of states. A tensile uniaxial strain along the x-direction has the
same effect. On the contrary, when a tensile uniaxial strain is induced along the ydirection, the HH band in the x-direction and the LH bands along the y-direction move
to lower energies. This causes a reduction in the density of states. A compressive strain
along the x-direction has the same effect [89].
2.4
kp Theory and the Quasi-Cubic Model
The conduction and valence bands of nitride semiconductors are comprised of s- and
p-like states, respectively. Unlike the conventional ZB III-N semiconductors and the
lack of a high degree of symmetry, the crystal field present removes the degeneracy at
the top of the conduction band. Moreover, unlike the ZB case, the spin–orbit splitting
is very small and makes all three bands in the valence band closely situated in energy.
Consequently, the three valence bands and the conduction band must be considered
in unison and this makes the use of an 8 · 8 kp Hamiltonian imperative. Because the
bandgaps of nitrides are very large, the coupling between the conduction and valence
bands can be treated as a second-order perturbation, which allows the 8 · 8
Hamiltonian to be split into one 6 · 6 Hamiltonian dealing with the valence band
and another 2 · 2 dealing with the conduction band [59]. As indicated above, the
conduction band is made of s-like states, which means that it can be treated as
parabolic with the dispersion relation
EðkÞ ¼ E c0 þ
2 k2z
h
==
2mc
þ
2 ðk2x þ k2y Þ
h
2m?
c
==
þ a?
c ðexx þ eyy Þ þ ac ðezz Þ;
ð2:26Þ
==
where a?
c and ac represent the in-plane and out-of-plane deformation potentials,
respectively.
For an isotropic parabolic conduction band, Equation 2.26 reduces to
EðkÞ ¼ E c0 þ
2 k2
h
þ ac e:
2mc
ð2:27Þ
2.4 kp Theory and the Quasi-Cubic Model
Ec0 is the conduction band energy at the k ¼ 0 point, e is the strain, and ac is the
deformation potential for the conduction band. The other terms have their usual
meanings. It should be pointed out that we are dealing with a linear system.
Using the basis jY 11 ">; jY 11 #>; jY 10 ">; jY 10 #>; jY 11 ">; jY 11 #>, the 6 6 Hamiltonians can be expressed as
1
0
0
K 0
F
0
H
B 0
G
D
H 0 K C
C
B
B H
D
l
0
I 0 C
C;
B
ð2:28Þ
B 0
H
0
l
D I C
B
C
@ K
0
I
D
G 0 A
0
K
0
I
0 F
where F, G, l, D, H, I, and K are defined as (two forms are given by Ren et al. [90] and
Albrecht et al. [91])
F ¼ D1 þ D2 þ l þ q;
G ¼ D1 D2 þ l þ q;
l ¼ A1 k2z þ A2 ðk2x þ k2y Þ þ D1 ezz þ D2 ðexx þ eyy Þ;
q ¼ A3 k2z þ A4 ðk2x þ k2y Þ þ D3 ezz þ D4 ðexx þ eyy Þ;
H ¼ iA6 kz ðk2x þ k2y Þ1=2 A7 ðk2x þ k2y Þ1=2 ;
H ¼ ðiA6 kz A7 Þðkx þ iky Þ þ iD6 ðexz þ ieyz Þ;
I ¼ iA6 kz ðk2x þ k2y Þ1=2 þ A7 ðk2x þ k2y Þ1=2 ;
I ¼ ðiA6 kz þ A7 Þðkx þ iky Þ þ iD6 ðexz þ ieyz Þ;
K ¼ A5 ðk2x þ k2y Þ;
K ¼p
A5ffiffiffiðkx þ iky Þ2 þ D5 ðexx eyy þ i2exy Þ;
D ¼ 2D3 ;
½91
½92
½91
½92
½91
½92
ð2:29Þ
where Ai is the valence band parameter corresponding to the Luttinger parameters in
the ZB system, D1 and D2,3 are the crystal field and spin–orbit splitting energies
(D2, ¼ D3 ¼ Dso), Di parameters represent the deformation potentials for the valence
band, and exx, eyy, and ezz are the strain tensors (also referred to as e11, e22, and e33 in
this book and many other publications). Both forms used by Albrecht et al. [91] and
Ren et al. [90] for H, I, and K parameters are given. The former uses the basis and
Hamiltonian in the (kx þ iky) form and contains all of the shear strain information.
The latter does the phase rotation to get two 3 · 3 and the relevant matrix elements are
then functions only of k-transverse and all of the directional phase information has
been compiled onto the basis states by a unitary transformation. For presentation of
the pseudomorphic strain, either form would be just as easy to use. Another
important point is that in the case where the shear strain is nonzero, such as when
uniaxial in-plane strain, the D5 and D6 terms must be included as represented by the
Albrecht et al. [91] notation above. If only biaxial strain is considered, the shear terms
of the strain tensor are zero and thus the D5 and D6 terms vanish. In the latter case,
the representation by Ren et al. [90] and others similar to it hold. A good description of
pertinent issues is discussed in Ref. [62].
Similar to the cubic system, the 6 · 6 matrix can be block diagonalized into two 3 · 3
matrices, and this can considerably simplify the band structure calculation. It should
j161
j 2 Electronic Band Structure and Polarization Effects
162
be mentioned that A7 can be assumed nearly zero due to symmetry considerations.
Chuang and Chang [58] derived the two 3 · 3 Hamiltonians when A7 is neglected and
obtained three doubly degenerate bands. Ren et al. [90] investigated the effect of A7
parameter on the valence band dispersion in wurtzitic crystals such as GaN. In fact,
Ren et al. [90] argued that theories forwarded by Chuang and Chang [58] replicates
that of Bir and Pikus [72] reported decades earlier. Choosing A7 ¼ 0 reduces the
results to that of Chuang and Chang [58] and Sirenko et al. [62].
To underscore the effect of A7 parameter, Ren et al. [90] compiled data from
empirical pseudopotential method (EPM) [67]. The same method was also applied to
wurtzitic and zinc blende phases of all the three binaries of nitrides by Fritsch
et al. [55]. Ren et al. [90] calculated the band structure for GaN for values of
A7 ¼ 93.7 meV Å and 0 as, shown in Figure 2.9. Clearly, inclusion of the A7 parameter
results in a much better fitting between the kp theory and EPM calculations. From the
(a)
0.00
Wz GaN
–0.01
HH
Energy (eV)
–0.02
LH
–0.03
–0.04
CH
–0.05
0.0
0.050
0.100
0.15
Wave vector, kx,( k// ) (1 Å–1)
Figure 2.9 (a) Valence band structures of
wurtzite GaN with the kp theory fitting including
the spin–orbit interaction with A7 ¼ 93.7 meVÅ
in the solid line. The dash-dotted line is the
result of fitting with A7 ¼ 0. The empirical
pseudopotential method (EPM) calculation data
(o) are from Ref. [67] ([90]). (b) Valence band
structure of wurtzite GaN, using the parameters
recommended by Ren et al. Courtesy of I.
Vurgaftman and J. Meyer and Ref. [90]. (c) The
same using the parameters recommended in
Ref. [152]. The dashed lines represent the case
for A7 ¼ 0. Courtesy of I. Vurgaftman and J. Meyer
and Ref. [90].
2.4 kp Theory and the Quasi-Cubic Model
(b)
0.00
Wz GaN
HH
–0.01
Energy(eV)
LH
–0.02
–0.03
–0.04
CH
–0.05
–0.06
–0.15
–0.10
k z , (k )
–0.050
0.0
0.050
0.100
0.15
-1
Wave vector (1 Å ) kx, (k//)
(c)
0.00
Wz GaN (c)
HH
–0.01
LH
Energy(eV)
–0.02
–0.03
–0.04
CH
–0.05
–0.06
–0.10
–0.15
k z , (k )
Figure 2.9 (Continued )
–0.050
0.0
0.050
Wave vector (1 Å-1)
kx, (k//)
0.100
0.15
j163
j 2 Electronic Band Structure and Polarization Effects
164
Table 2.7 Fitted splitting energies and Luttinger-like parameters
for the valence band of the wurtzite GaN: Di parameters are in unit
of meV, and the Rydberg terms are in units of h2 =2m0 except for
A7, which is in unit of meV Å.
D1
D2
D3
A1
A2
A3
A4
A5
A6
A7
21.1
3.61
3.61
7.21
0.440
6.68
3.46
3.40
4.9
93.7
quality of the fit, one can argue that the 93.7 value for the Luttinger-like parameter A7
is a good one. The other parameters giving this fit are listed in Table 2.7. In addition to
the cycled parametric values of A parameters by kp theorists, a good extraction of
A1–A7 parameters from empirical pseudopotential method band structures of AlN,
GaN, and InN can be found in Ref. [55]. Vurgaftman and Meyer [152] followed a nearly
identical path to that of Ren et al. [90] but calculated the band structure in both kx and ky
crystal directions, as shown in Figure 2.9. The dashed lines, as in the case of the paper
by Ren et al. [90], correspond to the case representing the case of A7 ¼ 0. In addition,
Vurgaftman and Meyer [152] and Vurgaftman et al. [92] mentioned the calculation of
the valence band structure for GaN using parameters representing the properties of
GaN that they prefer, the results of which are also shown in Figure 2.9 for comparison.
The effective masses are calculated using the parallel and perpendicular hole
masses m//and m? that can be expressed in terms of their dependence on the
Luttinger-like parameters Ai as follows:
==
m0 =mhh ¼ ðA1 þ A3 Þ;
==
m0 =mlh ¼ ðA1 þ A3 Þ;
==
m0 =mso
ð2:30Þ
¼ A1 :
?
Here, m represents the mass in the (kx, ky) plane, which means kz ¼ 0 and
m0 =m?
hh ¼ ðA2 þ A4 A5 Þ;
2
m0 =m?
lh ¼ ðA2 þ A4 A5 Þ 2A7 =jD1 j ;
m0 =mh?
so
¼
ð2:31Þ
A2 þ 2A27 =jD1 j;
where m//is along the kz-direction (kx ¼ ky ¼ 0) and m? in the (kx, ky) plane, which
means kz ¼ 0.
The effective masses are calculated using the parallel and perpendicular hole
masses m//and m? together with Luttinger-like parameters using Equations 2.30
and 2.31.
As indicated in the schematic of Figure 2.5, both the spin–orbit and the crystal field
splitting affect the structure of the valence band in wurtzitic crystals [93]. Typically, the
relevant parameters are correlated to one another as Dso ¼ 3D2 ¼ 3D3, in spite of the
fact that a small D2/D3 anisotropy has sometimes been reported [94,95] and Dcr ¼ D1.
Experimentally, the splitting parameters are obtained from the energy differences of
the A, B, and C free excitons, which have nonlinear dependencies on the various
splittings [96]. It should be pointed out that the nomenclature for the three valence
2.4 kp Theory and the Quasi-Cubic Model
bands for hexagonal system is A, B, and C for HH, LH, and SO (CH) bands when
including A7 terms, because spin splitting and strain can significantly alter as to which
band of eigenstates is heavy or light at various k-values, particularly in the c-plane.
An early experimental undertaking by Dingle et al. [97] led to Dcr ¼ 22 meV and
Dso ¼ 11 meV. An analysis by Gil et al. [98] yielded Dcr ¼ 10 meV and Dso ¼ 18 meV.
Chuang and Chang [58] attempting to rederive these parameter from the same data
but with what was termed as a more precise description of the effect of strain on the
valence band edge energies arrived at values of Dcr ¼ 16 meV and Dso ¼ 12 meV.
Reynolds et al. [99] obtained Dcr ¼ 25 meV and Dso ¼ 17 meV from a fit to exciton
energies, with A and B determined by photoluminescence (PL) and C determined by
reflection but with a geometry not fully ideal in terms in that some error is introduced
in the value of C exciton energy. Again, using exciton energies values of Dcr ¼ 22 meV
and Dso ¼ 15 meV were obtained by Shikanai et al. [100], Dcr ¼ 37.5 meV and
Dso ¼ 12 meV by Chen et al. [101], Dcr ¼ 9 meV and Dso ¼ 20 meV by Korona
et al. [102], and Dcr ¼ 9–13 meV and Dso ¼ 17–18 meV by Campo et al. [103] and
Julier et al. [104]. The values of Dcr ¼ 10 meV and Dso ¼ 17 meV were determined by
both Edwards et al. [105] and Yamaguchi et al. [96]. Noticeable is one of the smallest
reported crystal field splittings to date, Dcr ¼ 9 meV, along with Dso ffi 18 meV
reported by Rodina et al. [106] based on detailed experimental investigation. On the
theoretical side, an ab initio calculation by Wei and Zunger [107] overestimates the
crystal field splitting Dcr ¼ 42 meV, but arrives at a Dso ¼ 13 meV that agrees well with
the experimental data. Suzuki et al. [56] reported Dcr ¼ 40 meV and Dso ¼ 8 meV or
Dso ¼ 3D2 ¼ 3D3 1.16 mRy and Dcr ¼ D1 ¼ 5.36 mRy for these splittings. Many firstprinciples calculations focusing on the valence band splitting are available in the
literature [58,90,67,154,108]. The experimental data, however, appear to converge on
the splittings Dcr ffi 10 meV and Dso ffi 17 meV, as suggested by Vurgaftman and
Meyer and tabulated in Table 2.8 [152].
The spin splitting of the valence band of wurtzitic GaN can be determined via the A7
parameter, as shown in Figure 2.9a, which is derived by Vurgaftman and Meyer [152]
assuming the parameters of Ren et al. [90]. On the contrary, Figure 2.9b is derived by
Vurgaftman and Meyer [152] using A parameters from Ren et al. [90] combined with
what is believed to be the more representative spin–orbit and crystal field splittings.
Modification of the A parameters alone, but with the corrected values of the splitting
energies, does not allow the recovery of the band structure resembling that shown in
Figure 2.9a. This simply implies that the field is not yet settled on a set of reliable
parameters and more refinement is needed for the most appropriate values of the A
parameters to be arrived.
To be sure six distinct valence band deformation potentials, as well as the strain
tensor and the overall hydrostatic deformation potential, are necessary to describe the
band structure of GaN under strain. In the cubic approximation, these can be
expressed in terms of the more familiar av, b, and d potentials [93]. In terms of the
calculation and combined calculation and measurement efforts, Christensen and
Gorczyca [31] reported a hydrostatic deformation potential a ¼ 7.8 eV, which has
been shown to agree well with 8.16 eV obtained by Gil et al. [98]. On the contrary, a
somewhat lower value of a ¼ 6.9 eV was attained through an ab initio calculation by
j165
j 2 Electronic Band Structure and Polarization Effects
166
Table 2.8 Recommended band structure parameters for wurtzitic GaN from Ref. [152].
Parameter
Value
Parameter
Value
Parameter
Value
Eg (eV, low
temperature)
a (meV K1)
b (K)
3.510
A1
7.21
D1 (eV)
3.7
0.909 (1 in [119])
830 (1100 in
[119])
10
17
0.20
0.20
4.9
11.3
A2
A3
0.44
6.68
D2 (eV)
D3 (eV)
A4
A5
A6
A7 (meV Å)
d13 (pm V1)
d33 (pm V1)
d15 (pm V1)
Psp (C m2)
3.46
3.40
4.90
93.7
1.6b
3.1b
3.1b
0.034
D4 (eV)
D5 (eV)
D6 (eV)
c11 (GPa)
c12 (GPa)
c13 (GPa)
c33 (GPa)
c44 (GPa)
Dcr (meV)
Dso (meV)
==
me =m0
m?
e =m0
a1 (eV)
a2 (eV)
4.5
8.2
4.1
4.0
5.5
390
145
106
398
105
367a
135a
103a
405a
95a
See Tables 2.27 and 2.28 for details related to the elastic constants, piezoelectric constants, and
spontaneous polarization charge. Any dispersion among the tables is a reflection of the
uncertainty in the available parameters. See Volume 2, Chapter 5 for an extended discussion of
Varshni parameters.
a
The second column figures for the Cii parameters are from Table 2.28 where a more expanded
list of elastic coefficients is given.
b
Table 2.28 provides additional data on d-parameters.
Kim et al. [109]. Noting that the hydrostatic potential is anisotropic, owing to the
reduced symmetry of the wurtzite crystal, Wagner and Bechstedt [85] calculated values
of 4.09 and 8.87 eV for the two hydrostatic interband deformation potentials. On
the transport side, fits to the experimental mobility data [110,111] yielded a conduction
band deformation potential approaching 9 eV, a topic discussed in some detail in
Volume 2, Chapter 3. Using pressure-dependent optical transition energies with
pressure, Shan et al. [112] reported a1 ffi 6.5 eV and a2 ffi 11.8 eV and uniaxial
deformation potentials b1 ffi 5.3 eV and b2 ffi 2.7 eV for the two hydrostatic interband
components. Employing photoreflectance measurements on compressively strained
M-plane GaN films (grown along the h1 0 1 0i direction) grown on g-LiAlO2 (1 0 0),
Ghosh et al. [113] reported a1 ¼ 3.1 eV with a2 ¼ 11.2 eV. Again, using M-planeoriented GaN layers, Gil and Alemu [114] obtained a1 ¼ 5.22 eV with a2 ¼ 10.8 eV.
Vurgaftman and Meyer [152] recommend a set of a1 ¼ 4.9 eV and a2 ¼ 11.3 eV,
which represents an average of all the measured values.
Numerous sets of valence band deformation potentials have been derived from
both first-principles calculations [58,60,61,115,116] and fits to experimental
data [94,98,100,96,112–114,117]. The dispersion among the reported data is unacceptable, which calls for further work to resolve the discrepancies and converge on
accurate parameters. If one were to average the deformation potentials that are
most widely quoted, values of D1 ¼ 3.7 eV, D2 ¼ 4.5 eV, D3 ¼ 8.2 eV, D4 ¼ 4.1 eV,
D5 ¼ 4.0 eV, and D6 ¼ 5.5 eV, which satisfy the quasi-cubic approximation that is
about to be discussed [58], are obtained. The deformation potential values for GaN are
2.5 Quasi-Cubic Approximation
tabulated in Table 2.8, and their effect on optical transitions are discussed in detail in
Volume 2, Chapter 5.
2.5
Quasi-Cubic Approximation
The genesis of the quasi-cubic approximation relies on the fact that the Wz and ZB
structures are both tetrahedrally coordinated and hence are closely related.
The nearest neighbor coordination is the same for Wz and ZB structures but differs
at the next nearest neighbor positions. The basal plane (0 0 0 1) of the Wz structure
corresponds to one of the (1 1 1) planes of the ZB. When the in-plane hexagons
are lined up in Wz and ZB structures, the Wz [0 0 0 1], ½1 1 2 0, and ½1 1 0 0 planes are
parallel to the ZB [1 1 1], ½1 0 1, and ½1 2 1 planes, respectively. This, in turn, leads to
correlations between the symmetry direction and the k-points for the two polytypes.
There are, however, twice as many atoms in the Wz unit cell as there are in the ZB one.
In addition to the band structure similarities between the doubled ZB and Wz
structures, one can establish a correlation between the Luttinger parameters in the
ZB system and parameters of interest in the Wz system by taking the z-axis along the
[1 1 1] direction and the x- and y-axes along the ½1 1 2 and ½
1 1 0 directions. For details
regarding the symmetry relations between the ZB and Wz polytypes, refer to
Refs [32,118]. Doing so leads to
D 2 ¼ D3 ;
A1 ¼ A2 þ 2A4 ;pffiffiffi
A3 ¼ 2A4 ¼ 2A6 4A5 ;
A7 ¼ 0;
D1 ¼ D2 þ 2D4 ;pffiffiffi
D3 ¼ 2D4 ¼ 2D6 4D5 :
ð2:32Þ
The A parameters can be related to the classical Luttinger parameters gi through
A1
A2
A3
A4
A5
A6
¼ ðg 1 þ 4g 3 Þ;
¼ ðg 1 2g 3 Þ;
¼ 6g 3 ;
¼ 2g 3 ;
¼ p
ðg ffiffi2ffi þ 2g 3 Þ;
¼ 2ð2g 2 þ g 3 Þ:
ð2:33Þ
The calculated values of the spin–orbit and crystal field splitting parameters with
those deduced from the observation of A, B, and C excitons are listed in Tables 2.8
and 2.9, which will be presented shortly. The calculations agree well in terms of the
spin–orbit splitting, but the theoretical crystal field splitting is much too large
compared to experimental data. The discrepancy may be due to the unaccounted
residual strain and strain inhomogeneities present in GaN films, as well as the
inaccuracy of the parameter values. The debate will probably continue until strainfree or homogeneously strained films can be prepared.
j167
j 2 Electronic Band Structure and Polarization Effects
168
Table 2.9 Effective masses and band parameters for wurtzitic GaN.
Parameter Aniso
==
me
m?
e
==
mhh
==
mlh
==
mch
m?
hh
m?
lh
m?
ch
A1
A2
A3
A4
A5
A6
A7
D1
0.138
0.151
2.000
2.000
0.130
2.255
0.191
0.567
7.692
0.575
7.192
2.855
2.986
3.360
0.160
0.043
Iso
A
B
0.138
0.151
2.007
2.007
0.130
2.249
0.261
0.317
7.698
0.600
7.200
2.816
2.971
3.312
0.171
0.023
0.20
0.18
1.10
1.10
0.15
1.65
0.15
1.10
6.56
0.91
5.65
2.83
3.13
4.86
0.20
0.18
1.76
1.76
0.16
1.61
0.14
1.04
6.27
0.96
5.70
2.84
3.18
a
0.039
C
0.14
0.15
1.479
1.479
0.130
1.592
0.299
0.252
7.706
0.597
7.030
3.076
3.045
4.000
a
0.194
0.038 0.022
D
E
0.14
0.15
1.453
1.453
0.125
1.595
0.236
0.289
7.979
0.581
7.291
3.289
3.243
4.281
0.179
0.022
0.19
0.17
1.76
1.76
0.14
1.69
0.14
1.76
7.14
0.57
6.57
3.30
3.28
F
0.19
0.17
1.96
1.96
0.14
1.87
0.14
1.96
7.24
0.51
6.73
3.36
3.35
4.72
0
0
0.021 0.021
G
H
I
0.19
0.19
0.23
0.19
1.89
2.00
1.96
1.89
2.00
1.96
0.12
0.16
0.16
2.00
2.04
1.20
0.15
0.18
0.16
0.59
1.49
1.96
7.21 6.4
6.36
0.44 0.50 0.51
6.68
5.9
5.85
3.46 2.55 2.92
3.40
2.56
2.60
4.9
3.06
3.21
0.094 0.108 0
0.021 0.036
Effective masses in units of free-electron mass m0, Luttinger-like parameters Ai (i ¼ 1, . . ., 6) in units of
h2 =2m0 , and A7 in units of eV Å . The crystal field splitting energy D1 is given in units of meV. The term
aniso represents the values derived using a band structure calculation with anisotropically screened
model potentials, whereas the term iso describes a comparative band structure calculation on the basis
of isotropically screened model potentials using an averaged e0 value by taking the spur of the dielectric
==
tensor. Here, me and m?
e represent the effective electron masses along and perpendicular to the
c-axis [55]. Anisotropically screened and isotropically screened values are from Ref. [55]. A: FP-LAPW
band structure calculations are from Ref. [56], and effective mass parameters are obtained through a
3D fitting procedure within cubic approximation; B: FP-LAPW band structure calculations are from
Ref. [56], and effective mass parameters are obtained by direct line fit; C: Ai from Ref. [153] obtained
through a Monte Carlo fitting procedure to the band structure and effective masses calculated using
Equations 2.30 and 2.31; D: direct kp calculations for Ai from Ref. [153] and effective masses calculated
using Equations 2.30 and 2.31; E: effective masses and Ai from Ref. [67] obtained through a line fit to
the band structure; F: direct kp calculation in a 3D fit from Ref. [67]; G: Ai obtained through a direct fit
from Ref. [90] and effective masses calculated using Equations 2.30 and 2.31; H: direct fit of Ai to firstprinciples band structure calculations from Ref. [154]; I: Ai and effective masses obtained in the quasicubic model from zinc blende parameters from Ref. [154].
a
A7 in the range of 0.136 eV Å has been set to zero.
The parameters mentioned in bandgap-related discussion for wurtzitic GaN are
tabulated in Table 2.8 [152] for the wurtzitic phase GaN.
All conventional nitridesin the wurtzite phase exhibit a direct energy gap, and the next
satellite conduction valley, which is the M valley, is some 2 [31]to 5 eV [56]higher than the
G valley. In addition to the one made available here, the wurtzite indirect gap related
issues have been amply discussed in the literature, some of which can be found in
Refs [31,53,75,120–122]. Sufficing it to state the bottom of the conduction band in GaN
can be well approximated by a parabolic dispersion relation, although a slight anisotropy
is expected due to the reduced lattice symmetry [56]. In many devices, the pertinent
property of the band structure is the region near the bottom of the conduction band,
which can be represented to a great deal with effective mass. In relatively early
2.6 Temperature Dependence of Wurtzite GaN Bandgap
experimental studies in GaN grown by hydride VPE, Barker and Ilegems [123] obtained
an electron effective mass of mn ¼ 0.20m0 from reflectivity measurements. Again early
on, Rheinlander and Neumann [124] inferred 0.24–0.29m0 for the effective mass from a
Faraday-rotation investigation of heavily n-doped GaN. Using heavily doped samples,
which was the norm then, and fits to the thermoelectric power, Sidorov et al. [125]
obtainedelectroneffective masses of0.1–0.28m0, dependingon whatprimary scattering
channel was assumed. For a review of early investigations of these and other properties,
thereaderisreferredtoreviewsfromthe1970s,suchastheonebyPankoveetal.[126]and
that by Kesamanly [127]. Congruent with the increased activity in GaN, fuelled by the
device demonstrations, particularly LEDs and later on lasers, a substantial body of work
has since produced more precise estimations of the electron mass. Among them are the
works by Meyer et al. [128] and Witowski et al. [129] who obtained masses of 0.236m0 and
0.222m0, respectively, utilizing shallow donor transition energies; the latter is with the
smallest error bars quoted in the literature (0.2%). Underscoring the importance of the
polaron correction, which is about 8% in GaN and comes about because of the strong
polar nature of GaN, Drechsler et al. [130] derived a bare mass of 0.20m0 from cyclotron
resonance data. A similar result was obtained by Perlin et al. [131] using infrared
reflectivityandHalleffectmeasurements,whichalsoledtoananisotropyoflessthan1%.
For comparison, a slightly larger dressed mass of 0.23m0 has been obtained by Wang
et al. [132] and Knap et al. [133]. A small downward correction may be necessary in the
former,astheelectronswereconfinedataninterface.Thelatterauthors,however,appear
to have corrected for that effect. Using n-type bulk GaN, which does not require an
appreciable correction that is needed in confined systems and employing infrared
ellipsometry measurements, Kasic et al. [134] reported slightly anisotropic electron
masses of 0.237 0.006m0 and 0.228 0.008m0 along the two axes. Again, using
modulation-doped structures, a series of authors, Elhamri et al. [135], Saxler et al. [136],
Wong et al. [137], Wang et al. [138], and Hang et al. [139], also reported on the effective
mass, the values of masses for which ranged from 0.18m0 to 0.23m0 from Shubnikov–de
Haas data. Elhamri et al. [135] suggested that strain effects, which are somewhat difficult
to be certain of, could have compromised somewhat the masses reported in some of
these reports. A value of 0.20m0 is very commonly used for the bare electron effective
mass and 0.22m0 for the experimentally relevant dressed mass. A more in-depth
discussion of the cyclotron and Shubnikov–de Haas measurements can be found in
Volume 2, Chapter 3. This bare mass figure of 0.20m0 agrees reasonably well with a
number of estimates based on theory, as outlined in a list in Ref. [140]. Owing to the large
uncertainty, no attempt is made to specify an F parameter for wurtzite GaN. However,
the interband matrix element may be obtained from the relation between the electron
mass and the relevant zone center energies [92].
2.6
Temperature Dependence of Wurtzite GaN Bandgap
The temperature dependence of the bandgap in semiconductors is often described by
an imperial expression (assuming no localization)
j169
j 2 Electronic Band Structure and Polarization Effects
170
EðTÞ ¼ Eð0Þ aT 2 =ðb þ TÞ:
ð2:34Þ
In the case of localization, which can also be construed as band tail effect, the
temperature dependence deviates from the above equation. In the framework of
the band tail model and Gaussian-like distribution of the density of states for the
conduction and valence band, the temperature-dependent emission energy could be
described by the following modified expression [141], which is based on a model
developed for Stokes shift in GaAs/AlGaAs quantum wells [142].
EðTÞ ¼ Eð0Þ ½aT 2 =ðb þ TÞ ½s2 =ðkTÞ;
ð2:35Þ
where the last term represents the localization component with s indicating the
extent of localization or band tailing, which is nearly imperative for In-containing
alloys. The values of the parameters a (in units of energy over temperature) and b (in
units of temperature), for wurtzitic GaN, are listed in Volume 2, Table 5.1. Although a
detailed discussion of these parameters in very high-quality samples is deferred to
Volume 2, Chapter 5, the evolution of them is discussed here to give the reader a
flavor that when the sample quality is under question, the fits to experiments could
lead to varying if not erroneous parameters. In concert with this approach, the spread
in the values of a and b for A exciton is also discussed in the text surrounding
Volume 2, Table 5.3. Varshni parameters have been deduced from the measured
variation of the A, B, and C excitonic energies with temperature early on by
Monemar [143] with Varshni parameters of a ¼ 5.08 · 104 meV T1 and b ¼ 996 T,
the sign for the latter of which is contradictory to the agreed upon values deduced
from high-quality samples. In chronological order of the reports, using optical
absorption measurements on bulk single crystals and also epitaxial layers grown
on sapphire, Teisseyre et al. [144] reported a ¼ 0.939–1.08 meV K1 and b ¼ 745–772
K; note the positive sign of b. Shan et al. [145] reported a ¼ 0.832 meV K1 and
b ¼ 836 K deduced from the temperature variation of the A exciton resonance. Petalas
et al. [146] determined the Varshni parameters to be a ¼ 0.858 meV K1 and b ¼ 700
K using spectroscopic ellipsometry. Relying on PL measurements, Salvador
et al. [147] obtained a ¼ 0.732 meV K1 and b ¼ 700 K. Using absorption measurements, Manasreh [148] reported a ¼ 0.566–1.156 meV K1 and b ¼ 738–1187 K on
samples grown by MBE and OMVPE. Using a variation of electroreflectance, the
contactless electroreflectance, Li et al. [149] led to a ¼ 1.28 meV K1 and b ¼ 1190 K
for the A exciton transition energy. Utilizing PL spectra of excitonic transitions,
Zubrilov et al. [150] reported values of a ¼ 0.74 meV K1 and b ¼ 600 K based on
exciton luminescence spectra. PL data of free and bound excitons were fitted by
Reynolds et al. [151] to a modified Varshni-like form that resulted in a ¼ 0.5 meV K1
and b ¼ 1060. Some of the dispersion in the reported values can be attributed to the
difficulty in identifying and resolving various excitonic transitions. Vurgaftman and
Meyer [152], averaging what they term as more credible results, recommend
a ¼ 0.909 meV K1 and b ¼ 830 K. A detailed discussion of the temperature dependence of the bandgap of GaN along with the Varshni parameters for all three excitons
(A, B, and C) are listed in Volume 2, Table 5.3, where A exciton related parameters are
a ¼ 1 meV K1 and b ¼ 1100 K.
2.6 Temperature Dependence of Wurtzite GaN Bandgap
The complete Ai parameters calculated by Fritsch et al. [55] as well as others
deduced from alternative methods are tabulated in Table 2.9. In addition, a compilation of the dispersion in the effective mass for both the conduction band and various
valence bands as obtained by various computational methods as well as parameters
used in the description of the bandgap for wurtzitic GaN, particularly, in the context of
empirical pseudopotential method, as described in Ref. [55], are also included in
Table 2.9.
Suzuki and Uenoyama [59] have determined the deformation potentials by the fullpotential linearized augmented plane wave (FLAPW) calculations. The values recommended for GaN by Vurgaftman and Meyer [152] are tabulated in Table 2.8. In the
calculations of Suzuki and Uenoyama [59], biaxial and uniaxial strains have been
introduced and reduced shifts in the G point energy to which a linear fit in terms of
strain was obtained. From the linearfit,the deformation potential values for thevalence
band were deduced. The figures obtained from the quasi-cubic approximation are
listed too. The good agreement between the calculated values and those determined
from the quasi-cubic model is strikingly obvious, which is indicative of the excellence
of the quasi-cubic approximation.
Assuming an approximately spherical potential in the neighborhood of the N
atoms, of the two spin states, the higher energy is in the one in which the electron spin
and the orbital angular momentum are parallel. This result is also anticipated on the
basis of the atomic spin–orbit splitting in which the P1/2 state is known to have energy
higher than the P3/2 state. The contributions of spin–orbit interaction and the
crystal field perturbation to the experimentally observed splittings (E1,2 and E2,3)
have been calculated with different linear combination of atomic orbitals (LCAO)
approximations [155,156].
The large effective mass and the small dielectric constant of GaN, relative to more
conventional group III–V semiconductors, lead to relatively large exciton binding
energies and make excitons, together with large exciton recombination rates, clearly
observable even at room temperature. The bottom of the conduction band of GaN is
predominantly formed from the s levels of Ga, and the upper valence band states
from the p levels of N. Even though sophisticated methods have been introduced and
discussed on these pages, the method of Hopfield and Thomas [157], which treats the
wurtzite energy levels as a perturbation to the zinc blende structure is discussed
briefly, as it provides a physical picture of band splitting in the valence band. Using
the quasi-cubic model of Hopfield [98], one obtains
E 1 ¼ 0;
v"
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
#
u dþD u
dþD 2 2
t
dD ;
þ
E2 ¼
2
2
3
v"
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
#
u 2
dþD u
d
þ
D
2
E3 ¼
dD ;
t
2
2
3
ð2:36Þ
ð2:37Þ
ð2:38Þ
j171
j 2 Electronic Band Structure and Polarization Effects
172
where Dcr and Dso represent the contributions of uniaxial field and spin–orbit
interactions, respectively, to the splittings E1,2 and E2,3.
2.7
Sphalerite (Zinc blende) GaN
The zinc blende GaN crystal consists of two interpenetrating face-centered cubic
lattices, one having a group III element atom, for example, Ga, and the other a group
V element atom, for example, N. The matrix element of the momentum operator
between the conduction and valence bands has been expressed by Kane [92,158] in
terms of a single parameter P whose value is termed as EP, which is an important
parameter. The other parameter of importance is the F parameter, again defined by
Kane, which comes about from a second-order perturbation theory and takes into
account the higher order band contributions to the conduction band. Their importance aside [92] EP and F are inherently difficult to determine accurately, due to the
fact that the remote band effects can be calculated but are not measurable quantities.
One experimental technique relies on measuring the effective g factor, which is not as
influenced by remote bands as the effective mass.
A number of experimental and theoretical studies have determined energy gaps for
zinc blende GaN [69,70,146,159–163]. The term gaps is used, as there seems to be a
dispersion in the reported values. Typically, the excitonic transitions [164–166]
observed in low-temperature PL is used to infer the bandgap, provided the exciton
binding energy is known, which in this case is 26.5 meV. Although, low-temperature
bandgaps ranging from 3.2 to 3.5 eV have been measured, most of them tend to be
between 3.29 and 3.35 eV. It is therefore reasonable to use a low-temperature bandgap
of 3.3 eV for zinc blende GaN. A reasonable figure for the room-temperature
fundamental bandgap is 3.2 eV, although the range of 3.2–3.3 eV stated in Chapter
1 remains. As in the case of wurtzitic GaN, the temperature dependence of the energy
gap was also studied for zinc blende GaN, examples of which can be found in the
works of Petalas et al. [146] and Ramirez-Flores et al. [167]. Both group of authors
found b ¼ 600 K (using the more reliable model 1 in Ref. [146]), but the a parameters
differed somewhat. Not having any real basis for selecting one or the other, the
average value of 0.593 meV K1 is considered the default value. Although the indirect
gap energies have not been measured, for a calculation by Fan et al. [69], the X-valley
and L-valley minima had been put at 1.19 and 2.26 eV above the G valley, respectively.
These compare with an earlier prediction by Suzuki et al. [56] of about 1.4 and 1.6 eV,
respectively, as shown in Figure 2.6c. Ramirez-Flores et al. [167] measured the
spin–orbit splitting in zinc blende GaN to be 17 meV.
Electron spin resonance measurements on zinc blende GaN determined an
electron effective mass of 0.15m0 [168], which may represent the only experimental
results, and the value is similar to the G-valley masses derived from first-principles
calculations by Chow et al. [169] and Fan et al. [69]. Effective masses of ml ¼ 0:5m0
and mt ¼ 0:3m0 have been calculated for the X valleys in GaN [70], which are similar
to the theoretical results of Fan et al. [69].
2.7 Sphalerite (Zinc blende) GaN
0.00
HH
ZB GaN
LH
–0.01
Energy (eV)
j173
–0.02
SO
–0.03
–0.04
–0.05
–0.06
–0.15
–0.10
[1 1 1]
–0.050
0.0
0.050
Wave vector (1 Å-1)
0.100
[0 0 1]
Figure 2.10 Valence band structure of zinc blende GaN [152].
The valence band of zinc blende GaN has been the topic of various theoretical
efforts, and the E–k diagram by Vurgaftman and Meyer [152] is show in Figure 2.10.
Although the hole effective masses in zinc blende GaN have apparently not been
measured, a number of theoretical predictions of Luttinger parameters are available
in the literature [69,70,164–166,168,169,425,170,171]. Once the Luttinger parameters are known, the full picture in terms of the hole effective masses can be
determined. First, it should be pointed out that in polar semiconductors such as the
III–Vcompounds in general and GaN in particular, it is the nonresonant polaron [172]
mass that is actually measured. The polaron mass exceeds the bare electron mass by
about 1–2%, the exact value of which depends on the strength of the electron–phonon
interaction. Because the band structure is governed by the bare electron mass, this is
the quantity that is typically reported whenever available.
At the valence band edge, the heavy hole (hh) effective masses in the different
crystallographic directions are related to the free mass by the Luttinger parameters in
the following manner [92]:
mzhh ¼
m0
2m0
m0
½110
½111
; mlh ¼
; mlh ¼
:
g 1 2g 2
2g 1 g 2 3g 3
g 1 2g 3
ð2:39Þ
0.15
j 2 Electronic Band Structure and Polarization Effects
174
Here, the z-direction is perpendicular to the growth plane of (0 0 1). These expressions described by Equation 2.39 show the relationship of the Luttinger parameters to
the hh effective masses that can typically be measured in a more direct manner. The
light hole (lh) and so hole effective masses are given by
mzlh ¼
m0
2m0
m0
½110
½111
; mlh ¼
; mlh ¼
;
g 1 þ 2g 2
2g 1 þ g 2 þ 3g 3
g 1 þ 2g 3
m0
E P Dso
:
¼ g1 mso
3E g ðE g þ Dso Þ
ð2:40Þ
ð2:41Þ
Equation 2.41, which relates the split-off hole mass to the Luttinger parameters, should
in principle contain an additional parameter to account for the effects of remote bands
that is analogous to the F parameter [92], but the remote bands are not necessarily the
same ones that cause the largest correction to the electron mass. Due to the dominance
of the wurtzitic GaN, insufficient effort and thus data exist for zinc blende GaN, which
is also true for even the well-investigated III–V materials to describe the effect of the
interaction with remote bands on the split-off hole mass quantitatively.
To restate, although the hole effective masses in zinc blende GaN have apparently not
been measured, a number of theoretical predictions of Luttinger parameters are
available in the literature. The values are based on averages of the heavy-hole and lighthole masses along [0 0 1], as well as the degree of anisotropy in g3–g2. Doing so leads to
the parameter set as g1 ¼ 2.70, g2 ¼ 0.76, and g3 ¼ 1.11. Similarly, averaging all the
reported split-off masses [69,70,163,171,173] leads to mso ¼ 0:29m0 . In its simplest
form, the Luttinger parameters can be used to quickly determine the effective masses
in various valence bands both in equilibrium and also under biaxial strain. In fact, with
biaxial strain, the valence band degeneracy can be removed, and most strikingly the
heavy-hole in-plane mass can be made smaller by compressing strain, a notion that has
been exploited in the InGaAs/GaAs system very successfully.
An average of the two theoretical values for EP in zinc blende GaN [163,173] yields
EP ¼ 25.0 eV, which in turn implies F ¼ 0.95. Caution is advised because these
values have not been verified experimentally.
In conjunction with calculations of the electronic band structure of binary nitrides
and specifically effective masses in the valence band, Fritsch et al. [55] also arrive at the
Luttinger-like kp parameters by empirical fits for the effective masses at the G point.
These Luttinger-like parameters for the valence band of zinc blende GaN are listed in
Table 2.10. Those for zinc blende AlN and InN will be given in Sections 2.8.2
Sections 2.9.2. The Ai parameters transformed from the Luttinger parameters
obtained with the help of the quasi-cubic approximation can be found in Tables 2.8
and 2.9. It has been argued that the Ai parameters calculated in this manner are in good
agreement with the calculated values that have been used to support the value and
validity of the quasi-cubic approximation, which greatly simplifies the calculations.
Fritsch et al. [55] obtained the valence band effective masses for zinc blende
binaries by solving the eigenvalues of the kp matrix while taking the spin–orbit
interaction into account. The effective masses so calculated for the conduction and
valence bands, the latter involving the light and heavy holes, as well as the spin–orbit
2.7 Sphalerite (Zinc blende) GaN
Table 2.10 Luttinger parameters g1, g2, and g3 for zinc blende GaN
obtained from a fit along the [1 1 0] direction along with those
available in the literature, as compiled in Ref. [55].
Parameter
A
B
C
D
E
c1
c2
c3
2.89
0.85
1.20
2.96
0.90
1.20
2.70
0.76
1.07
3.07
0.86
1.26
2.67
0.75
1.10
A: empirical pseudopotential calculation by Fritsch et al. [55]; B: self-consistent FP-LAPW method
within local density approximation from Ref. [171]; C: first-principles band structure calculations
from Ref. [170]; D: empirical pseudopotential calculation from Ref. [174]; E: recommended
values taken from Ref. [152].
split-off mass, and the anisotropy taken into account are listed in Table 2.11 for zinc
blende GaN. The data contain those obtained by full potential linearized plane waves
(FP-LAPW), empirical pseudopotential method (EPM) calculations and those calculated with Luttinger parameters employing
ðm0 =mhh=lh Þ½100 ¼ g 1
ðm0 =mhh=lh Þ
2g 2 ;
½111
¼ g 1 2g 3 ;
2g
g2
ðm0 =mhh=lh Þ½1100 ¼ 1
2
g3
ð2:42Þ
;
where subscripts hh and lh represent the heavy-hole and light-hole effective masses,
respectively. The spin–orbit split-off-hole effective mass, mso, is isotropic in all the
three directions and is given by
ðm0 =mso Þ ¼ g 1 :
ð2:43Þ
Table 2.11 Effective masses for electrons (e), heavy holes (hh),
light holes (lh), and spin–orbit split-off (so) holes in units of the
free-electron mass m0 along the [1 0 0], [1 1 1], and [1 1 0]
directions for zinc blende GaN.
[100]
[100]
[111]
[111]
[110]
[110]
Reference
me
mhh
mlh
mhh
mlh
mhh
mlh
mso
A
B
C
D
E
F
0.14
0.14
0.13
0.17
0.15
0.12
0.84
0.86
0.76
0.85
0.85
1.34
0.22
0.21
0.21
0.24
0.24
0.70
2.07
2.09
1.93
1.79
2.13
1.06
0.19
0.19
0.18
0.21
0.21
0.63
1.52
1.65
1.51
1.40
1.55
1.44
0.20
0.19
0.19
0.21
0.21
0.58
0.35
0.30
0.32
0.37
0.29
0.20
Compiled by Fritsch et al. [55]. A: after Ref. [55]; B: self-consistent FP-LAPW method within local
density approximation from Ref. [171]; C: empirical pseudopotential calculation from Ref. [69]; D:
calculated from Luttinger parameters from Ref. [170], using Equations 2.42 and 2.43; E:
calculated from recommended Luttinger parameters from Ref. [152], using Equations 2.42
and 2.43; F: empirical pseudopotential calculation from Ref. [70].
j175
j 2 Electronic Band Structure and Polarization Effects
176
Because various calculations place the hydrostatic deformation potential for zinc
blende GaN in the range 6.4 to 8.5 eV [31,69,70,104,169,175], for the lack of a better
choice, the average value of the hydrostatic deformation potential a ¼ 7.4 eV is
chosen. It should be mentioned that due to the nature of the atomic bonding in III–V
materials, the bandgap increases for a compressive strain. Under positive hydrostatic
pressure that produces negative strain because the lattice constant gets smaller, the
change in the bandgap corresponding to a change in volume, DV/V ¼ (exx þ exx þ ezz),
given by DEg ¼ a(exx þ exx þ ezz) is positive, which necessitates the sign of the
deformation potential to be negative. The deformation potential is the sum of the
conduction and valence band deformation potentials, a ¼ ac þ av. Under pressure, the
conduction band edge is believed to move upward in energy while the valence band
moves downward, with most of the change being in the conduction band edge. Similar
to the total bandgap change, the change in valence band under pressure can be
described as DEg ¼ av(exx þ exx þ ezz). Naturally, it is much easier to measure the
change in the total bandgap due to strain than its component effects on the conduction
and valence bands. The valence band deformation potential value reported by Wei and
Zunger [176], namely, av ¼ 0.69 eV, is suggested. The sign convention for av is
adopted from Vurgaftman et al. [92]. To restate, the shrinking volume and negative
strain cause the valence band to move down. The values reported in the literature are in
the range of 0.69 to 13.6 eV. The suggested value is consistent with the expectation
that most of the strain shift should occur in the conduction band.
As for the shear deformation potentials b and d, the same is applied, which yields a
suggestion that b ¼ 2.0 eV with the full range of reported values being 1.6 to
3.6 eV. The recommendation d ¼ 3.7 eV is an average of the published results
from Ohtoshi et al. [175], Van de Walle and Neugebauer [177], and Binggeli et al. [178].
No experimental confirmations of any of these deformation potentials for zinc blende
GaN appear to exist. Turning to elastic constants, the values of C11 ¼ 293 GPa,
C12 ¼ 159 GPa, and C44 ¼ 155 GPa have been taken from the theoretical calculations
of Wright [179]. Very similar sets have also been calculated by Kim et al. [109,180] and
Bechstedt et al. [181]. For more details of elastic constants for all three binaries, refer
to Tables 2.25–2.27 and 2.28 that will follow later on in this chapter. The parameters in
conjunction with the band structure for zinc blende GaN are compiled in Table 2.12.
2.8
AlN
AlN forms the larger bandgap binary used in conjunction with GaN for increasing the
bandgap for heterostructures. As in the case of GaN, AlN also has wurtzitic and zinc
blende polytypes, the latter being very unstable and hard to synthesize. Owing to
increasing interest in solar blind devices and expectations that larger AlGaN with
large mole fractions of AlN would have large breakdown properties, this material has
been steadily gaining interest. It should also be mentioned that the N overpressure on
Al is the smallest among those over Ga and In, paving the way for equilibrium growth
of AlN bulk crystals, albeit not without O contamination.
2.8 AlN
Table 2.12 Parameters associated with the band structure for zinc blende GaN.
Parameter
alc (Å) at T ¼ 300 K
E Gg (eV)
a (G) (meV K1)
b (G) (K)
E Xg ðeVÞ
a (X) (K)
b (X) (meV K1)
E Lg ðeVÞ
a (L) (meV K1)
b (L) (K)
Value
4.50
3.3
0.593
600
4.52
0.593
600
5.59
0.593
600
Parameter
Value
Parameter
Value
Dso (eV)
me ðGÞ
ml ðXÞ
mt ðXÞ
c1
c2
c3
mso
0.017
0.15
0.5
0.3
2.70
0.76
1.11
0.29
EP (eV)
F
VBO (eV)
ac (eV)
av (eV)
b (eV)
d (eV)
c11 (GPa)
c12 (GPa)
c44 (GPa)
25.0
0.95
2.64
6.71
0.69
2.0
3.7
293
159
155
Bandgaps are for low temperature [152].
2.8.1
Wurtzite AlN
Wurtzite AlN is a direct bandgap semiconductor with a bandgap near 6.1 eV and still
considered to be semiconductor. The zinc blende polytype is not stable with a
predicted indirect bandgap, as will be discussed in the next section. The AlN derives
its technological importance from providing the large bandgap binary component of
the AlGaN alloy, which is commonly employed both in optoelectronic and electronic
devices based on the GaN semiconductor system. Early on, the absorption measurements carried out by Yim et al. [182] and later by Perry and Rutz [183] indicated a large
energy gap for wurtzite AlN of 6.28 eVat 5 K to 6.2 eVat room temperature. The actual
figures are converging at values about 0.1 eV below those contained in those early
reports. As for the dependence of the bandgap on temperature, the Varshni parameters of a ¼ 1.799 meV K1 and b ¼ 1462 K were reported by Guo and Yoshida [184],
who also found the low-temperature gap to be 6.13 eV, which is similar to that
reported by Vispute et al. [185], a value closer to the values observed of late. Tang
et al. [186] resolved what they believed to be the free or shallow impurity bound
exciton in their cathodoluminescence (CL) data, at an energy of 6.11 eV at 300 K.
Brunner et al. reported a variation from 6.19 eV at 7 K to 6.13 eV at 300 K [187]. A
group of same authors Wethkamp et al. [188] used spectroscopic ellipsometry and
determined that the energy gap varies from 6.20 eV at 120 K to 6.13 eV at 300 K.
Kuokstis et al. [189] resolved a low-temperature free-exciton transition at 6.07 eV. Guo
et al. [190] reported the temperature dependence of the reflectance spectra, while
fitting it to the Bose–Einstein expression.
Using the low-temperature data in conjunction with the Varshni parameters of
Guo and Yoshida [184], leads to an intermediate value of 6.23 eV for the lowtemperature bandgap. The Varshni parameters reported by Brunner et al. [187]
indicate no significant divergence from GaN for the entire AlGaN alloy composition
range, which may bring the accuracy of the data into question. As in any semiconductor, the quality and strain nature of the films can alter the results. The availability
j177
j 2 Electronic Band Structure and Polarization Effects
178
of high-quality homoepitaxial AlN with presumably no strain has shed the much
needed light onto the issues surrounding the actual bandgap of AlN [191]. However,
even then O contamination could cause the near-band emission peak observed for
shift, as it has done so for high-quality bulk substrates reported by Slack et al. [192]. In
the experiments of Silviera et al. [191], the epitaxial layer was 0.5 mm, plenty
considering the small penetration depth of 10 keV electrons used for the CL
experiments, and the substrate was 287 mm thick with O concentrations of about
5 · 1019 cm3 as measured by Neutron activation. The homoepitaxial AlN films have
been grown by organometallic vapor-phase epitaxy on the single-crystal AlN substrates and efforts were undertaken to reduce the oxygen content of the film. The lowtemperature near band edge CL spectrum of the AlN film is shown in Figure 2.11.
The open squares correspond to the experimental data obtained at 5 K, while the full
line representing the best fit to the experimental data using Corinthian line shapes
with transitions are indicated with dashed lines. The assignments shown are a result
of thermal quenching behavior. The full widths at half maxima (FWHM) of the
narrowest emission line at 6.023 eV is about 1.0 meV and was reported to be perhaps
limited by the slit size used during the experiment. A measurement of the 253.65 nm
emission line of a low-pressure Hg lamp using the same slits size set resulted in a
FWHM of about 0.7 meV, as tabulated in Table 2.13.
Figure 2.12 shows the temperature-dependent CL spectra for the AlN film. A rapid
decrease in the intensities of the four peaks initially observed between 5.98 and
6.01 eV with the increase in temperature is evident, which is consistent with
recombination processes involving excitons bound to shallow neutral centers. The
peaks at 6.023 and 6.036 eV remain intense with increasing temperature, which lets
them gain the free exciton A (FXA) and free exciton B (FXB) assignments, respectively,
Figure 2.11 High-resolution CL spectrum of an AlN
homoepitaxial film. The full line represents our best fit using
Lorentzian line shapes, and the dashed lines are the transitions
composing the full line [191].
2.8 AlN
Table 2.13 Energy positions, full widths at half maxima (FWHM),
and preliminary assignments associated with the transitions
shown in Figure 2.11 [191].
Energy (eV)
FWHM (meV)
Assignment
5.98
6.000
6.008
6.01
6.023
6.036
49.4
11.0
1.5
44.0
1.0
8.0
A07 X A
D027 X A
D017 X A
D017 X B
FXA
FXB
due to their large binding energies. The line around 6.07 eV, shown as FX2A , is some 2
orders of magnitude weaker than the most intense bands observed in the spectrum.
On the basis of the similarity in the luminescence spectra of both GaN and AlN, the
peak at 6.07 eV is attributed to the first excited state of the FXA. This assignment
allows an estimation of the FXA binding energy as 63 meV, which is about twice the
value for GaN. This leads to an estimated low-temperature bandgap of AlN of
6.086 eV (the sum of the FXA energy and its binding energy).
Returning to the band structure of AlN, of considerable significance, the crystal field
splitting in wurtzitic AlN is believed to be negative. The ramification of this is that the
topmost valence band is the crystal hole band. Calculations have yielded a range of
crystal field splittings, namely, Dcr ¼ 58 meV by Suzuki et al. [56], Dcr ¼ 217 meV
by Wei and Zunger [107], Dcr ¼ 176 meV by Shimada et al. [193], and Dcr ¼ 244 meV
by Wagner and Bechstedt [85]. Moreover, splittings of Dcr ¼ 104 and 169 meV
were obtained from first-principles and semiempirical pseudopotential calculations,
respectively, by Pugh et al. [163] and Dcr ¼ 215 meV by Kim et al. [154]. Averaging all of
the available theoretical crystal field splittings, one obtains a value of Dcr ¼ 169 meV.
Silveira et al. [194], using optical reflectance data performed on a- and c-plane bulk AlN
and a quasi-cubic model developed for the wurzite crystal structure, determined the
crystal field splitting to be D ¼ 225 meV. Note that the negative sign for the crystal
Figure 2.12 Temperature-dependent CL spectra of 0.5 mm thick
AlN film deposited on 287 mm bulk AlN substrate [191].
j179
j 2 Electronic Band Structure and Polarization Effects
180
field splitting has important implications, namely, that the G7 valence band is on the top
of G9 valence band, which is opposite of that in GaN.
As for the spin–orbit splitting, the literature values range from 11 [163] to
20 meV [124]. Silveira et al. [194] again using the optical reflectance spectra in bulk
AlN determined the spin–orbit splitting energy to be d ¼ 36 meV. In view of the
experiments in high-quality bulk AlN, the value of 36 meV is recommended even
though it is much larger than the calculated value of 19 meV recommended by Wei
and Zunger [107].
As in the case of GaN, the region of the energy band near the bottom of the
conduction band, as it manifests itself in devices, can be represented by the effective
mass. The same of course applies for the top of the valence band. A number of
investigators have calculated the AlN electron effective mass [154,124,163,195,196],
with the prediction that it displays a greater anisotropy than that for wurtzitic
==
GaN [56]. The bare mass values of m?
e ¼ 0:30m0 and m e ¼ 0:32m 0 obtained by
averaging the available theoretical masses may represent a good set of default values
as this stage. It should again be underscored that further experimental studies are
needed to verify the calculations. As for the valence band, a number of theoretical sets
of valence band parameters are available [56,58,154]. There is an apparent disagreement in the signs for A5 and A6 among these reports, which may be irrelevant,
because only absolute values of those parameters enter the Hamiltonian [154,163].
The A parameters given by Kim et al. [154] are suggested because the crystal field and
spin–orbit splittings reported by these authors are closest to the ones suggested here.
The hydrostatic deformation potential for wurtzite AlN has been reported to be in
the range of 7.1 and 9.5 eV [31,180], which is consistent with the observation
that the bandgap pressure coefficients for AlGaN alloys have little dependence on
composition, as reported by Shan et al. [197]. The calculated values of a1 ¼ 3.4 eV
and a2 ¼ 11.8 meV reported by Wagner and Bechstedt [85] are assumed to represent
the material well. Theoretical values are also available for a few of the valence
band deformation potentials such as D3 ¼ 9.6 eV, and D4 ¼ 4.8 eV [180]. However,
the complete set D1 ¼ 17.1 eV, D2 ¼ 7.9 eV, D3 ¼ 8.8 eV, D4 ¼ 3.9 eV, D5 ¼ 3.4
eV, and D6 ¼ 3.4 eV with the last value derived using the quasi-cubic approximation
presented by Shimada et al. [193] can be used in the absence of any other reliable data.
For mere availability reasons and few other issues, the mechanical properties of
AlN have seen a good deal of experimental activity very early on, which later
was followed by theory. Tsubouchi et al. [198], McNeil et al. [199], and Deger
et al. [200] measured the elastic constants of wurtzitic AlN. A good many theoretical
papers have also been reported [154,193,201–204]. The values suggested by
Wright [179], who also provided a detailed discussion of their expected accuracy,
namely, C11 ¼ 396 GPa, C12 ¼ 137 GPa, C13 ¼ 108 GPa, C33 ¼ 373 GPa, and C44
116 GPa, are recommended.
Several piezoelectric coefficients [205,206] for early AlN at least in part can be
found in Ref. [207]. The result for d33 ¼ 5.6 pm V1 reported in Ref. [207] is in
reasonably good agreement with the previous determinations but differs somewhat
from d33 ¼ 5.1 pm V1 measured by Lueng et al. [208]. While these experiments
focused on only d33, both d33 and d13 can be determined from first-principles
2.8 AlN
Table 2.14 Recommended band structure parameters for wurtzitic AlN [152].
Parameter
Value
Parameter
Value
Parameter
Value
Eg (eV, low temperature)
a (meV K1)
b (K)
Dcr (meV)
Dso (meV)
==
me =m0
m?
e =m0
a1 (eV)
a2 (eV)
6.077 [194]
1.799, 0.9a
1462, 1000a
225 [194]
36 [194]
0.32
0.30
3.4
11.8
A1
A2
A3
A4
A5
A6
A7 (meV Å)
d13 (pm V1)
d33 (pm V1)
d15 (pm V1)
Psp (C m2)
3.86
0.25
3.58
1.32
1.47
1.64
0 (default)
2.1
5.4
3.6
0.090
D1 (eV)
D2 (eV)
D3 (eV)
D4 (eV)
D5 (eV)
D6 (eV)
C11 (GPa)
C12 (GPa)
C13 (GPa)
C33 (GPa)
C44 (GPa)
17.1
7.9
8.8
3.9
3.4
3.4
396
137
108
373
116
See Tables 2.27 and 2.28 for details related to the elastic constants, piezoelectric constants, and
spontaneous polarization charge. Any dispersion among the tables is a reflection of the
uncertainty in the available parameters. Note that the G7 valence band is above the G9 valence
band, which is opposite of GaN. It is also similar to that in ZnO, which is somewhat controversial.
See Zinc Oxide: Materials Preparation, Properties, and Devices, by H. Morkoç and Ü. Özg€
ur, Wiley
(2008) regarding the valence band ordering in ZnO.
a
Obtained using the temperature dependence of the A exciton energy reported in Ref. [194].
calculation [193,209–212]. The recent theoretical values of Bernardini and Fiorentini,
d33 ¼ 5.4 pm V1 and d13 ¼ 2.1 pm V1 [210], are suggested although the elastic
coefficients given in that reference are somewhat larger than the recommended
values. On the basis of recent measurements [206,207] and a calculation [210] of the
shear piezoelectric coefficient, Vurgaftman and Meyer [152] recommend d15 ¼ 3.6
pm V1. The parameters concerning the bandgap-related issues for wurtzitic AlN
recommended by Vurgaftman and Meyer [152] are tabulated in Table 2.14.
A compilation of the dispersion in the effective mass for both the conduction band
and various valence bands as obtained by various computational methods as well as
parameters used in the description of the bandgap for wurtzitic InN, particularly, in
the context of empirical pseudopotential method, as described in Ref. [55], are
tabulated in Table 2.15.
Even though a detailed discussion of polarization is reserved (Section 2.12), a
succinct treatment of the topic is given here as it is relevant to the topic under
discussion. The difference between the GaN and AlN spontaneous polarizations
strongly causes a net polarization at the interface between the two materials that
extends to the GaN/AlGaN interfaces as well. This charge, which is bound, influences
the band profiles and energy levels in GaN/AlN quantum heterostructures. Although
rigorous calculations [84,181,209,213] of the spontaneous polarization Psp(AlN) have
produced results spanning a fairly broad range, from 0.09 to 0.12 C m2, values
for the difference Psp(AlN)Psp(GaN) have tended to be more consistent, with most
falling between 0.046 and 0.056 C m2. Experimentally, for some time the majority of
workers on the GaN/AlGaN system reported somewhat smaller Psp(AlN)Psp(GaN).
For example, Leroux et al. [214,215] derived 0.051 < Psp < 0.036 C m2 for AlN.
A study of the charging of GaN/AlGaN field effect transistors led to a similar
j181
j 2 Electronic Band Structure and Polarization Effects
182
Table 2.15 Effective masses and band parameters for wurtzitic AlN.
Parameter
==
aniso
iso
A
B
C
D
E
F
me
m?
e
0.231
0.242
0.232
0.242
0.33
0.25
0.33
0.25
0.24
0.25
0.24
0.25
0.35
0.33
mhh
2.370
2.382
3.68
3.53
1.949
1.869
3.53
4.41
2.370
2.382
3.68
3.53
1.949
1.869
3.53
4.41
0.209
3.058
0.285
1.204
4.789
0.550
4.368
1.511
1.734
1.816
0.134
0.128
0.209
3.040
0.287
1.157
4.794
0.571
4.374
1.484
1.726
1.788
0.153
0.160
0.25
10.42
0.24
3.81
4.06
0.26
3.78
1.86
2.02
0.229
2.584
0.350
0.709
4.367
0.518
3.854
1.549
1.680
2.103
0.204
0.093
0.212
2.421
0.252
1.484
4.711
0.476
4.176
1.816
1.879
2.355
0.096
0.093
0.26
11.14
0.33
4.05
3.86
0.25
3.58
1.32
1.47
1.64
0.27
2.18
0.29
4.41
3.74
0.23
3.51
1.76
1.52
1.83
0
==
==
mlh
==
mch
m?
hh
m?
lh
m?
ch
A1
A2
A3
A4
A5
A6
A7
D1
0.25
6.33
0.25
3.68
3.95
0.27
3.68
1.84
1.95
2.91
0
0.059
0
0.059
0.215
Effective masses in units of free-electron mass m0, Luttinger-like parameters Ai (i ¼ 1, . . ., 6) in
units of h2 =2m0 , and A7 in units of eV Å. The crystal field splitting energy D1 is given in units of
meV. The term aniso represents the values derived using a band structure calculation with
anisotropically screened model potentials, whereas the term iso describes a comparative band
structure calculation on the basis of isotropically screened model potentials using an averaged e0
value by taking the spur of the dielectric tensor [55]. Anisotropically screened and isotropically
screened values are from Ref. [55]. A: FP-LAPW band structure calculations are from Ref. [170], and
effective mass parameters are obtained through a 3D fitting procedure within cubic approximation;
B: FP-LAPW band structure calculations are from Ref. [170], and effective mass parameters are
obtained by a direct line fit; C: Ai from Ref. [153] obtained through a Monte Carlo fitting procedure
to the band structure and effective masses calculated using Equations 2.30 and 2.31; D: direct kp
calculations for Ai from Ref. [153] and effective masses obtained from Ai using Equations 2.30
and 2.31; E: direct fit of Ai to first-principles band structures from Ref. [154]; F: Ai and effective
masses obtained in the quasi-cubic model from zinc blende parameters from Ref. [154].
conclusion [216], and Hogg et al. [217] were able to fit their luminescence data by
assuming negligible spontaneous polarization. Park and Chuang [218] required
Psp ¼ 0.040 C m2 to reproduce their GaN/AlGaN quantum well data. On the
contrary, Cingolani et al. [219] reported good agreement with experiment using a
higher value derived from the original Bernardini et al. [209] calculations.
A significant step toward resolving this discrepancy has been the realization that
the AlGaN spontaneous polarization cannot be linearly interpolated between the
values at the binary end points [220–222]. In combination with an improved
nonlinear strain treatment of the piezoelectric effect, the discrepancy between theory
and experiment for GaN/AlGaN quantum wells has been largely eliminated [85]. For
additional details, see Section 3.14 and the text dealing with Tables 2.25, 2.27
and 2.28. We adopt Psp ¼ 0.090 C m2 as the recommended value for AlN, in
conjunction with Psp(GaN) ¼ 0.034 C m2. The recommended band structure
parameters for wurtzite AlN are compiled in Table 2.14.
2.8 AlN
2.8.2
Zinc Blende AlN
Only a handful of purportedly successful growths of zinc blende AlN on zinc blend
substrates, such as GaAs and 3-C SiC, and Si substrates following low-temperature
zinc blende GaN buffer layers have been reported [223–226]. Consequently, much of
the discussion here relies primarily on theoretical projections and so do the parameter
set recommended for this polytype. The only quantitative experimental study of the
bandgap indicated a G-valley indirect gap of 5.34 eV at room temperature [224].
Assuming that the Varshni parameters for the wurtzitic AlN hold for the zinc blende
polytype, the aforementioned room-temperature bandgap translates to a low-temperature gap of 5.4 eV. Vurgaftman and Meyer [152] recommend 4.9 and 9.3 eV for the
X- and L-valley gaps, respectively [31,69,163]. The spin–orbit splitting is expected to be
nearly the same as in wurtzite AlN at 19 meV [154,107,171,227]. Averaging
the theoretical results from a number of different publications [69,154,163,170,173],
one arrives at a G-valley effective mass of 0.25m0. The longitudinal and transverse
masses for the X valley have been predicted to be 0.53m0 and 0.31m0, respectively [69].
If the method used previously for the GaN is applied to zinc blende AlN, one arrives at
recommended Luttinger parameters of g1 ¼ 1.92, g2 ¼ 0.47, and g3 ¼ 0.85, and mso
0.47m0 [69,154,170,173]. These as well as the other literature values of the Luttinger
parameters are listed in Table 2.16.
Fritsch et al. [55] calculated the effective masses for conduction and valence bands,
the latter involving the light and heavy holes, as well as the spin–orbit split-off mass,
which with the anisotropy taken into account are listed in Table 2.17 for zinc blende
AlN. The data contain those obtained by FP-LAPW, EPM calculations and those
calculated with Luttinger parameters (Equations 2.42 and 2.43).
When the calculated [163,173] values for the momentum matrix are averaged,
a value of EP ¼ 27.1 eV (with F ¼ 101) is obtained. Hydrostatic deformation
potentials of 9.0 eV [31] and 9.8 eV [69] have been reported. The deformation
potential values, a ¼ 9.4 eV, av ¼ 4.9 eV [69,107], b ¼ 1.7 eV [69,177,178], and
d ¼ 5.5 eV [180,177,178], have been suggested [152]. The elastic constants of
C11 ¼ 304 GPa, C12 ¼ 160 GPa, and C44 ¼ 193 GPa calculated by Wright [179] are
Table 2.16 Luttinger parameters g1, g2, and g3 for zinc blende AlN
obtained from a fit along the [1 1 0] direction along with those
available in the literature, as compiled in Ref. [55].
Parameter
A
B
C
D
E
c1
c2
c3
1.85
0.43
0.74
1.54
0.42
0.64
1.50
0.39
0.62
1.91
0.48
0.74
1.92
0.47
0.85
A: empirical pseudopotential calculation by Ref. [55]; B: self-consistent FP-LAPW method within
local density approximation from Ref. [171]; C: first principles band structure calculations from
Ref. [170]; D: empirical pseudopotential calculation from Ref. [174]; E: recommended values
taken from Ref. [152].
j183
j 2 Electronic Band Structure and Polarization Effects
184
Table 2.17 Effective masses for electrons (e), heavy holes (hh),
light holes (lh), and spin–orbit split-off (so) holes in units of the
free-electron mass m0 along the [1 0 0], [1 1 1], and [1 1 0]
directions for zinc blende AlN.
[100]
[100]
[111]
[111]
[110]
[110]
Reference
me
mhh
mlh
mhh
mlh
mhh
mlh
mso
A
B
C
D
E
0.23
0.28
0.21
0.30
0.25
1.02
1.44
1.05
1.39
1.02
0.37
0.42
0.35
0.44
0.35
2.64
4.24
2.73
3.85
4.55
0.30
0.36
0.30
0.36
0.28
1.89
3.03
2.16
2.67
2.44
0.32
0.37
0.31
0.38
0.29
0.54
0.63
0.51
0.67
0.47
Compiled by Fritsch et al. [55]. A: after Ref. [55]; B: self-consistent FP-LAPW method within local
density approximation from Ref. [171]; C: empirical pseudopotential calculation from Ref. [69]; D:
calculated from Luttinger parameters from Ref. [170], using Equations 2.42 and 2.43; E:
calculated from recommended Luttinger parameters from Ref. [152], using Equations 2.42
and 2.43.
similar to the sets quoted in other theoretical works [180,181,228] and are therefore
suggested. These and other band structure parameters recommended for zinc blende
AlN are tabulated in Table 2.18 [152].
2.9
InN
As in the case of AlN, the interest in InN has so far been not necessary because of its
properties, but because of the InGaN alloy that is used in lasers and LEDs operative in
the visible and violet regions of the optical spectrum. In fact, if and when the
technological issues are overcome, the InGaN channel FETs may also be superior to
the GaN channel varieties [229], some details of which are discussed in Volume 3,
Table 2.18 Parameters associated with the band structure for zinc blende AlN.
Parameter
Value
Parameter
Value
Parameter
Value
alc (Å) at T ¼ 300 K
E Gg ðeVÞ
a (G) (meV K1)
b (G) (K)
E Xg ðeVÞ
a (X) (K)
b (X) (meV K1)
E Lg ðeVÞ
a (L) (meV K1)
b (L) (K)
4.38
5.4
0.593
600
4.9
0.593
600
9.3
0.593
600
Dso (eV)
me ðGÞ
ml ðXÞ
mt ðXÞ
c1
c2
c3
mso
0.019
0.25
0.53
0.31
1.92
0.47
0.85
0.47
EP (eV)
F
VBO (eV)
ac (eV)
av (eV)
b (eV)
d (eV)
c11 (GPa)
c12 (GPa)
c44 (GPa)
27.1
1.01
3.44
4.5
4.9
1.7
5.5
304
160
193
Bandgaps are for low temperature [152].
2.9 InN
Chapter 3. The properties, particularly the fundamental parameters of InGaN for a
given composition, depend very much on the InN parameters, particularly its
bandgap. It is therefore important to understand the properties of bulk InN in its
wurtzitic form.
InN, however, is not all that easy, even considering the general difficulties
encountered in the nitride semiconductor system, to synthesize. The somewhat
intractable problem with InN is the enormous difference in the ionic size of its
constituent atoms in that the atomic radii for In and N are largely different, which
leads to highly distorted interatomic distances, interatomic bonding charges, tendency to form metallic clusters of the group III constituent, and inhomogeneous
strain. All of these could, in principle, lead to pronounced anomalies in all the
properties of InN, inclusive of measured bandgap and nature of defects. To make
matters worse, the InN layers are grown at best on GaN epitaxial layer with large
lattice mismatch (lm), aggravating many of the aforementioned problems. In spite of
all these, progress is steadily made.
It should be added that after having been accepted as the bandgap of InN, the
1.98 eV figure came under new scrutiny in that a plethora of reports concluded
the actual bandgap to be 0.7–0.8 eV. Just at a time when a good many got convinced of
the newer data, questions have been raised about the models along with heightened
level of scrutiny of the new low-bandgap data. Assuming that interpretation of
experiments pointing to the small bandgap figures are impeccable, theories also
begin to be developed, even though long-standing understanding such as cation rule
would be broken by the small bandgap figure.
2.9.1
Wurtzitic InN
The bandgap of InN has been a point of controversy dating back to the early days of
InGaN development [1,230,231]. Early absorption studies on sputtered thin films
concluded that the InN bandgap is in the 1.7–2.2 eV range [232–236]. However, no
band-to-band PL could be observed in the samples prepared by sputtering in early
developmental stages or later on in films grown by OMVPE and MBE. A review [237]
of various crystal growth related issues and resultant properties as well as a
proceedings [238] of a meeting devoted to debating these issues is available in the
literature. In contrast to earlier reports of no near band edge emission, Davydov
et al. [239–242] and others [243–245] reported near band edge emission but at much
lower energies near 0.6–0.8 eV, depending on the report. Also see the comment,
Ref. [246], on Ref. [239] and reply to that comment [247]. These reports relied to
various extents on absorption, photoluminescence, and photoluminescence excitation experiments that showed the experimental evidence for the bandgap of InN to be
overwhelmingly in the range of 0.7–0.8 eV and recommended a zero-temperature
gap of 0.78 eV [248] and 692 2 meV [249], and in fact values as low as 0.65 eV [239]
and 0.67 eV [250] have been reported as well. InN films of N-polarity grown by RF
molecular beam epitaxy exhibited a bandgap value of about 0.7–0.75 eV as measured
by optical transmission and reflection measurements [251–256]. The PL emission
j185
j 2 Electronic Band Structure and Polarization Effects
186
line appeared at 0.7 eV with no shift in energy between 300 and 77 K. These particular
samples also exhibited room-temperature electron mobilities in the range of
m ¼ 1750–2000 cm2 V1 s1 and an electron concentration in the range of n ¼ 2–3
· 1018 cm3 at room temperature. Some details about the growth of these films are
provided in Section 3.5.13.
The polycrystalline or nanocrystalline nature of those early thin films associated
with high electron densities and low mobilities led the proponents of the small
bandgap for InN to suggest that those early films most likely contained a good deal of
oxygen coupled with Moss–Burstein effect that could push the apparent bandgap
upward. Another possible explanation for the dispersion in the reported values of the
bandgap and also in support of the smaller bandgap may have to do with blue shift
caused by any quantum size effects. At least the correct trend has been established by
Lan et al. [257] who reported PL emission at 1.9 eV in nanorods of diameter between
30 and 50 nm (dubbed the brown InN), whereas 0.766 eV emission (measured at
20 K) was observed in rods with a diameter in the range of 50–100 nm (dubbed the
black InN). Further refinement of the work led to the observation that the samples
with fine (10 nm) or containing very high carrier concentrations exhibit the visible
emission. In fact, both IR and visible peaks could be observed in the same sample
when the samples show bimodal distribution of grain size [258].
The InN nanorods catalytically formed in the upstream portion of the substrate
were of the brown type while those downstream were of the black variety. The Au
catalyst that floats on the top of the InN nanorods as the growth progresses were
shown to be encapsulated with In2O3, which is somewhat unexpected and may be
made possible owing to close epitaxial relationship between wurtzitic InN and In2O3,
indicating the participation of O in the catalytic process. The source of O, in this case,
was attributed to residual O in the reactor as well as the quartz tube used. Furthermore, the black InN photon emission in a PL experiment quenched above 150 K while
that from the brown InN exhibited strong emission at room temperature, albeit
broad. The variation in the observed emission wavelength has been attributed to a
variety of sources including O incorporation, Moss–Burstein shift due to high
electron concentration, and quantum size effect. The quantum size effect would
require diameters of less than 5 nm that is much larger than the 20–50 nm brown InN
rods, which cannot therefore explain the bandgap shift to 1.9 eV from about 0.7 eV. It
should be stated that the characteristic PL peak in O-implanted InN is different from
the broad visible peak.
The possibility of InN quantum dot formation in InGaN as being responsible for
bandgap variations has not been of as much use in that even intentional dot
formation, albeit limited to possibly only one report, did not lead to any blue
shift [259]. The results are not precise but speak to the trend and demonstrate that
great care must be taken to make sure that all the samples are grown under identical
conditions, and doping levels are the same and not very large. Another issue of
paramount importance that comes into the equation is the degree of crystallinity of
the InN films. For example, Anderson et al. [260] produced polycrystalline InN
with 0.8 eV luminescence present but did not identify the size of polycrystalline
grains.
2.9 InN
Proponents of the earlier and larger bandgap for InN bring on the table several
arguments undermining the validity of the low-bandgap figure. Among theme is that
the absorption squared versus energy plots used to obtain the apparent bandgap in a
semiconductor with very high carrier concentration underestimates the bandgap due
to band tailing [261]. In addition, the 0.7–0.8 eV peak ascribed by some to the bandgap
of InN is attributed to defects caused by nonstoichiometry of the films, which are
grown under extremely In reach conditions and far away from equilibrium condition [261]. Moreover, The Moss–Burstein blue shift used by proponents of the small
bandgap to account for the large bandgap reported earlier is not consistent among all
the samples in that the sample with low carrier concentrations in the past confirmed
the large bandgap of InN, albeit they were most likely heavily compensated. Finally, the
opponents of the larger bandgap argue that the observed transition at 0.7–0.8 eV is due
to Mie resonance that in turn is due to scattering or absorption of light in InNcontaining clusters of metallic In, which may have been mistaken for the low bandgap,
as only the In cluster containing samples do show the 0.7–0.8 eV peak [262].
Let us now discuss the evolution of InN, and in particular, its perceived and
admittedly controversial perceived bandgap. The early attempts to produce InN relied
on not well-developed methods, as compared to modern crystal growth techniques,
and as such produced mostly powder and nanopowders [263–265]. Naturally, the
early work on thermodynamic decomposition studies and X-ray diffraction (advantageous for power diffractometry due to the nature of the films) were performed on
powder films. Even in these very powder samples, there was some observed variation
in the color of the material in that some have been noted to be black or blackbrown [264–266] as opposed to the deep red that one would expect for the 1.8 eV
bandgap originally estimated [265]. For a witty historical account of these early efforts
through the controversy, the reader is referred to Refs [267,268]. Below a discussion of
the role of oxygen, possible defects that might be responsible for the 0.7 eV emission,
the Moss–Burstein shift, particulars of carrier absorption, and Mie resonance is given
in an effort to give an overall appreciation of issues causing the controversy in
determining the bandgap parameter of InN.
Focusing on the issue of O, because In2O3 has a bandgap of 3.75 eV [269], the
argument goes that if InN films are heavily O contaminated, the bandgap would be
pushed upward. This is one of the points used by the proponents of the small bandgap.
However, as shown in Figure 1.34, to bring the bandgap from 0.7 to 1.9 eV would
require some 30% O in the film assuming that the Vegards law holds. It should also be
noted that the stated O must form an alloy for increasing the bandgap not just as surface
contamination or inclusion of O at grain boundaries. One of the samples used to
support the InN–In2O3 alloy formation was an RF-sputtered film with poor mobility
and high carrier concentration [270]. Auger analysis used to determine the O content in
the aforementioned layer is sensitive to only O and cannot determine whether it is
alloyed in the semiconductor InN. In addition, any depth profiling accompanied by that
technique requires argon ion etching, which is known to result in severe nitrogen loss
and lead to overestimation of the atomic oxygen concentration owing to the recycling of
sputtered oxygen on the film surface coupled with the strong bond between oxygen and
surface indium [271]. An alternative method, Rutherford backscattering (RBS), was
j187
j 2 Electronic Band Structure and Polarization Effects
188
also used by Davydov et al. [270] with the sample mounted on glass, which in
consideration of the sample thickness may not have allowed a definitive determination
whether the O signal is due to O in InN or the glass substrate used. Nevertheless, the O
content so measured was 20%, which is to be contrasted to 10% obtained [272] from
measurements performed in similar layers using elastic recoil, which is accurate for
elemental analysis. It should again be mentioned that elemental O in the film, even if
present in stated quantities, is not the same as InN:In2O3 alloy, which would increase
the bandgap, not to mention the fact that such alloying is not favored by temperatures
employed in RF sputter deposited InN. One can surmise that amorphous InON, NO2
and surface hydroxide species are implicated as being responsible for the 1.8–2 eV
bandgap reported. These species are only as surface species, and there has not as yet
been any evidence reported of any other form of InN–In2O3 alloy species during the
growth of InN, as can be discerned from Figure 1.34 that 10% or even 20% oxygen
could not account for the bandgap of 1.9 eV if a bandgap of 0.7 eV is assumed for InN.
Figure 1.34 clearly indicates that an alloy with about 37 at.% oxygen, which translates to
44% In2O3, would be needed to provide a bandgap of 2.0 eV if the bandgap of pure InN
is 0.7 eV. Consequently, 10% (at.) oxygen, even if all were alloyed with InN, would
account for a blue shift of about only 0.3 eV, much less than some 1 eV for consistency.
If the bandgap of the alloy follows a bowing parameter, unless it is positive, which is
unlikely, the blue shift caused by alloyed O would even be smaller. This view is
supported by the results of Yoshimoto et al. [273] that reported a bandgap value of
1.8–2.0 eV for MBE InN grown on quartz with 3% atomic oxygen present in the film.
Evidently, this level of O contamination does not prevent one from arriving at the
longstanding bandgap of InN, as any blue shift caused by this level of elemental O even
if all is in the alloy form does amount to much.
The role of O in InN had been investigated early on in sputtered films. Among
them is the work of Westra et al. [274] who produced InN with carrier concentrations
between 7 · 1019 and 2 · l020 cm3 and mobility of 4–10 cm2 V1 s1. Rutherford
backscattering data indicated 11% atomic oxygen, and indium-to-nitrogen ratios
slightly above a value of 1. No evidence of oxygen or oxynitride phases was observed in
the X-ray diffraction spectra, which led those authors to propose that oxygen is in the
form of an amorphous indium oxynitride, similar to the observations of Foley and
Lyngdal [275]. In this case the structure would maintain the stoichiometry, while the
oxygen would not be detected by X-ray diffraction. Contribution from NO2 has been
observed for samples containing higher oxygen content in a polycrystalline InN
matrix with no evidence for O in X-ray diffraction or infrared absorption spectra [271],
wherein grain boundaries were proposed to be the host for O. Raman data for
sputtered InN with 10% atomic oxygen concentration reveals only InN-related
phonon peaks, implying the lack of alloy formation [272,276]. The moral of the
aforementioned discussion is that any O present in at least polycrystalline InN that
has been examined with the role of O in mind did not seem to be incorporated as an
alloy, which explicitly leads to the conclusion that could not contribute to the bandgap.
An in-depth discussion of the topic can be found in Ref. [267].
Moss–Burstein effect has been forwarded as a plausible argument for observing
1.8–2 eV absorption if the 0.7 eV bandgap is assumed. This shift occurs when carrier
2.9 InN
concentration is above the Mott critical density, meaning larger than the conduction
band density, that is, the Fermi level lies in the conduction band. In such a case,
electrons fill the bottom of the conduction band so that the apparent bandgap
measured by optical absorption is increased by an amount of extension of the Fermi
level. As a result, extra photon energy is required to excite electrons from the top of the
valence band to the Fermi-level position within the conduction band suggested by
Trainor and Rose [277]. But the required large electron concentrations produces
bandgap renormalization, which is in competition with the Moss–Burstein effect.
High doping concentrations cause band tailing effect, which acts to reduce the
apparent bandgap. Layers of InN with large variation in electron concentration have
been reported along with the measured bandgap, as compiled in Figure 1.35, as
function of electron concentration. The measured bandgap is the convolution of
Moss–Burstein shift and bandgap renormalization culminating in the measurement
of EG þ EF, the EF being measured with respect to the conduction band edge. If the
plot is limited to MBE films only, the data appear to support the argument for small
bandgap and variation attributed to Moss–Burstein shift. However, when all the data
in films prepared by any growth method in aggregate are plotted, there seems to be
quite a scatter, including large bandgap associated with relatively lower impurity
levels.
The data basically do not show any trend in that a good many of the samples with
high and low electron concentration both do exhibit the large bandgap value. The
critical data having to do with the samples of relatively lower electron concentrations
are associated with compensated samples. This is an important issue in that material
compositional, or stain-related nonuniformities may also hamper efforts to determine the bandgap accurately. A critical view may raise question about the quality of
the samples. What is clear is that the bandgap of the nondegenerate InN is still
unknown and will require considerably more investigation [272].
What about some yet unknown defect being responsible for or the source of the
0.7 eV emission and absorption? Ask Butcher [267] who considered this possibility
and provided arguments backed by experimental data in support of it. Butcher [267]
suggested that the evidence for a 0.7 eV bandgap is also consistent with the presence
of a 0.7 eV deep-level trap in that the available absorption data for the best published
apparently 0.7 eV material (without the Moss–Burstein shift due to low carrier
concentrations) exhibit an energy dependence consistent with a deep-level trap of
|si-like orbital symmetry and was inconsistent with direct band-to-band transitions.
Butcher [267] also made the case that the slope dependence of absorption coefficient
plots is consistent with variations in the density of such a deep-level trap. Further, the
0.7 eV material showed that emission to be emanating from regions of indium-rich
aggregates [270]. If one wants to be skeptical, one could argue that it is likely that the
said emission could be a result of surface states at the metal–semiconductor interface
rather than being associated with the InN band edge. It was also shown that material
grown without such aggregates had an absorption edge nearer 1.4 eV [278]. The early
InN data were produced by N-rich material, whereas the 0.7 eV material is grown
under In-rich conditions and the same probably holds for OMVPE-grown layers. The
early samples grown under N-rich conditions exhibited a higher than usual unit cell
j189
j 2 Electronic Band Structure and Polarization Effects
190
volume [279]. Such variation in the unit cell volume with stoichiometry would be
consistent with that observed for GaN [280]. Being forced to grow InN at very low
temperatures, typically below 550 C, with the caveat that the temperature measurements are not absolute, the driving force for desorption of excess In is not all that
potent. The potency of In droplet formation has been noted by Yamaguchi et al. [281]
who indicated that as little as a 20 C increase in temperature from optimum growth
conditions can result in indium droplets. Clearly the stoichiometry, specifically In
inclusion issue, is something that would need to be dealt with.
With the evidence for indium-rich aggregates in the InN matrix and suggestion
that they are responsible for the 0.7 eV luminescence properties of InN, the data of
Wu et al. [282] has been discussed under a different light [267]. The samples
investigated by Wu et al. had undergone an irradiation with 2 MeV protons to a
dose of 2.23 · 1014 protons cm2 that resulted in a factor of 2 increase in the intensity
of 0.7 eV PL peak, which was interpreted as radiation hardness. This increase in PL
peak intensity can be ascribed to increased density of radiative defects, as has been
well documented for III–V materials by Lang [283]. This is typically accompanied by
band-edge photoluminescence that is either quenched or left unperturbed by
radiation damage. Another interpretation of the observations of Wu et al. [282] might
therefore be an increase in radiation-induced defects although not being promoted [267]. An issue is the fact that InN is particularly susceptible to nitrogen loss when
bombarded with ions [284–287], leading to possible indium-rich aggregates. The
aforementioned discussion lays the groundwork for a plausible connection between
the 0.7 eV peak and In-rich aggregates, but does not quite attempt to discuss the
nature of that emission.
Shubina et al. [288] and also Ivanov et al. [278] attempted to do just the same,
meaning shed some light into the nature of the 0.7 eV emission. The efforts of those
authors utilizing microcathodoluminescence studies coupled with imaging of
metallic In have shown that bright infrared emission at 0.7–0.8 eV arises in the
close vicinity of In inclusions and is likely associated with surface states at the
metal–InN interfaces. Employing thermally detected optical absorption (TDOA)
measurements, Shubina et al. [288] suggested a bandgap near 1.5 eV, reserving a
more definitive judgment until after more accurate measurements could be performed. Shubina et al. [288] have actually broadened the range of samples, examining
various substrates including sapphire by choosing two representative sets of InN
epilayers grown by both plasma-assisted molecular beam epitaxy and organometallic
VPE methods. The dominant IR emission in these samples were observed to be in the
range of 0.7–0.8 eV, independent of the growth technique used to prepare them and
of excitation, such as optical with different laser lines, or by an electron beam in
conjunction with a CL performed at 5 K in an analytical scanning electron microscope. No correlation between the IR emission and the electron concentration, which
ranged from 2.1 up to 8 : 4 · 1019 cm3 (determined from IR ellipsometry measurements using an effective electron mass of me ¼ 0 : 11m0) [234] and measurement
temperature was discernable, consistent with other reports [289]. Not all the 18
samples studied emitted light, and all the samples emitting IR PL emission did
contain In-rich aggregates. Analytical microscopy sensitive to atomic weight in
2.9 InN
backscattered electrons (BSE) geometry, and energy dispersive X-ray (EDX) analysis
along with CL were employed to establish a definite correlation between the In-rich
aggregates and IR PL emission. Again, the bright 0.7–0.8 eV IR emission in both
MBE and OMVPE sets of the samples was found to be associated with the In
aggregates.
Total optical extinction losses in a semiconductor matrix with metallic clusters have
been established. In addition to the interband absorption in the matrix, those losses
contain two additional components, namely, a bipolar absorption of radiation energy
and its conversion into heat in small particles, and resonant scattering on plasmon
excitations, which is important for larger particles [290]. Both characteristic components have been observed in the optical spectra obtained by Shubina et al. [288]. Those
authors also observed emission in the range of 0.8–1.4 eV that was attributed to the
scattered background signal, the root cause of which was most likely associated with
nonchromatic spontaneous emission of the semiconductor laser used or the fluorescence of all optical components at high excitation power levels. Owing to electron
beam excitation, the scattered signal was absent in the CL spectrum. Unless the
aforementioned spurious signals are accounted for, erroneous conclusions could be
drawn. Citing the absence of such signals, Shubina et al. [288] employed thermally
detected optical absorption technique performed at 0.35 K. The method is based on
the detection of a small increase in the sample temperature caused by phonons
produced by nonradiative recombination processes as a result of optical absorption
and bipolar absorption of light in In-rich aggregates. The TDOA spectra contained
additional peaks below the principle absorption edge in films containing In-rich
aggregates but not others, owing to absorption within those aggregates. The
sharpness of the observed feature is related to the resonance at the extremely low
measurement temperature that prevents thermal broadening induced by electron
acoustic phonon scattering [288]. A natural conclusion of the aforementioned
observations is that the strong IR absorbance is most likely associated with Mie [291]
resonances due to scattering or absorption of light in InN-containing In aggregates or
metallic In clusters.
In the Mie theory [291], the extinction losses for a metallic sphere depend on the
complex dielectric functions of both the matrix material, e, which is InN in this case,
and metal, em, which is In in this case. Consequently, the resonance energy of the In
clusters in InN matrix with a high-frequency dielectric constant of e0 ¼ 8.4 (based on
stoichiometric InN, which is the case here as the InN films studied were In-rich) is
considerably smaller than that in vacuum. For a treatment of the dielectric constant in
InN, the reader is referred to Ref. [292]. Shubina et al. [288] argued that the In
inclusions are predominantly formed either between columns or initiated at the
interface with the substrate. Not knowing the shape and density of those clusters
accurately, the authors employed a model developed for nonspherical metal particles
in an absorbing matrix [293], which is based on the Maxwell–Garnett approximation
for an effective medium [294], to demonstrate that the resonance absorption energy
in the InN–In composite can shift down to the IR range depending on the In content
and the shape of the In clusters. A lack of accurate knowledge of the InN complex
dielectric constant and the specifics of In clusters prevented the authors to arrive at a
j191
j 2 Electronic Band Structure and Polarization Effects
192
shape and density of In clusters responsible for the IR absorption. Suffice it to say that
given the available parametric data, it is very plausible that the observations are
related to Mie resonance absorption caused by In-rich aggregates in the InN matrix.
Clearly, additional experiments are imperative and will surely be available in due
time. Even with the reported available data and analysis, Shubina et al. [288] argue that
it is unlikely that the bandgap of InN is at 0.7–0.8 eV range.
Confirming to a large extent the results obtained in InN reported in 1980s, using
epitaxially grown wurtzite InN, Guo and Yoshida [184] measured low-temperature
and room-temperature gaps of 1.994 and 1.97 eV, respectively, along with Varshni
parameters of a ¼ 0.245 meV K1 and b ¼ 624 K. Estimates of the crystal field
splitting in wurtzite InN range from 17 to 301 meV [66,107,163]. A value of 40 meV
can be adopted. On the basis of the calculation, spin–orbit splittings vary from 1 to
13 meV [107,163], but a value of Dso ¼ 5 meV is recommended by Vurgaftman and
Meyer [152].
Ironically, the small bandgap of InN actually goes against the long held cation rule,
in which for isovalent, meaning common-cation semiconductors, the direct gap at
the G point increases as the anion atomic number decreases. This implies, for
example, that the bandgap of InN should be larger than that of InP, which is 1.4 eV,
which is consistent with the values of 1.5 eV and higher reported for InN. It should,
however, be stated that the breakdown of the common-cation rule is not unusual in
ionic semiconductors. This is articulated in an interesting observation where in
Nag [246] pointed out that this gap is unusually low in the context of trends exhibited
by other semiconductor materials. Fore example, the bandgap of ZnO is also smaller
than that of ZnS. This unexpected behavior has been attributed to two effects. Wei
et al. [295] argued that a much lower 2s atomic orbital energy of N (18.49 eV)
compared with P (14.09 eV) and other group V elements lowers the conduction
band minimum at the G point. Moreover, the smaller bandgap deformation potential
of InN (3.7 eV) in comparison to InP (5.9 eV) weakens the atomic size effect. The
atomic size effect is the one that forms the basis of the common-cation rule. Similarly,
the effect of the lower orbital energy of O as compared to the other group VI elements
such as S is responsible for ZnO bandgap being smaller than that of ZnS. The
controversy on the theory side matches that of the experiments. It could be argued
that in the end carefully thought of experimental evidence will carry the day and
theories with appropriate approximations will be developed to support the general
direction of experiments.
Tsen et al. [296] studying nonequilibrium optical phonons in a high-quality singlecrystal MBE-grown InN with picosecond Raman spectroscopy reached the conclusion that their results are not consistent with the large bandgap of InN. The basis for
this argument is as follows: above gap photons cause creation of electron and hole
pairs that very quickly relax toward the bottom/top of conduction/valence band by
emitting phonons. For wurtzitic InN, using me ¼ 0:14 me [297], mh ¼ 1:63 me [298],
hwL ¼ 2:34 eV, and Eg ¼ 1.89 eV gives an index of refraction n ¼ 3.0 (measured by
Tsen et al. [296]) and hwLO ¼ 73:4 meV (corresponding to A1(LO) phonon mode
energy); the phonon wave vector in the experiments of Tsen et al. [296] is q ¼ 2nkL
7.08 · 107 m1, where kL is wave vector of the excitation laser with photon energy
2.9 InN
2.34 eV. Owing to a much larger associated hole mass, the phonons emitted by holes
can be ignored. In a parabolic band, excess electron energy is given by
mh
DE ¼ ðhwL E g Þx
ffi 0:41 eV;
ð2:44Þ
me þ mh
which is about five times the LO phonon energy hwLO ¼ 73:4 meV, which means that
the energetic electrons are capable of emitting five LO phonons during their
thermalization to the bottom of the conduction band. However, because of energy
and momentum conservation during the electron–LO phonon interaction, there
exists a range of LO phonon wave vectors that electrons can emit. As depicted in
Volume 2, Figure 3.7, for an electron with wave vector ~
ke and excess energy DE, the
minimum and maximum LO phonon wave vectors it can emit are given by [299]
pffiffiffiffiffiffiffiffiffi
2me pffiffiffiffiffiffi pffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
ð DE DE hwLO Þ;
kmin ðor qmin Þ ¼
h
and
kmax ðor qmax Þ ¼
pffiffiffiffiffiffiffiffiffi
2me pffiffiffiffiffiffi pffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
ð DE þ DE hwLO Þ:
h
ð2:45Þ
Because of the nature of E–k relationship of the electron, the lower the electron
energy, the larger kmin (also referred to as qmin) and the smaller kmax (also referred to
as qmax) are. Therefore, at some electron energy, kmin of the LO phonon will be larger
than the wave vector probed by the Raman scattering experiment, which is q ¼ 7.08
· 107 m1. When this occurs, the energetic electrons can no longer emit LO phonons
with wave vectors detectable in the Raman scattering experiments. These conditions
corresponds to kmin ffi 1.1 · 108 m1 and kmax ffi 2.4 · 109 m1 in the experiments of
Tsen et al. [296]. Although the effect of nonparabolicity of the conduction band for
InN is currently not known, the general trend is that the effective electron mass
increases with the increase in energy in the conduction band for a typical semiconductor. By taking the nonparabolicity into account, the detectable kmin can be revised
as being larger than 1.1 · 108 m1. Consequently, no nonequilibrium A1(LO) phonon
population should be detected in the Raman experiments of Tsen et al. [296] when the
effect of nonparabolicity of the conduction band is considered. This means that if the
bandgap energy of InN were 1.89 eV, there would be no detectable nonequilibrium
A1(LO) phonon population with the excitation laser photon energy of 2.34 eV. This
contradicts the fact that nonequilibrium A1(LO) phonons have been observed and
argues against the large InN bandgap.
A similar argument can be applied to the scenario that the bandgap of InN is
ffi0.8 eV, in the case of which the same laser photon energy (2.34 eV) can excite the
electrons up in the conduction band so that it would take 6 A1(LO) phonons to be
emitted for full thermalization. No A1(LO) phonons should be detected with 1.96 eV
laser excitation. Both of the above arguments are consistent with the experimental
observations. Earlier, it was stated that the proponents of the larger bandgap InN
argue that the small bandgap reported for InN could be due to incorrect attribution of
the deep-level emission to the band-edge emission as defects capture electrons or
j193
j 2 Electronic Band Structure and Polarization Effects
194
holes and emit photons (through a radiative relaxation process) of lower energy than
the bandgap. This capture process can also emit phonons through a nonradiative
relaxation process. Let us for a moment suppose that the 0.8 eV luminescence reported
in the literature is due to such a capture process by some unknown deep-level defect(s)
in InN. If so, the observations of Tsen et al. [296] of nonequilibrium A1(LO) phonons in
InN with excitation laser having photon energy 2.34 eV suggests that nonradiative
relaxation processes also play a role in the capture. However, due to deep-level defects
having very localized wave functions, their momenta are widely spread due to
the uncertainty principle. This suggests that electron–phonon interaction during the
capture process does not need to conserve momentum. Therefore, phonon wave
vectors of almost every magnitude can be emitted. In other words, the defect-model
predicts that if one detects nonequilibrium A1(LO) phonons with 532 nm excitation
then one should also detect nonequilibrium A1(LO) phonons with 634 nm excitation.
This is in contradiction with the experimental observation of Tsen et al. [296]. In short,
the nonequilibrium phonon experiments are consistent with the 0.8 eV bandgap.
In a renewed effort to determine the fundamental gap of InN, Arnaudov et al. [249]
studied the shape and energy position of near band edge photoluminescence spectra
of InN epitaxial layers with different doping levels. They implied that samples with
high doping concentration have been used to infer the bandgap of InN without
properly accounting for the effects of band filling, band nonparabolicity, and
electron–electron and electron–impurity interactions. In addition to usual difficulties
associated with highly doped samples, another aggravating factor is a clear lack of
convergence in the value of the electron effective mass. Properly accounting for the
aforementioned effects, Arnaudov et al. suggest a bandgap of Eg ¼ 692 2 meV for an
effective mass at the conduction band minimum mn0 ¼ 0.042m0. They also argue that
the value of Eg ¼ 0.69 eV reported in Ref. [270] extracted from the absorption and
photoluminescence spectra of samples with carrier concentration n > 6 · 1018 cm3
within the band-to-band recombination model while taking into account the
Burstein–Moss shift and bandgap renormalization due to many-body effects agrees
with the data; the value used for the effective mass at the bottom of the conduction
band, mn0 ¼ 0.1m0, is inconsistent with the universal Kanes relation (mn0; Eg). This
relation predicts a bandgap energy of 1.7 eV for mn0 ¼ 0.1m0, and to obtain near to
0.7 eV bandgap one must use a much smaller effective mass of mn0 ¼ 0.042m0.
Interestingly, the same value of Eg ¼ 0.69 eV was obtained by fitting the absorption
spectra, this time in a lightly doped sample with n ¼ 3.5 · 1017 cm3, with a sigmoidal
function that includes only the band tailing effect and does not involve any value for the
effective mass [300]. Taking advantage of improved crystalline quality and utilizing
samples with electron concentrations in the range of 7.7 · 1017–6 · 1018 cm3
Arnaudov et al. [249] undertook the task of determining the bandgap on InN from
optical data but with the interpretation of the emission spectra in such highly
conducting layers in terms of the free-electron recombination band (FERB) model,
which has been previously reported in the context of GaAs [301,302], InP [303],
InSb [304], and GaN [305]. By analyzing not only the emission energy position but
also the shape of the spectra simultaneously and taking into consideration the
specifics associated with both high and low electron concentrations, Arnaudov
2.9 InN
E
1.0
Ec
0.8
PL intensity (au)
n = 1.7 × 1018cm–3
E F = E Fn
Degenerate band
tails
G* = E
0.6
Ev
0.4
n = 6 × 1018 cm–3
Fp
gn ,gp
0.2
Eg -G2*Eg-G1*
Eg EF2
0.0
0.5
0.7
0.6
EF 3
0.8
Energy (eV)
Figure 2.13 Experimental, depicted with
symbols, and calculated, depicted with solid
lines, PL spectra of samples with 1.7 · 1018 and
6.0 · 1018 electron concentrations as measured
by Hall effect. The inset schematically depicts the
recombination mechanism between the
degenerate electrons in the conduction band
DOS to the level G* in the valence band tails as
relied on in modeling. The energy positions
representing the best fits for Eg, EF, and the
intrinsic, or the unperturbed, bottom of the
conduction band Eg–G* are also indicated [249].
et al. [249] are able to determine the fundamental bandgap for the electron effective
mass in InN as Eg ¼ 692 2 meV for an effective mass at the bottom of the
conduction band mn0 ¼ 0.042m0, which is consistent with Kanes relation.
PL spectra of two samples with electron concentrations of 1.7 · 1018 cm3 6 · 1018
cm3, as determined by Hall measurements, by Arnodov et al. [249] are shown in
Figure 2.13 (points)). Both samples exhibit a broad emission band with a maximum at
685 meV and 705 meV, respectively, the former for the lower doped and the latter for
the higher doped sample due to a larger Burstein–Moss shift. Noteworthy, however, is
that the emission band of the sample with a higher Hall concentration is broader and
more asymmetric, and its PL peak is at a higher energy due to the larger Burstein–Moss shift. The low-energy side of the spectral band of the sample with higher
electron concentration shifts to lower energy compared to the sample with lower Hall
concentration, which tends to narrow the apparent optical bandgap, consistent with
emission spectra from highly doped semiconductors. In addition, these observations
are characteristic of free to bound recombination of degenerate conduction electrons
with nonequilibrium valence holes in the valence band tail, as shown in the inset of
Figure 2.13 [301–305]. Moreover, the shape of the emission bands follows the energy
distribution of electrons in the conduction band, all the while their energy positions
are determined by the interplay of the equilibrium Burstein–Moss shift (blue shift)
and the effective bandgap renormalization (red shift).
j195
j 2 Electronic Band Structure and Polarization Effects
196
To deduce the bandgap energy Arnaudov et al. [249] fit the experimental emission
spectra with that obtained from the general expression for the intensity versus the
photon energy I(hn) given by (neglecting the energy dependence of the probability for
radiative transitions, as was done for heavily doped GaN [305])
ð¥ ð¥
IðhnÞ
g n ðE Fn Þf n ðE n E Fn Þg p ðE p Þf p ðE p E Fp ÞdðE n E p hnÞdE n dE p ;
00
ð2:46Þ
where gn(En) and gp(Ep) are the density of states in the conduction and valence bands
at electron and hole energies En and Ep, respectively. The terms fn and fp represent the
associated nonequilibrium Fermi–Dirac functions, and EFn and EFp are the quasiFermi levels for electrons and holes. The conduction band density of states gn(En), as
well as the electron effective mass mn(En), can be calculated in the framework of
Kanes two-band kp model as described in Ref. [300]. Although the bandgap
information is implicit in the En Ep term of the delta Dirac function, the bandgap
term Eg is sometimes explicitly subtracted from En Ep for emphasis, as done by
Arnaudov et al. [249]. The reader is referred to Volume 3, Chapter 2 for a detailed
discussion of spontaneous emission intensity calculations.
Because the nonequilibrium holes are situated in a relatively narrow energy window
deep in the band tails, they do not significantly affect the spectral distribution of the
emitted light. Therefore, the quantity gn(En) fn(En EFn) in Equation 2.46 roughly
reproduces the shape of the FERB shown in the upper part of the inset in Figure 2.13.
Likewise, the term gp(Ep) fp(Ep EFp) in Equation 2.46 determines primarily the energy
position of the emission band associated with the unperturbed fundamental bandgap
Eg via Equation 2.47, as depicted in the lower part of the inset in Figure 2.13. It should be
pointed out that the FERB model includes a calculation of the spectral shape as well as
analytical renormalization of the bandgap due to the presence of ionized impurities. In
this case, the energy positions of both the low- and high-energy slopes of the emission
band are sensitive to the electron concentration induced by ionized impurities.
In conjunction with the nonequilibrium Fermi–Dirac function of electrons, fn,
Arnodov et al. [249] used the Fermi level EFn ffi EF corrected for the temperature of
electrons y, which can differ from the lattice temperature T, electron–electron and
electron–impurity interactions [305], and the nonparabolicity of the conduction band
density of states (DOS), which can be determined in framework of the two-band kp
model. The valence band DOS gp(Ep) is replaced by a Gaussian, determining the tails
deep in the bandgap through the root mean square (rms) impurity potential G as
detailed in Ref [305]:
pffiffiffi 4pe2
ðN i R3s Þ1=2 ;
G¼2 p
eRs
with Rs ffi
ð2:47Þ
aBe
e
h2
ðna3Be Þ 1=6 and aBe ffi
;
2
4pe2 4pmn0
where the terms e represent in order the electric permittivity, Rs the Thomas–Fermi
screening length, Ni ¼ [(1 þ K)/(1 K)]n the total ionized impurity concentration,
2.9 InN
K the compensation ratio, n the extrinsic electron concentration, and aBe the effective
Bohr radius of electrons. The value of Rs is smaller than aBe and thus the equilibrium
and nonequilibrium degenerate electrons are free above the bottom of the conduction band [305]. The situation with the nonequilibrium holes is quite different in that
in III–V materials the effective Bohr radius aBh is much smaller than Rs due to the
relatively large hole effective mass mp. Moreover, holes are classically localized, at
least for not extremely high impurity concentrations and high temperatures [306], at
the potential minima
of the valence band tails near the thermal equilibrium level
pffiffiffi
G ¼ E v þ 2G kT=2. As shown for heavily doped GaN [305], the level G* plays
the role of the quasi-Fermi level in the nonquasi-equilibrium recombination FERB
model. Thus, we can replace the value of EFp in the Fermi–Dirac function for holes fp
can be replaced by G*, meaning one case set EFp ¼ G*.
To comment on the electron effective mass, the FERB emission spectra of samples
with 1.7 · 1018 and 6.0 · 1018 cm3 Hall electron concentration can be calculated with
varying n, y, and K and using the value of mn0 ¼ 0.042m0 suggested in Ref. [307]. For
the relative static permittivity, a value of e ¼ 14.61 was used by Arnaudov et al. [249].
Assuming a zero-compensation ratio, which is a good first-order approximation,
the best fits of the spectra are obtained with bandgap values of 690 and 692 meV, for the
sample with 1.7 · 1018 cm3 electron concentration and the sample with 6.0 · 1018
cm3 electron concentration, respectively. To reconcile the small difference in the
aforementioned bandgap values, a small compensation ratio of K ¼ 0.06 for sample
with 1.7 · 1018 cm3 electron concentration and K-value of 0.01 for sample with
6.0 · 1018 cm3 electron concentration was introduced. Doing so resulted in a
bandgap valued of Eg ¼ 692 meV for both samples. The best fit values for the two
samples are shown in Figure 2.13 (solid lines). It should be noted that best fit values of
the electron concentration nopt are noticeably lower than those deduced by the
measured Hall effect. This dispersion may perhaps be related to the inhomogeneities
in the films. In spite of this both represent degenerate cases because the Motts
transition concentration is estimated to be about nMott ¼ 5 · 1016 cm3 for mn0 ¼ 0.042
m0. The calculated curves agree very well with the experimental spectra, with
the exception for the low-energy range. In this region, an additional contribution
from a deeper emission center could in principle be possible, which is not included
in the model. In the high-energy portion of the spectrum, the accuracy is more
reliable.
To summarize the above discussion, using the shape as well as the energy position
of the near band edge PL spectra of InN epitaxial layers with different doping levels,
Arnaudov et al. [249] concluded that the radiative transition is between the degenerate
electrons in the conduction band and nonequilibrium holes in the valence band
tails and that the fundamental bandgap of InN is Eg ¼ 692 2 meV for an effective
mass at the conduction band minimum of 0.042m0, which is consistent with the Kane
model. The optical transmission and reflection data obtained from a high-quality
InN film grown by MBE with N-polarity support the smaller bandgap figures of
InN as shown in Figure 2.14. The growth details and transport properties of InN
layers similar to the one that led to the data presented in Figure 2.14 are discussed in
Section 3.5.13.
j197
j 2 Electronic Band Structure and Polarization Effects
198
70
100
80
65
60
60
40
55
20
Reflection (%)
Transmission (%)
E g = 0.7– 0.75 eV
50
0
45
1.0
1.5
2.0
2.5
3.0
3.5
Wavelength (μm)
Figure 2.14 Optical reflection and transmission data obtained in
an N-polarity InN film grown by MBE indicative of a bandgap
between 0.7 and 0.75 eV. Courtesy of A. Yoshikawa.
As for the true value of bandgap of wurtzitic InN, although the data for small
bandgap data are convincing and the arguments for the small bandgap are compelling, for some reason, some controversy remains. This controversy is expected to
evaporate when and if the large bandgap observed is earlier and some InN samples
are explained satisfactorily. We should reiterate that the data in high-quality samples
converge on the small bandgap. However, there is still some dispersion in the exact
bandgap value that seems to be between 0.65 and 0.8 eV. In this chapter, the
arguments for both the large and small bandgap as well as the pitfalls for each in
terms of the reliability of the data and a historical review are provided for the reader to
be abreast with the conflicting issues surrounding the matter.
Turning our attention momentarily to other electronic properties affected by the
band structure, measurements of the electron effective mass in wurtzitic InN
produced values of 0.11m0 [234], 0.12m0 [308], and 0.14m0 [297], as well as
0.24m0, for the mass perpendicular to the c-axis [309]. Kasic et al. [297] used infrared
spectroscopic ellipsometry and micro-Raman scattering to study vibrational and
electronic properties of wurtzitic in 0.22 mm thick InN layers grown by RF MBE, as
well as Hall effect measurements, and arrived at the isotropically averaged effective
electron mass of 0.14m0. The mass value of 0.14m0 closely matches at least one
theoretical projection [53]. It should be mentioned that all the InN films used for these
investigations featured very high electron concentrations, which are endemic to InN,
in the 1018 cm3 or higher, and causes the Fermi level to degenerate well in the
conduction. Consequently, any nonparabolicity in the conduction band would affect
the effective mass measurements. However, the realization that the InN bandgap is
narrower than that previously thought prompted a reexamination of the effective
mass issue also [310]. Accounting for the substantial nonparabolicity that can cause
2.9 InN
an overestimate of the mass because high doping leads to a band-edge effective mass
of 0.07m0, which is what is recommended here as was done by Vurgaftman and
Meyer [152]. Turning our attention to holes, valence band mass parameters have been
calculated by Yeo et al. [67] using the empirical pseudopotential method, and also by
Pugh et al. [163] and Dugdale et al. [153] using more or less the same technique. The
results of the first two investigations are quite similar, Pugh et al. [163] employed
three different levels of computation comprising first-principles total energy calculations, semiempirical pseudopotential calculations and kp calculations. Band structures were obtained from each method in a consistent manner and were used to
provide effective masses and kp parameters. These parameters are useful in
investigating the electronic structure of alloys and quantum well heterostructures.
These valence band parameters are the recommended values with the caveat in that
the lower InN energy gap may require a downward revision of the light-hole mass.
The parameters concerning the bandgap-related issues for wurtzitic InN recommended by Vurgaftman and Meyer [152] are tabulated in Table 2.19.
A compilation of the dispersion in the effective mass for both the conduction band
and various valence bands as obtained by various computational methods, as well as
parameters used in the description of the bandgap for wurtzitic InN, particularly, in
the context of empirical pseudopotential method, as described in Ref. [55], are
tabulated in Table 2.20.
Christensen and Gorczyca [31] predicted a hydrostatic deformation potential of
4.1 eV for wurtzite InN, which compares to a smaller value of 2.8 eV calculated by
Kim etal. [180].Inthe absence of any predilection forany of the tworeports, averaging the
two leads to a ¼ 3.5 eV. In the absence of any calculations of the valence band
deformation potentials, appropriating the parameter set specified above for GaN could
be a good default at this point. The elastic constants measured by Sheleg and Savastenko [81], early on more refined values arrivedby calculations suchasthe set reported by
Wright [179], are available, which are C11 ¼ 223 GPa, C12 ¼ 115 GPa, C13 ¼ 92 GPa,
Table 2.19 Recommended band structure parameters for wurtzitic InN [152].
Parameter
Value
Parameter
Value
Parameter
Value
Eg (eV, low temperature)
a (meV K1)
b (K)
Dcr (meV)
Dso (meV)
==
me =m0
m?
e =m0
a1 (eV)
a2 (eV)
1.5–1.8
0.245
624
0.040
0.005
0.07
0.07
3.5
3.5
A1
A2
A3
A4
A5
A6
A7 (meV Å)
d13 (pm V1)
d33 (pm V1)
d15 (pm V1)
Psp(C m2)
8.21
0.68
7.57
5.23
5.11
5.96
0 (default)
3.5
7.6
5.5
0.042 (0.041)
D1 (eV)
D2 (eV)
D3 (eV)
D4 (eV)
D5 (eV)
D6 (eV)
c11 (GPa)
c12 (GPa)
c13 (GPa)
c33 (GPa)
c44 (GPa)
3.7
4.5
8.2
4.1
4.0
5.5
223
115
92
224
48
See Tables 2.27 and 2.28 for details related to the elastic constants, piezoelectric constants, and
spontaneous polarization charge. Any dispersion among the tables is a reflection of the
uncertainty in the available parameters.
j199
j 2 Electronic Band Structure and Polarization Effects
200
Table 2.20 Effective masses and band parameters for wurtzitic InN.
Parameter
==
me
m?
e
==
mhh
==
mlh
==
mch
m?
hh
m?
lh
m?
ch
A1
A2
A3
A4
A5
A6
A7
D1
anisotropic
isotropic
A
B
C
D
0.138
0.141
2.438
2.438
0.140
2.661
0.148
3.422
7.156
0.244
6.746
3.340
3.208
4.303
0.072
0.214
0.137
0.140
2.493
2.493
0.137
2.599
0.157
1.446
7.298
0.441
6.896
3.064
3.120
3.948
0.103
0.084
0.11
0.10
1.56
1.56
0.10
1.68
0.11
1.39
9.62
0.72
8.97
4.22
4.35
0.11
0.10
1.67
1.67
0.10
1.61
0.11
1.67
9.28
0.60
8.68
4.34
4.32
6.08
0
0.10
0.10
1.431
1.431
0.106
1.410
0.196
0.209
9.470
0.641
8.771
4.332
4.264
5.546
0.278
0.0375
0.10
0.10
1.350
1.350
0.092
1.449
0.165
0.202
10.841
0.651
10.100
4.864
4.825
6.556
0.283
0.0375
0
Effective masses in units of free-electron mass m0, Luttinger-like parameters Ai (i ¼ 1, . . ., 6) in
units of h2 =2m0 , and A7 in units of eV Å. The crystal field splitting energy D1 is given in units of
meV. The term aniso represents the values derived using a band structure calculation with
anisotropically screened model potentials, whereas the term iso describes a comparative band
structure calculation on the basis of isotropically screened model potentials using an averaged e0
value by taking the spur of the dielectric tensor [55]. Anisotropically screened and isotropically
screened values are from Ref. [55]. A: effective masses and Ai are from Ref. [67] obtained through
a line fit to the band structure; B: direct kp calculation in a 3D fit from Ref. [67]; C: Ai from
Ref. [153] obtained through a Monte Carlo fitting procedure to the band structure and effective
masses calculated using Equations 2.30 and 2.31; D: direct kp calculations for Ai from Ref. [153]
and effective masses calculated using Equations 2.30 and 2.31.
C33 ¼ 224 GPa, and C44 ¼ 48 GPa. Other sets of parameters calculated by Kim et al. [180]
and Davydov [201] are also available in the literature. For a more detailed discussion of
elastic and piezoelectric coefficients as well as polarization issue, refer to Section 2.12
and Tables 2.25–2.27 and 2.28. Owing to the fact that the piezoelectric coefficients in InN
have apparently not been measured, for consistency the theoretical values of Bernardini
and Fiorentini [210], d33 ¼ 7.6 pm V1, d13 ¼ 3.5 pm V1, and d15 ¼ 5.5 pm V1, are
suggested. In spite of the fact that the spontaneous polarization data for GaN/GaInN
structures are not as conclusive as one would like at this point, most likely owing to
relatively poor material quality, the value Psp(InN) ¼ 0.042 C m2 is consistent with a
thorough comparison of experiment and theory [84]. Recommended band structure
parameters for wurtzite InN are compiled in Table 2.19 [152].
2.9.2
Zinc Blende InN
Experiments on zinc blend InN are very rare although this polytype has been
reported [311]. The bulk of the reports comprise theoretical estimates of its band
2.9 InN
Table 2.21 Luttinger parameters g1, g2, and g3 for zinc blende InN
obtained from a fit along the [1 1 0] direction along with those
available in the literature, as compiled in Ref. [55].
Parameter
A
B
c1
c2
c3
7
0.97
1.22
3.27
1.26
1.63
A: empirical pseudopotential calculation by Fritsch et al. [55]; B: recommended values taken from
Ref. [152].
parameters. The zinc blende variety has been projected to have a direct band
alignment, with G-, X-, and L-valley gaps of 1.94, 2.51, and 5.82 eV, respectively [70].
However, this particular calculation was performed before the controversy in the
bandgap of wurtzitic InN. If the arguments available in the literature and
presented in the Section 2.9.1 dealing with wurtzitic InN were to hold in favor
of the large bandgap, then the aforementioned bandgap values will be more
credible. For the valence band, spin–orbit splittings in the range 3–13 meV
have been projected [107,171,227]. Vurgaftman and Meyer [152] recommends
the 5 meV value among them. As in the case of the bandgap, the effective mass
value for the wurtzitic polytype is recommended for the zinc blende variety, that is,
0.07m0. As mentioned in the previous section, this value is arrived at after the
nonparabolicity effects are accounted for. To reiterate, the range of values reported
for the Wz InN is 0.10–0.14m0 [70,163,173]. The longitudinal and transverse
masses for the X valley have been calculated to be 0.48m0 and 0.27m0, respectively [70]. The recommended Luttinger parameter set by Vurgaftman and Meyer [152]
is g1 ¼ 3.72, g2 ¼ 1.26, and g3 ¼ 1.63, which is derived from the work of Pugh
et al. [163], and the split-off mass is chosen to be mso ¼ 0.3m0 [70,163]. These as
well as the other literature values of the Luttinger parameters for zinc blende InN
are listed in Table 2.21.
Fritsch et al. [55] calculated the effective masses for conduction and valence bands,
the latter involving the light and heavy holes, as well as the spin–orbit split-off mass,
which along with the anisotropy taken into account are listed in Table 2.22 for zinc
blende InN. The data contain those obtained by FP-LAPW, EPM calculations, and
those calculated with Luttinger parameters Equations 2.42 and 2.43.
The EP parameter value given by Meney et al. [173] is more likely because the
alternative value by Pugh et al. [163] implies too large a value for F. The resulting
parameter set is EP ¼ 17.2 eV and F ¼ 4.36. For the hydrostatic deformation
potential, an average value of 3.35 eV from the theoretical [31,180] candidates of
2.2 to 4.85 eV has been recommended [152]. The valence band deformation
potentials listed in Table 2.19 are compiled from the calculations of Wei and
Zunger [107], Kim et al. [180], Tadjer et al. [70], and Van de Walle and Neugebauer [177]
are av ¼ 0.7 eV, b ¼ 1.2 eV, and d ¼ 9.3 eV. Elastic constants of C11 ¼ 187 GPa,
C12 ¼ 125 GPa, and C44 ¼ 86 GPa are assumed from the calculations of Wright [179],
j201
j 2 Electronic Band Structure and Polarization Effects
202
Table 2.22 Effective masses for electrons (e), heavy holes (hh),
light holes (lh), and spin–orbit split-off holes (so) in units of the
free-electron mass m0 along the [1 0 0], [1 1 1], and [1 1 0]
directions for zinc blende InN.
[100]
[100]
[111]
[111]
[110]
[110]
Reference
me
mhh
mlh
mhh
mlh
mhh
mlh
mso
A
B
C
0.13
0.12
0.10
1.18
0.83
2.18
0.21
0.16
0.89
2.89
0.83
2.29
0.19
0.16
0.93
2.12
1.55
3.10
0.20
0.15
0.79
0.36
0.30
0.30
Compiled by Fritsch et al. [55]. A: after Ref. [55]; B: calculated from recommended Luttinger
parameters from Ref. [152], using Equations 2.42 and 2.43; C: empirical pseudopotential
calculation from Ref. [70].
Table 2.23 Parameters associated with the band structure for zinc
blende InN bandgaps are for low temperature [152].
Parameter
Value
Parameter
Value
Parameter
Value
alc (Å) at T ¼ 300 K
E Gg ðeVÞ
a (G) (meV K1)
b (G) (K)
E Xg ðeVÞ
a (X) (K)
b (X) (meV K1)
E Lg ðeVÞ
a (L) (meV K1)
b (L) (K)
4.98
0.78 1.5–1.8
0.245
624
2.51
0.245
624
5.82
0.245
624
Dso (eV)
me ðGÞ
ml ðX Þ
mt ðX Þ
c1
c2
c3
mso
0.005
0.07
0.48
0.27
3.72
1.26
1.63
0.3
EP (eV)
F
VBO (eV)
ac (eV)
av (eV)
b (eV)
d (eV)
c11 (GPa)
c12 (GPa)
c44 (GPa)
17.2
4.36
2.34
2.65
0.7
1.2
9.3
187
125
86
which are similar to other calculated sets [180,181]. The recommended band
structure parameters for zinc blende InN are compiled in Table 2.23.
2.10
Band Parameters for Dilute Nitrides
Dilute nitrides can be described as conventional III–V compound semiconductors,
wherein a small N fraction on the order of a small percentage is added. Addition of
even very small amounts of N causes substantial changes to the bandgap and the
lattice contact of the host compound to the point that standard bowing parameters for
the bandgap variation linear interpolation of lattice constants between the host
material and zinc blende GaN or GaInN do not apply. A single bowing parameter is
inadequate even if the goal is only to describe the energy gap for a relatively wide
range of compositions [312]. The technological driving force is really compelling in
2.10 Band Parameters for Dilute Nitrides
that wavelengths of interest for short and long haul communications systems can be
obtained on GaAs technology by adding small fractions of N into the GaAs or InGaAs
lattice. The motivation here is in materials incorporating only a small percentage of
nitrogen [313], because it is highly questionable whether more than 10–16% N can be
incorporated stably. Even in cases when one can, the quality is very inferior.
Annealing techniques employed to improve layer quality end up causing N segregation, reducing the fraction in the bulk. Having mentioned this, it should also be
pointed out that N isoelectronic doping of compound GaP, which has an indirect
bandgap, for LEDs [314] predates the flurry of activity aimed at imparting substantial
changes in the band structure, which is the topic of this section.
The band structure of dilute nitride compound semiconductors has been reviewed
in the literature by Vurgaftman and Meyer [152]. The treatise here follows the same
philosophy in that the N-containing GaAs followed InP is treated before segueing
onto other compound semiconductors such as those based on Sb anion. The
properties of the conduction band in dilute nitride semiconductors can be described
in terms of the band anticrossing (BAC) model [315]. It should be mentioned that a
small percentage of N usually has little effect on the valence bands. This twoparameter anticrossing model can be spun formally in terms of the many-impurity
Anderson model within the coherent potential approximation. It can also be thought
of as the interaction between a single, spatially localized N level and the conduction
band of the underlying traditional As-, P-, Sb-based compound semiconductors.
Alternatively, Lindsay et al. [316] predicted the identical fundamental bandgap by
assuming that the interaction involves a weighted average of perturbed upper states
as opposed to a single N level.
If one neglects the effect on the valence bands completely, the energy dispersion
relation for the two-coupled bands within the BAC model can be expressed as
qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
1 C
ð2:48Þ
E ðkÞ ¼
E ðkÞ þ E N
½E C ðkÞ þ E N 2 þ 4xV 2 :
2
Here, EC(k) is the conduction band dispersion of the nominal nonnitride semiconductor (e.g., GaAs in the case of GaAsN), EN is the position of the nitrogen isoelectronic
impurity level in the nonnitride semiconductor, V is the interaction potential between
the two bands, and x is the N mole fraction. The energy dispersion relation for the twocoupled conduction bands in GaAs0.99N0.01 showing the characteristic anticrossing is
plotted in Figure 2.15. As can be gleaned from the band structure treatment for
standard nitride semiconductors, any temperature dependence arises from the shift of
the conduction band dispersion EC(k) not that of the valence band. In the dilute
semiconductor case also, the bandgap is assumed to follow the Varshni formula of
Equations 2.34 and 2.35, but with EN taken temperature independent. One consequence of this assumption is a considerable weakening of the variation of the
fundamental energy with temperature [317]. The weak shift of EN with applied
pressure necessitates a fresh look at the deformation theory. The strain dependence
of the E transitions can be determined by substituting the applicable deformation
potentials of the host nonnitride semiconductor to obtain EC(k), followed by deriving
E (k) from Equation 2.48. The implementation of Equation 2.48 requires input of
j203
j 2 Electronic Band Structure and Polarization Effects
204
2.4
T = 300 K
GaAs0.99N0.01
Energy (eV)
2.2
E+
2.0
1.8
EN
1.6
EC
E–
1.4
1.2
–0.15
–0.10
–0.050
0.0
0.050
0.100
0.15
Wave vector (1 Å–1)
Figure 2.15 Conduction band dispersion relations for
GaAs0.99N0.01 at 300 K from the band anticrossing model
(BAC) [319] (solid curves). For comparison, the unperturbed
GaAs conduction band and the position of the nitrogen level are
shown as the dotted and dashed curves, respectively [152].
band parameters of the host semiconductor. Although, we do not reproduce the
nonnitride parameters in this work to preserve space, all of the required values are
tabulated in a review [92]. The more detailed kp theory can also be applied.
The BAC model can be extended to treat spin-doubled conduction bands, valence
band, and nitrogen impurity bands by modifying the eight-band kp theory [318–321].
Coupling of the nitrogen band to the X and L valleys have also been introduced [322,323] but at the expense of additional complexity, which may not be
warranted. Unless compelled, it is preferable to stick to the simple two-parameter fit
of Equation 2.48. It has been pointed out that the fixed position of the nitrogen level
with respect to the vacuum level implies a tandem shift with the valence band
maximum of the host nonnitride material. To account for the experimental observation of nonnegligible deviations from referencing to the valence band offset, a
separate nitrogen level for each host material is typically specified. Vurgaftman and
Meyer [152] suggest that the valence band offset for an unstrained dilute nitride be set
equal to that of the host semiconductor. As mentioned above, within the realm of the
BAC model, the primary effects of the nitrogen are on the conduction band. Further,
2.10 Band Parameters for Dilute Nitrides
even the 10-band model does not shift the valence band maximum in the absence of
strain or quantum confinement, while of course influencing the hole dispersion
relations. Although a finite type-I or type-II offset in strained structures have been
reported on the basis of experiments, they are not sufficiently compelling to deviate
from a null offset relative to the host in the absence of strain.
Despite its simplicity, Equation 2.48 provides a basis for describing material
properties, such as the fundamental energy gap, which are governed by the transition
from E to the top of the valence band, the temperature dependence of the gap, the
electron effective mass, and the characteristics of the upper band Eþ. It must be kept
in mind that within the theory of Lindsay et al. [316], there is not necessarily a single
well-defined Eþ band. Somewhat in similar vein, the extent to which the BAC
representation may be considered fundamentally realistic is still a matter of active
discussion [315,324]. Clearly, the BAC model considers only a single nitrogen level on
a substitutional lattice site or a narrow impurity band formed from such levels. In the
process, it neglects mixing with the L and X valleys, and more complex nitrogen
behavior in the semiconductor such as the formation of nitrogen pairs and clusters.
In contrast, the more complicated pseudopotential calculations that consider some of
these issues are computationally demanding [325–327]. In addition, the numerical
results do not lend themselves to simple formalism such as Equation 2.48. Buttressing the simpler BAC approach is the discovery by Lindsay et al. [316] that the
verifiable prediction of the dependence of the bandgap on the N content may be
unaffected by generalizing to a multiplicity of higher lying states. Let us now turn our
attention to specific dilute nitride semiconductor systems.
2.10.1
GaAsN
In conjunction with early investigations motivated by visible LED development, it has
been known that small quantities of nitrogen in GaAs and GaP form deep-level
impurities [314]. However, it has only been after the advent of nitride semiconductors
that also paved the way for investigating traditional compound semiconductors, such
as GaAs with N content definitely beyond the quantities ( 1% or more) used for
doping experiments [328,329]. On the flip side of this, reports [330] of the incorporation of small amounts of As into GaN are neither common nor easy. The presence
of As in GaN has been stated to cause modified surface reconstruction and/or act as
surfactant or be a source of dopant impurities.
Even though care must be exercised, PL measurements can be used to determine
the bandgap, provided the relative position of the particular transition energy is
known, and have been applied to GaAsN with N fractions up to 1.5% in an effort to
determine the dependence of the energy gap on N composition [331]. As compared to
other ternaries, a large bandgap bowing parameter of 18 eV was found, which for
small compositions is equivalent to a linear model [332]. Early theoretical studies
projected bandgap bowing parameters based primarily on the dilute nitride semiconductor with large N content [333–341]. Although the large bowing parameter
was originally supposed to produce a semimetallic overlap at intermediate
j205
j 2 Electronic Band Structure and Polarization Effects
206
compositions [336], more detailed investigations that followed led to reduced bowing
parameter with increasing composition [338,339], with experimental backing based
on investigations of Bi and Tu [342], who studied N compositions as large as 15%.
In discord with the above-mentioned linear dependence of the bandgap for small N
fractions, subsequent investigations pointed to a highly nonlinear reduction in the
energy gap for small N compositions [343–349]. Another noteworthy discovery was
the realization of a significant weakening of the temperature and pressure dependencies of the bandgap for GaAsN and also GaInAsN with small In fractions [346,350,351]. In aggregate, it is clear that a simple bowing approximation
could not adequately describe the GaAsN alloy. In this vein, Shan et al. [352] proposed
the band anticrossing model and confirmed a weak pressure dependence for the
nitrogen band transitions with a deduced deformation potential of 1.2 eV. To put
matters in context, the density functional calculation of Jones et al. [353,354] also
predicted reduced pressure dependence without invoking the BAC model.
Additional report supporting the BAC model has been the finding by Skierbiszewski et al. and others pointing to a significantly heavy electron mass in GaInAsN
[355–361]. On the anticorrelation side is another set of measurements by Young
et al. [362] who found a reduction in the effective mass with increasing N content, in
direct conflict with the BAC model that predicts an increase even at the zone center.
Increasing mass with increasing N content, however, appears to hold. Consistent
with the overall expected behavior of dilute nitride semiconductors based on the BAC
model, the temperature dependence of the bandgap has been confirmed to be notably
weaker in GaAsN than in GaAs [317,363]. In a similar vein, GaAsN film electroreflectance [364] resolved both the E and Eþ transitions, the band description of which
can be seen in Figure 2.15. Extending to a ternary, the bandgap reduction was also
observed in nitrogen-implanted Al0.27Ga0.73As samples [365]. The transition between
the doped and alloyed materials was studied by Zhang et al. [366,367]. Here, doped
implies quantities manifesting themselves as dopants without radical changes
incurring on the host material. The alloy material implies that the N concentration
is sufficiently high to cause substantial changes in the host material. Zhang et al.
observed evidence for impurity banding at N concentrations as small as 0.1% N.
Zhang et al. also proposed an alternative method for the characterization of the
bandgap energy, which is not based on the BAC model [368]. This transition point was
quantified as 0.2% by Klar et al. [369].
Figure 2.16 depicts the fundamental energy bandgap that is between the valence
band maximum and the E conduction band minimum, as a function of N fraction,
x, for GaAs1xNx at 300 K. Also shown is a curve with a constant bowing parameter of
18 eV (dotted line) along with another incorporating a variable bowing parameter of
(20.4–100x) eV, as suggested in a review by Vurgaftman et al. [92] (dashed line).
Ostentatiously, the BAC model predicts a substantially higher energy gap beyond the
N fraction of 1.5%. The available experimental data (points in Figure 2.16), compiled
in Ref. [315], show much better consistency with the BAC model than with either of
the two utilizing bowing parameters.
It should be reiterated that GaAs1xNx alloys with x > 5% become increasingly
difficult to grow while retaining quality. As such, compositions with large N fraction
2.10 Band Parameters for Dilute Nitrides
GaAs
1.4
Bandgapenergy (eV)
GaAs1–xNx
1.2
1.0
BAC
0.8
C = 20.4 –100 xeV
C = 18 eV
Experimental data
0.6
0.00
0.01
0.02
0.03
0.04
0.05
N mole fraction, x
Figure 2.16 Energy of the fundamental bandgap in GaAsN as a
function of nitrogen concentration x (a) from the BAC model
(solid curve), (b) using the variable bowing parameter from the
review of Vurgaftman et al. [92] (dashed curve), and (c) using a
constant bowing parameter (dotted curve). For comparison, the
available experimental data as compiled in Ref. [315] are also
plotted (circles) [92,152].
may not have the technological significance [313]. On the theory side, no upper limit
on the N compositions beyond which the BAC model becomes invalid has been
reported. Despite some spread in the reported values for the two primary parameters
EN, 1.65–1.71 eV, referenced with respect to the GaAs valence band maximum, and V
(2.3–2.7 eV), the values reported by Shan et al. [352], namely, EN ¼ 1.65 eV, V ¼ 2.7 eV,
are suggested.
In discord with the conventional BAC model, which assumes that the addition of N
has little effect on the valence bands, two reports noted a larger than expected heavy-/
light-hole splitting in GaAsN containing a small percentage nitrogen [370,371]. This
implies a strong bowing in the valence band shear deformation potential b, although
the increase in the deformation potential is inconsistent in these two reports.
Expanding their earlier work, Egorov et al. [372] again observed strain-induced
splitting of light-hole and heavy-hole bands of tensile-strained GaAsN. The observed
dependence of the bandgap in unstrained GaAsN on the nitrogen content differed
substantially from that predicted by the theory assuming that the bandgap in GaAsN
j207
j 2 Electronic Band Structure and Polarization Effects
208
can be reduced to zero. In cases like this when there is discrepancy, the default
position is to underscore the importance of additional investigations.
An issue of importance in device applications is the nature and magnitude of the
band alignment at a GaAs/GaAsN heterojunction. While the BAC model implies that
the GaAsN conduction band minimum must lie below that in GaAs, it is not clear
whether the valence band maximum in GaAsN should exhibit any relative shift.
Various predictions disagree on this issue [334,341]. On the experimental side, X-ray
photoelectron spectroscopy data suggest a type-II band alignment [373] but with quite
large error bars on the valence band offset (VBO). This type of band alignment was
supported by PL measurements [374]. Follow-up investigations utilizing not just
optical but electrical characterizations as well have concluded a definite type-I
alignment [375–379]. A point of discord may lie in the fact that the built-in strain
was not completely relaxed in any of the heterostructures covered in the aforementioned investigations. Egorov et al. [377] accounted for strain effects and deduced that
the band offset for unstrained GaAs0.98N0.02 with respect to GaAs. The value of the
valence band discontinuity between GaAs and GaAsN0.02 at 18 K was evaluated to be
15 5 meV, taking into account the bandgap of GaAsN alloy [372]. On the contrary
Egorov et al. [379] reported the band alignment of InxGa1xAs/GaAsN heterojunctions to be type I or type II, depending on the In content x, a point that needs to be
confirmed by additional measurements. The BAC model parameters recommended
by Vurgaftman and Meyer [152] for GaAsN and all of the other dilute nitrides for
which information is available are summarized in Table 2.24.
2.10.2
InAsN
Another binary compounded with N is InAsN, which garnered early theoretical
interest [334,380], followed by tight binding calculation focused on the effects of
nitrogen clustering in the alloy [381]. Experimental investigations of this dilute
nitride have been reported [382–386]. Measurements of the electron effective mass in
this alloy indicated a large increase [380,390], analogous to that in GaInAsN [355].
Table 2.24 Band anticrossing (BAC) model parameters for some
of the dilute nitride semiconductors [152].
Parameters
EN w.r.t. VBM (eV)
V (eV)
GaAsN
InAsN
Ga1xInxAsN
GaPN
InPN
Ga1xInxPN
InSbN
1.65
1.44
1.65(1 x) þ 1.44x 0.38x(1 x)
2.18
1.79
2.18(1 x) þ 1.79x
0.65
2.7
2.0
2.7(1 x) þ 2.0x 3.5x(1 x)
3.05
3.0
3.05(1 x) þ 3.0x 3.3x(1 x)
3.0
2.10 Band Parameters for Dilute Nitrides
While the authors [385,386] appear to argue that the BAC model could not account for
any increase greater than doubling of the mass in the nitrogen-free host material,
upon closer examination of the model it becomes evident that their view is contradicted, and also by the results available for GaInAsN [355], clearly displaying a
similarly large increase in mass. Consequently, it is reasonable to assume that the
BAC model is applicable to InAsN. At the same time, however, it should be stated that
the available information is somewhat incomplete and future investigations may
alter this assertion. Extracting the position of the nitrogen level with respect to the
valence band maximum in the host InAs from the valence band offsets tabulated in a
review article [92], a value for EN ¼ 1.44 eV is recommended. The measurements of
Naoi et al. [382] are consistent with values for the potential V ranging between 1.9 and
2.3 eV. A round figure of V ¼ 2.0 eV could be a good default.
2.10.3
InPN
A few experimental studies of InP1xNx are available in the literature [387,388].
Because the bandgaps and valence band offsets of GaAs and InP differ only slightly, it
would follow that the BAC model would apply equally well to InPN. Yu et al. [388]
derived band structure parameters in which the GaAs/InP VBO was assumed to be
0.35 eV. However, Vurgaftman et al. [92] recommended a value of 0.14 eV, but
considering the result EN ¼ 2.0 eV by Yu et al. led them to update their recommended
value to EN ¼ 1.79 eV, both being with respect to the valence band maximum of
InP. [92] A reexamination of the coupling potential that is most consistent with the
data of Yu et al. [388] led Vurgaftman et al. [92] to recommend V ¼ 3.0 eV.
2.10.4
InSbN
Even though InSb has the smallest bandgap for the conventional III–V binaries,
addition of N has attracted some attention because of the potential that even longer
wavelengths, beyond those accessible by InSb, can be accessed. Murdin et al. [389–
391] made experimental observations that the effective mass in InSb1xNx increases
despite a considerable reduction in the bandgap, which is consistent with all the other
dilute nitride semiconductors discussed above. The authors derived EN ¼ 0.65 eV.
This compares with EN ¼ 0.85 eV above the top of the valence band. At this stage, the
experimental values are recommended as default. The same authors also reported
V ¼ 2.2 eV and supplemented the minimal BAC dispersion relation of Equation 2.48
with an additional shift of the level of nitrogen position with increasing N fraction [389,390]. It should, however, be pointed out that it appears that their value for V
underestimates the observed bandgap decrease [389]. This led Vurgaftman et al. [152]
to recommend V ¼ 3.0 eV, which should yield better consistency with the available
data, albeit little. Because the effect of nitrogen on the band structure of InSbN is
significant in terms of the light-hole dispersion, the full 10-band kp model may be
required for any realistic calculations.
j209
j 2 Electronic Band Structure and Polarization Effects
210
2.10.5
GaPN
The early LED [314] developments utilized N-doped GaP because of the unique band
structure of this binary. It is an indirect gap semiconductor with both X and L valleys
lower in energy than the G point. Nitrogen acts as an isoelectronic impurity in GaP
and has been employed as the active material of visible LEDs [314] until the advent of
other conventional direct bandgap and III–V and nitride-based varieties. Initial
studies of GaP with an alloy-like concentration of N were reported by Baillargeon
et al. [392,393]. Miyoshi et al. [394] investigated the transmogrification of the GaPN
luminescence spectrum with increasing N content and were able to observe the
emission from excitons bound to nitrogen pairs for x < 0.5%. Bi and Tu [395]
reported GaP1xNx with x as large as 16% using gas-source molecular beam epitaxy.
A number of theoretical reports predicted that GaPN retains its indirect gap nature up
to relatively large (beyond dilute) N concentrations [334,339,341].
Shan et al. [396,397] reexamined this view of indirect bandgap within the BAC
model, and in the process found out that the anticrossing between G-valley states and
the N impurity band moves E below the X valley for arbitrarily small values of x. They
derived V ¼ 3.05 eV employing the well-established value of EN ¼ 2.18 eV, relative to
the valence band maximum. This places the nitrogen level slightly below the
conduction X valley. As in the case of GaAsN whose band structure is less dependent
on pressure and temperature than its host material GaAs, GaPN too sports a decrease
in the pressure dependence of the fundamental transition with a deformation
potential of 1.2 eV [396]. The temperature dependence of the fundamental gap
also sees a reduction [398,399]. As in the case of all the dilute nitrides, a large increase
in the electron effective mass is observed [400]. Congruous with popular LED
materials for decades, strong luminescence for small N fractions [401,402], occurring
despite the indirect gap of the GaP host, is another feature of this material.
The evolution of transitions due to isolated N centers, N pairs, and N clusters that
hasbeenobservedforx < 1%,andthemixingwithX-andL-valleystates,however,cannot
be described by the simple BAC model [403–405]. For example, Buyanova et al. observed
a sudden reduction in the radiative lifetime of the fundamental transition for x > 0.5%,
which they attributed to an effective indirect-to-direct crossover [406]. The wealth of
phenomena reported for GaPN can perhaps beexplainedwith a more flexible theoretical
approach such as the supercell pseudopotential formulations of Kent and Zunger [324–
326]. Even though the accuracy of the BAC model is more limited in GaPN than in the
other dilute nitride alloys due to the proximity of the X valley and the plethora of
complex experimental observations for intermediate compositions, the parameter set
EN ¼ 2.18 eV and V ¼ 3.05 eV of Shan et al. [398] can be used as default values.
2.10.6
GaInAsN
Let us now extend our treatment of the effect of N to ternary alloys. Having established
BAC parameters for GaAs1xNx and InAs1xNx, they need to be determined for the
2.10 Band Parameters for Dilute Nitrides
Ga1yInyAs1xNx alloy whose host material is InGaAs. Owing to applications in long
haul communications systems, most of the technological interest among all the dilute
nitrides has so far focused on this quaternary. Additional impetus can be found in its
applications to solar cells and photovoltaics that can be grown on GaAs substrates
rather than InP, the former having a more established and less expensive technology
base. Addition of N in InGaN narrows the bandgap for In concentrations, while at the
same time providing tensile strain compensation far less than those required in
conventional GaInAs quantum wells that are under compressive strain, countering the
composition-induced reduction in the bandgap. Moreover, the wavelengths near
1.55 mm are not accessible with standard InGaAs-based quantum wells on GaAs
substrates, which necessitates the use of InP substrates, which allow employment of
larger concentrations of In. Incorporation of N in InGaAs can mitigate the situation
and allow the use of GaAs substrates even for the extended wavelength for long haul
fiber-based communications systems.
Specific to the discussion of the bandgap, the applicability of the BAC model to the
case of GaInAsN is quite well established [320,321,351,352,355,357] in that much of
what is reported for the dilute nitrated binaries discussed above has also been found
to be applicable to this quaternary. Investigations employing very low In fractions [351,352,355], for example, on the order of 10%, typically found no differences
of significance from GaAsN except of course the decrease in the InGaAs energy
gap [92]. For materials with larger In fractions, prepared on both GaAs [407,408] and
InP [409] substrates, Zhukov et al. [410] proposed an alternative model. Pan et al. [359]
took EN to be independent of the In concentration and employed V ¼ 2.5 eV.
Although Choulis et al. [320,321,411] utilized the same assumption with respect to
EN, their value for the coupling potential was considerably lower: V ¼ 1.675 eV. A
similar value (V ¼ 1.7 eV) was independently deduced by Polimeni et al. [408] for In
compositions ranging from 25 to 41%. In contrast, Sun et al. [412] found that,
depending on the particular transition between the conduction and valence subbands, V in the range of 2.8–3.0 eV was imperative to account for the luminescence
data for InGaAsN/GaAs quantum wells having an In content of 27.2%.
The finding of a smaller bandgap reduction in GaInAsN than in GaAsN with N
fraction is in fact expected and is due to the ordering of the nitrogen atoms in the
InGaAs matrix [413,414]. There is some experimental evidence for carrier localization in the presence of both In and N in the quaternary alloy [415,416]. In one
investigation [416] a series of five distinct transitions, which were attributed to five
different environments for the N atom in the alloy, have been reported. Vurgaftman
et al. [92] attempted to provide the best available parameters for the InGaAsN
quaternary, while maintaining consistency with the parameters recommended above
for GaAsN and InAsN. Accordingly, the position of the nitrogen band EN should be
determined from the shift of the valence band offset in GaInAs [92]. This should
include the small, yet nonnegligible, bandgap bowing with composition. This leads to
a smooth variation of EN between 1.65 eV (for GaAsN) and 1.44 eV (for InAsN) and is
consistent with the intuitive expectation, the basis of which is in no contradiction with
any definitive experiments in that the position of the nitrogen level should not vary
with respect to vacuum. Within this framework, Vurgaftman et al. [92] proposed a
j211
j 2 Electronic Band Structure and Polarization Effects
212
bowing of the coupling potential V in the Ga1yInyAsN alloy: V ¼ 2.7(1 y) þ 2.0y
3.5y(1 y) eV. Admittedly, this parameterization does not fully agree with the
experimental results for this quaternary [320,321,351,352,355,359,408]. But, it is
nearly consistent with the median values and the recommended binary end points.
As in the case of the ternaries, any strain must be added to the host semiconductor
properties in the basic BAC model while employing these parameters.
2.10.7
GaInPN
It must be mentioned at the get go that there are not as much data available in this
quaternary system. One report of the energy gap in bulk Ga0.46In0.54PN is available in
the literature [417]. According to estimates based on the VBO dependence in GaInP,
the quaternary alloy under discussion represents a special case in that one would
expect a proximity of the nitrogen energy level and the host conduction band edge,
perhaps within as little as 10–20 meV [92]. If one presumes, for the sake of
discussion, that the BAC model remains valid in this limit, the reported energy
gaps allow one to obtain a coupling potential in the 2.1–2.3 eV range. Considering that
this is considerably smaller than the recommended values for both InPN and GaPN,
by analogy with GaInAsN one could surmise a bowing of the Ga1yInyPN interaction
potential: V ¼ 3.05(1 x) þ 3.0x–3.3x(1 x) eV. It is noteworthy that the bowing
parameters derived for the two quaternaries are quite similar. It should again be
underscored that further studies are needed to confirm and/or update this value.
2.10.8
GaAsSbN
In addition to the mixed cation ternary host materials, mixed anion host materials
also present opportunities. Among them is the alloy host GaAsSb that produces the
quaternary alloy GaAs1xySbyNx [418–420]. This quaternary has the potential for
reaching long wavelengths also on GaAs substrates. Unfortunately, because of the
sparse nature of the data for this material, and the total lack of any reports
on GaSb1xNx, one can only conjecture that the procedure recommended above
for GaInAsN be followed. The material-specific parameters, that is, the constant
V ¼ 2.7 eV at least for Sb fractions 20% should be assumed.
2.11
Confined States
If the physical size of the semiconductor in any direction is comparable to the de
Broglie wavelength for electrons in electronic processes and exciton diameter in lowtemperature optical processes, the size effects become important. The constriction can
form one side, two sides, and three sides in the case of which the system represents
three-dimensional, two-dimensional, one-dimensional, and zero-dimensional, as
2.11 Confined States
Figure 2.17 Schematic representation of three-dimensional, twodimensional, one-dimensional, and zero-dimensional systems in
real space.
shown in Figure 2.17. The two-dimensional, one-dimensional, and zero-dimensional
correspond to quantum wells, wires, and dots, respectively. From the transport point of
view, when the size of the well in a quantum well structure is comparable to the de
Broglie wavelength in the semiconductor forming the well, the conduction and valence
bands are modified noticeably in that the density of states in both the conduction and
valence bands are discretized. From the excitonic optical transitions point of view, the
length scale is the Bohr radius. This picture is depicted schematically in Figure 2.17a
and b for a wurtzite semiconductor bulk (3D system), quantum well (2D system),
quantum wire (1D system), and quantum dot (0D system). Moreover, equally important is the modification of these energies in the semiconductor caused by strain effects.
In short, eigenstates of strained-layer superlattices require the consideration of both
strain and quantum size effects. Eigenstates applicable to the early versions of
compound semiconductor based quantum wells without strain were modeled by the
envelope function formalism of Bastard [421]. If this method is to be used, one has to
calculate the strain-dependent bandgap first. Bastards formalism can then be utilized
to determine the transition energies in quantum wells, which must be added to the
strain component, as shown by Marzin [422]. Marzin developed a method for
calculating the bandgap of a strained cubic semiconductor that, when employed in
conjunction with the envelope function approximation, leads to eigenstates in quantized structures of strained systems (Figure 2.18).
In Bastards method revised by Marzin, an 8 · 8 Kane Hamiltonian matrix [423]
is employed to give an accurate description of a strained quantum structure. The
j213
j 2 Electronic Band Structure and Polarization Effects
214
Figure 2.18 Schematic representation of (a) a GaN/AlGaN
multiple quantum well structure and the conduction and valence
band edges at the G point, and (b) valence and conduction band
diagrams of the same with the confined states indicated.
size of the Hamiltonian is justified because there are three valence bands, in
addition to the conduction band, with spin-up and spin-down for each band.
Similar to cubic semiconductors, the wurtzite phase also has one conduction band
and three valence bands, heavy- and light-hole states as well as spin–orbit bands,
each with spin-up and spin-down, necessitating the use of an 8 · 8 Hamiltonian.
The strain effects in the GaN system have been treated by Gil et al. [98,424] and
others, as reviewed in Ref. [59]. However, until the time when uniformly strained
films can be grown, correlations to experiments will remain weak and reduce the
level of confidence in the predictions especially and the analysis as a whole. This is
particularly true for InGaN wells, as there is also phase separation to deal with, in
addition to nonuniform strain.
We shall once again emphasize that there exist many discrepancies concerning the
determination of some important parameters for the relevant bulk material properties
such as hole masses, bandgap energy bowing parameters, shear and deformation
potentials, and band offsets that are necessary for determining and understanding the
properties of confined structures. Calculations, using ab initio methods, have been
applied to estimate the unknown but necessary parameters for the band structure
calculations. Later on, Sirenko et al. [62] have performed envelope function calculations
of the valence band in wurtzite quantum wells following the formalism of Rashba–
Sheka–Pikus (RSP) developed for bulk wurtzite semiconductors. Employing a 6 · 6
Luttinger–Kohn model, Ahn [425] studied the effect of a very strong spin–orbit split-off
band coupling on the valence band structure of GaN-based materials. Considering that
the spin–orbit band is extremely important for GaN because of its very narrow
2.11 Confined States
spin–orbit splitting (10 meV), the spin–orbit split-off coupling was taken into consideration in the calculations. In addition, it was assumed that the electrons in QW are
confined by the conduction band offset (DEc) and the holes by the valence band offset
(DEv), the values of which are also of some controversy.
When the size of the well is comparable to the Bohr radius of an exciton in the
semiconductor forming the well, the exciton transition energies are modified. The
Bohr radius is given by
a0 ¼
4pes h2
;
m q2
ð2:49Þ
where es is the dielectric constant of the semiconductor. In GaN, due primarily to its
large effective mass, this radius is about 28 Å, necessitating very small physical
dimensions before noticeable quantization can occur. In an experimental quantum
well, the wave function is constricted along the growth direction, which we shall term
as the z-direction. In the orthogonal directions, the system is free.
The problem is similar to that of a vibrating string with the ends held stationary.
The vibration wavelengths are given by
ln ¼
2L
n
with n ¼ 1; 2; 3;
ð2:50Þ
where L represents the length of the string.
In a quantum well, the rapidly varying Bloch waves will be affected by the barriers
in the z-direction, and the effect can be lumped into an envelope function that is
slowly varying. The wave function can be expressed as [49,426]
Cn ðk? ; zÞ ¼
X
n
f n ðk? ; zÞu n expðjk? r ? Þ;
ð2:51Þ
where fn(k?, z) and uv represent the envelope and Bloch functions, respectively, and n
is the subband index. The summation is performed for spin-up and spin-down of
the conduction band where the value of six is assumed for the three valence bands.
The wave function expression must be solved for the conduction band and the
valence band with the envelope function satisfying Schr€
odingers equation for the
particular potential barrier height [59]. For the case where the conduction band is
almost s-like, the G7 state suffices. However, for the valence band, due to band mixing,
a 6 · 6 Hamiltonian including all the three uppermost valence bands must be used.
Moreover, if strain is present, which is the case in almost all nitride-based structures,
the Hamiltonian must include the effect of strain as well. To complicate matters
further, the piezoelectric effect induces large electric fields at the heterointerfaces,
particularly, in samples utilizing InGaN wells.
The envelope function must satisfy
X
h q
Hnn0 k? ;
þ V n ðzÞdnn þ Hvn0 ðeÞ yv;n0 ðk? ; zÞ
j qz
ð2:52Þ
v0
¼ E n ðk? Þyv;n0 ðk? ; zÞ;
j215
j 2 Electronic Band Structure and Polarization Effects
216
where m ¼ 1, 2 for the conduction band state and n ¼ 1, . . . , 6 for the valence band
state. The details of how the dispersion of the conduction and valence band states can
be found in Ref. [59]. Suffice it to say that the conduction band is formed of nearly slike states and can be considered nearly parabolic, ameliorating the confined state
calculations, as will be discussed in the next section. In finding the total energy
between the confined conduction and valence band states, one may assume that the
majority of the contribution is due to the confinement energy in the conduction band
because of the large disparity between the electron and hole effective masses in favor
of the conduction band. However, when gain in semiconductor lasers is considered,
the dispersion of valence band states must be taken into consideration.
2.11.1
Conduction Band
If the potential barrier is infinitely high, the wave vector in the z-direction will be
quantized and assumes the discrete values of
2p
np
; n ¼ 1; 2; 3;
ð2:53Þ
¼
kzn ¼
ln
L
where L is the thickness of the quantum well. Assuming a parabolic band structure
that satisfactorily describes the s-like conduction band, the confinement energy can
be expressed as
DE conf ¼
2 k2zn
h
h2 np
¼
2m 2m L
2
n ¼ 1; 2; 3:
ð2:54Þ
Taking into account the energy dispersion relationship in the x- and y-directions for a
parabolic conduction band, we have
h2
np 2
n ¼ 1; 2; 3;
ð2:55Þ
þ k2x þ k2y
E ¼ Ec þ
L
2m
with n ¼ 1, 2, 3, and Ec representing the conduction band edge.
For a true one-dimensional wire along the x-direction, discretization along the ydirection would also occur in addition to the z-direction, giving rise to confined
energy states (with complete confinement) of
h2
np 2
þ k2x þ k2y ; n; m ¼ 1; 2; 3 . . . :
ð2:56Þ
E ¼ Ec þ
L
2m
If we consider a semiconductor whose constant energy surface for conduction
band in k-space is a sphere, such as the case in GaN, the volume of that sphere and the
number of available states in k-space are proportional to k3 in terms of momentum
and E3/2 in terms of energy, as shown in Figure 2.19a. The density of states per unit
energy associated with that system is proportional to E1/2, again as shown in
Figure 2.19a. The area and the number of available states in k-space in an ideal
system confined in one direction only (representing quantum wells), which is often
the z or the growth direction, is proportional to k2 or E, as shown in Figure 2.19b. The
2.11 Confined States
density of states in this case is given by m =ph2 and forms a staircase as shown in
Figure 2.19b. If we continue and place a confinement in the x-direction in addition to
the z-direction, which represents quantum wires, the line length and the number of
available states in k-space is proportional to k in terms of momentum and E1/2 in
terms of energy as shown in Figure 2.19c. The corresponding density of states takes
the dependence of E1/2, again as shown in Figure 2.19c. If confinement is imposed
in all three directions, which represent the pseudoatomic or quantum dot state, the
energy is discretized in all directions and the resultant density of states takes a deltalike function in energy, as shown in Figure 2.19d. The case for quantum dots where
(a)
kz
dN/dE
~E 1/2
3
N ~ k (N-E3/2)
ky
E
kx
(b)
kz
dN/dE
N ~ k 2(N-E)
~Constant
ky
E
kx
Figure 2.19 Constant energy surfaces and density of states for 3D
in (a) , 2D in (b) , 1D in (c) , and 0D in (d) systems, respectively.
The constant energy surface are represented by a sphere, circle, a
line, and a point in 3D, 2D, 1D, and 0D systems, respectively, in
semiconductor such as GaN conduction band that has a spherical
constant energy surface in 3D.
j217
j 2 Electronic Band Structure and Polarization Effects
218
(c)
kz
Bulk
dN/dE ~ E–1/2
N ~ k(N-E 1/2 )
n=4
n=3
n=2
n=1
ky
E
kx
(d)
dN/dE ~ δ (E)
kz
Bulk
n=4
n=3
n=2
n=1
ky
E
kx
Figure 2.19 (Continued )
physical dimension in all directions are scaled to the size of the wave function or
smaller is treated in Volume 2, Chapter 5.
Considering quantum wells formed in growth along the z-direction, the confinement occurs in the same direction and thus the wave vector is quantized. The
component of the wave vector experiencing confinement is typically referred to as the
z-component, out-of-plane component, or simply kz. In-plane components of the
wave vector kx and ky, however, are not quantized and the usual energy momentum
dispersion (E–k) diagram would apply. A pictorial description of the E–k diagram in a
two-dimensional system with confinement along the z-direction for two quantum
energy levels is shown in Figure 2.20.
If the barrier potential is large but not infinite, the wave function outside the well
decays exponentially, which is called the evanescent wave. No analytical solution exits
for the subband energies when the potential barrier is not infinitely high. Graphical
solutions treated in many textbooks on quantum mechanics and numerical solutions
as well do exist. The problem is made even more complicated in semiconductors in
2.11 Confined States
E
E
E2
E1
π/Lz 2π /L z
kx ,k y
kz
kx
Figure 2.20 Energy momentum dispersion relation in a twodimensional system with confinement along the z-direction.
Shown on the left is the three-dimensional view while that on the
right represent two slices depicting the E–kin-plane relationships for
quantum levels.
that not only is the barrier not infinite but also the barrier and well materials do not
have the same carrier mass. In this case, the boundary condition must be changed
from the continuity of the derivative of the wave function in the z-direction to the
continuity of the particle flux in the z-direction, that is,
1 qyB
1 qyW
¼ ;
ð2:57Þ
mB qz
mW qz
at Z L/2, assuming the origin of the z-axis to be in the middle of the well. The terms
mB and mW represent the effective masses in the barrier and well materials,
respectively. Likewise, fnB and fnW depict the envelope wave functions [427] in the
barrier and well materials, respectively. The solution for the subband energies can be
computed numerically.
The conduction band minimum for GaN as well as AlN is at the zone center and
twofold degenerate. The confinement energies for the GaN/AlGaN quantum wells
can reasonably be estimated by means of the envelope function approximation in the
same manner as that extensively used for the GaAs/AlGaAs material system [427,428]. Following the Weisbuch and Vinter notation, the low-lying conduction
electron state can be represented by [427,428]
X
½expðik? ðrÞucj ðrÞf n ðzÞ;
ð2:58Þ
CðrÞ ¼
j¼W;B
j
where W and B represent the well and barrier materials, uc ðrÞ is the conduction band
zone center Bloch wave function of GaN or AlGaN, fn(z) is a slowly varying envelope
function, k? is the transverse (in-plane) wave vector, is the envelop wave function
j
[427], and the growth direction is along the z-axis. Because uc ðrÞ is the same for GaN
and Al(Ga)N, Schr€odingers equation reduces to
h2 q2
þ VðzÞ f n ðzÞ ¼ E n f n ðzÞ:
ð2:59Þ
2m0 qz2
j219
j 2 Electronic Band Structure and Polarization Effects
220
In the above equation, m*(z) is the corresponding effective mass of the conduction
electron, V(z) represents the profile of the minimum of the conduction band along
the growth direction, and E(z) is the confinement energy. Assuming no doping in
either regions, V(z) has a rectangular well-like profile. The solution of the confinement energies is similar to a particle in a box problem. The boundary conditions are
1 df n ðzÞ
that fn(z) and mðzÞ
dz be continuous across the interface. The latter is necessary to
ensure the conservation of the particle current.
Al x Ga1–x N/GaN, x = 0.2, L B = L W = 2 nm
0.25
Energy (eV)
0.20
0.15
0.10
0.05
0.00
–2
–1
0
1
2
3
4
Distance (nm)
(a)
Figure 2.21 (a) The conduction band edge
potential profile for a representative Al0.2Ga0.8N/
GaN single quantum well with a barrier and well
thickness of 2 nm (or 20 Å) each for a Ga polar
sample with the [0 0 0 1] direction pointing to the
left. Polarization charge causes deviation from
the square well and no screening due to free
carriers is accounted for. The scales for the
vertical and horizontal axes are in terms of eV and
nm, respectively. Courtesy of V. Litvinov. (b) The
conduction band edge potential profile for a
representative Al0.3Ga0.7N/GaN single quantum
well with a barrier and well thickness of 2 and
4 nm, respectively for a Ga-polar sample with the
[0 0 0 1] direction pointing to the right.
Polarization charge causes deviation from the
square well and no screening due to free carriers
is accounted for r. The scales for the vertical and
horizontal axes are in terms of eV and nm,
respectively. Courtesy of V. Litvinov. (c) The
conduction band edge potential profile for a
representative In0.3Ga0.7N/GaN single quantum
well with a barrier and well thickness of 2 and
4 nm, respectively, for a Ga-polar sample with the
[0 0 0 1] direction pointing to the right.
Polarization charge causes deviation from the
square well and no screening due to free carriers
is accounted for. The scales for the vertical and
horizontal axes are in terms of eV and nm,
respectively. Courtesy of V. Litvinov.
2.11 Confined States
Al x Ga1-x N/GaN, x = 0.3, LB = 4 nm, L W = 2 nm
0.5
Energy (eV)
0.4
0.3
0.2
0.1
0.0
–4
–2
0
2
4
Distance (nm)
(b)
In x Ga
1–x
N/GaN, x = 0.3, LB = 4 nm, LW = 2 nm
1.4
1.2
Energy (eV)
1.0
0.8
0.6
0.4
0.2
0.0
–4
(c)
–2
0
2
4
Distance (nm)
Figure 2.21 (Continued )
The case of quantum wells in the nitride system requires not just confinement
effects due to barriers but also polarization-induced charge. The latter tends to distort
the band profile due to the induced electric field. The details of the polarization charge
and its effect on the quantum wells can be found in Section 2.12.5 and Volume 2,
j221
j 2 Electronic Band Structure and Polarization Effects
222
Chapter 5. Essentially, the conduction and valence band edges get skewed due to the
field induced by polarization, as shown in Figure 2.21a and b for representative
Al0.2Ga0.8N/GaN (Ga-polar) and Al0.3Ga0.7N/GaN (Ga-polar) single quantum well
structures, the former with barrier and well thicknesses of 2 nm (or 20 Å) each, with
[0 0 0 1] direction pointing to the left, and the latter with barrier and well thickness of 2
and 4 nm and [0 0 0 1] direction pointing to the right. The same for a representative
In0.3Ga0.7N/GaN single quantum well with a barrier and well thickness of 2 and 4 nm,
respectively, for a Ga-polar sample with the [0 0 0 1] direction pointing to the right is
shown in Figure 2.21c. The band profile depicts the case with no screening. Free
carrier induced screening would alter the profile. The calculated eigenstates, only the
ground levels exist, for Al0.1Ga0.9N/GaN and Al0.2Ga0.8N/GaN as a function of
quantum well thickness for a barrier thickness of 2 nm are shown in Figure 2.22.
Also shown in dashed lines are the energy levels without polarization such as the case
on a-plane (nonpolar surface) quantum wells discussed in Volume 3, Chapter 1. The
calculated eigenstates for Al0.3Ga0.7N/GaN ground and the first excited states (only
two states are available) for a barrier thickness of 4 nm as a function of quantum well
thickness are shown in Figure 2.23. For increased activity in deeper UV devices, both
0.30
x = 0.1, level 1
x = 0.2, level 1
x = 0.2, level 2
LB = 2 nm
Alx Ga1–x N/GaN
Dashed lines:
without polarization
0.25
Energy (eV)
0.20
0.15
0.10
0.05
0.00
1
2
3
4
5
LW (nm)
Figure 2.22 Calculated eigenstates, only the ground levels exist,
for Al0.1Ga0.9N/GaN (lower curve) and Al0.2Ga0.8N/GaN (upper
curve) for a barrier width of 2 nm as a function of quantum well
thickness. Also shown in dashed lines are the energy levels
without polarization such as the case on a-plane (nonpolar
surface) quantum wells. Courtesy of V. Litvinov.
6
7
2.11 Confined States
in terms of emitters and detectors, it is necessary to consider structures where
AlxGa1xN is used as the active part of the device in the form of a quantum well.
Choosing AlN, arbitrarily, as the barrier for AlxGa1xN, the eigenstates for two
representative cases are presented. Also shown in dashed lines are the energy levels
without polarization such as the case on a-plane (nonpolar surface) quantum wells
discussed in Volume 3, Chapter 1. Shown in Figure 2.24 are the ground and first
excited states as a function of well width for an AlN/Al0.4Ga0.6N quantum having a
barrier width of 2 nm. It should be noted that only the ground and first excited states
are available. Also shown in dashed lines are the energy levels without polarization
such as the case on a-plane (nonpolar surface) quantum wells discussed in Volume 3,
Chapter 1. The same structure but with AlN/Al0.5Ga0.5N, representing nearly the
solar blind region of the Suns spectrum, is shown in Figure 2.25. Also shown in
dashed lines are the energy levels without polarization, such as the case of a-plane
(nonpolar surface) quantum wells discussed in Volume 3, Chapter 1.
For the visible or nearly visible part of the spectrum, InGaN quantum wells
typically with GaN barriers are employed. This is in part due to not only benefits
gained by heterostructure devices but also technological reasons, the latter due to the
decomposition of thick InGaN layers and inordinate amounts of ammonia required
0.45
0.40
0.35
AlxGa1–x N/GaN
level 1
level 2
L B = 4 nm, x = 0.3
Dashed lines:
without polarization
Energy (eV)
0.30
0.25
0.20
0.15
0.10
0.05
0.00
1
2
3
4
LW ( nm )
Figure 2.23 Calculated eigenstates for Al0.3Ga0.7N/GaN for
ground and the first excited states (only two states are available)
for a barrier thickness of 4 nm as a function of quantum well
thickness. Also shown in dashed lines are the energy levels
without polarization such as the case on a-plane (nonpolar
surface) quantum wells. Courtesy of V. Litvinov.
5
6
j223
j 2 Electronic Band Structure and Polarization Effects
224
1.0
level 1
level 2
x = 0.4
L B = 2 nm
AlN/Al x Ga 1–x N
Dashed lines:
without polarization
Energy (eV)
0.8
0.6
0.4
0.2
0.0
1.0
1.5
2.0
2.5
3.0
3.5
4.0
L W (nm)
Figure 2.24 Calculated ground and first excited states (only two
states are available) as a function of well width for an AlN/
Al0.4Ga0.6N quantum having a barrier width of 2 nm. Also shown in
dashed lines are the energy levels without polarization such as the
case on a-plane (nonpolar surface) quantum wells. Courtesy of
V. Litvinov.
to grow thick layers. In this vein, the calculated ground state (for x ¼ 0.1) and ground
and excited states (for x ¼ 0.2) as a function of well width for an InxGa1xN/GaN
quantum well having a barrier thickness of 2 nm are presented in Figure 2.26.
2.11.2
Valence Band
In an attempt to determine the valence band subband structure, Suzuki and
Uenoyama [59] calculated the band discontinuities from first-principles calculations
and found them to be 0.11 and 0.43 eV for the valence and conduction bands of GaN/
Al0.2Ga0.8N. The elastic stiffness constants, taken from prior experimental data,
employed for GaN were, in units of 1011 dyn cm2, 29.6, 13.0, 15.8, 26.7, and 2.41 for
C11, C12, C13, C33, and C44, respectively (see Table 1.24). For illustrative purposes, the
valence band structure in unstrained 30 and 50 Å GaN/Al0.2Ga0.8N quantum wells is
exhibited in Figures 2.27a and 2.28 where the strain due to the lattice and thermal
mismatch are neglected. Bandgap discontinuities of 0.11 and 0.43 eV were adopted
for the valence and conduction bands, respectively. The confinement energies in deep
wells are inversely proportional to the effective mass in the growth direction and
2.11 Confined States
1.4
AlN/Al x Ga1–xN
1.2
Dashed lines:
without polarization
level 1
level 2
x = 0.5
LB = 2 nm
Energy (eV)
1.0
0.8
0.6
0.4
0.2
0.0
1.0
1.5
2.0
2.5
3.0
3.5
4.0
LW (nm)
Figure 2.25 Calculated ground and first excited states (only two
states are available) as a function of well width for an AlN/
Al0.5Ga0.5N quantum well having a barrier width of 2 nm,
representing nearly the solar blind region of the Suns spectrum.
Also shown in dashed lines are the energy levels without
polarization such as the case on a-plane (nonpolar surface)
quantum wells. Courtesy of V. Litvinov.
directly proportional to the square of the well length. The HH and LH bands are not
coupled. Consequently, the HH band can be construed as parabolic with little, if any,
change in strain. This is because the C6v crystal symmetry of the bulk remains. The
upper
pffiffiffi (LH) and lower (CH) bands are coupled with a constant coupling coefficient
2D3 , which means that coupling of these bands is independent of kz and strain. The
effective masses of the HH and LH bands are too heavy to cause substantial
confinement energy, whereas the CH band with a lighter mass causes more split
due to quantization and, in a sense, makes the crystal splitting (Dcr) larger. For
comparison, in ZB structures the coupling between the light hole and the spin–orbit
band is dependent on kZ and thus the bands change considerably with strain, and the
in-plane heavy-hole mass becomes light and the in-plane light-hole mass becomes
heavy.
The effect of strain on the quantum well was also considered by Suzuki and
Uenoyama [60], assuming coherently strained quantum wells. Consequently, the inplane lattice constant of the layers is assumed to be that of the substrate with a
substantial effect on the subband structure of the quantum wells. The beneficial
effects of strain in electronic and optoelectronic devices based on ZB crystals have
j225
j 2 Electronic Band Structure and Polarization Effects
226
0.8
0.7
Dashed lines:
without polarization
In x Ga1–x N/GaN
x = 0.1
x = 0.2, first
x = 0.2, second
L B = 2 nm
Energy (eV)
0.6
0.5
0.4
0.3
0.2
0.1
0.0
1
2
3
4
5
6
LW (nm)
Figure 2.26 Calculated ground state (for x ¼ 0.1) and ground and
excited states (for x ¼ 0.2) as a function of well width for an
InxGa1xN/GaN quantum wells having a barrier width of 2 nm. For
x ¼ 0.2, the ground and the first excited states are available. Also
shown in dashed lines are the energy levels without polarization,
such as the case on a-plane (nonpolar surface) quantum wells.
Courtesy of V. Litvinov.
been documented well [88]. This and the fact that there is some lattice mismatch
between GaN and its ternaries make it imperative that the effect of strain on the
properties of Wz quantum wells be considered. Figures 2.27b and 2.28b exhibit the
valence band subband structure for 30 and 50 Å GaN/Al0.2Ga0.8N quantum wells.
The biaxial strain was assumed to be 0.5% in the (0 0 0 1) c-plane. Superimposed is the
valence band structure for 30 and 50 Å Wz GaN/Al0.2Ga0.8N quantum wells with 0.5%
tensile strain in the c-plane. Results indicate that the wells get deeper for compressive
strain and shallower for tensile strain. The density of states at the valence band
maximum gets smaller for the compressive and larger for tensile strain. However, the
change is very small as the symmetry remains as in the bulk with no further removal
of the degeneracy. It must be mentioned though that uniaxial strain along the x- or the
y-direction only causes the HH band to move to a higher energy and leads to a reduced
density of states. Consequently, the effects of this type of strain resemble those in bulk
GaN. In a sense, the Q well alone does not result in any special characteristic that
would lead to much improved results for lasers [429].
Valence band confinement energies for relaxed and 0.5% compressively strained
GaN/Al0.2Ga0.8N quantum wells in the c-plane are depicted in Figure 2.29. The data
2.11 Confined States
50
L = 30 Å
100
No strain
50
0
–50
0
Energy (meV)
Energy (meV)
HH1
LH1
LH2
–100
HH2
–150
–200
0
–50
L = 30 Å
j227
With 0.5 %
compressive
strain
HH1
LH1
LH2
HH2
–100
–150
20
10
15
5
kx ,ky ,Wave number (× 106 cm –1)
–200
0
(a)
20
10
15
5
kx ,ky ,Wave number (× 10 6 cm –1)
(b)
Figure 2.27 Plot of the upper valence band structure (HH and LH
bands) in a 30 Å GaN/Al0.2Ga0.8N quantum well with bandgap
discontinuities of 0.11 and 0.43 eV for the valence and conduction
bands, respectively; (a) without strain and (b) with 0.5
compressive strain in the c-plane [60].
have been deduced from the calculations of Suzuki and Uenoyama [59] for 30, 40, 50,
and 60 Å well thicknesses and band discontinuities of 0.11 and 0.43 eV for the valence
and conduction bands, respectively. The other parameters utilized can be found in
the tables presented in Chapter 1.
2.11.3
Exciton Binding Energy in Quantum Wells
Exciton binding energies in reduced dimensional systems vary from those of the
bulk [430]. In the GaN/AlGaN system, one would expect the binding energy to go up,
as the confinement gets stronger. If the well thickness is continually reduced, at some
point the overlap with AlGaN becomes very noticeable. Toward zero well thickness,
the binding energy should approach the value of AlGaN. Considering the strong
localization and possible role of excitons in optical processes even at room temperature, it is imperative that the binding energy be known. Bigenwald et al. [431]
considered the very problem of exciton binding energy in a GaN/Al0.2Ga0.8N system
with results leading to the obvious conclusion that the effect of confinement is large
and cannot be ignored. They calculated the exciton binding energies and oscillator
strengths with the formalism developed [432] prior to the aforementioned investigation by applying a two-parameter trial function. Due to the anisotropy of the structure,
the dielectric constant was globalized and the particle masses were weighted with the
j 2 Electronic Band Structure and Polarization Effects
228
50
100
L = 50 Å
Strain free
L = 50 Å
HH1
50
0
0.5 % Compressive
strain
LH1
E n e r g y (m e V )
HH1
–50
HH2
–100
LH1
0
LH2
HH2
–50
–100
HH3
LH2
–150
–150
–200
0
–200
0
5
10
15
20
5
10
15
20
k x ,k y ,Wave number (× 106 cm–1)
kx,ky,Wave number (× 106 cm–1)
(a)
(b)
Figure 2.28 Plot of the upper valence band structure (HH and LH
bands) in a 50 Å GaN/Al0.2Ga0.8N quantum well with bandgap
discontinuities of 0.11 and 0.43 eV for the valence and conduction
bands, respectively; (a) without strain and (b) with a compressive
strain of 0.5 in the c-plane [60].
0
HH
LH
CH
100
CH 1
Strained b ul k GaN
50
LH 2
Bulk Al 0.2Ga 0.8N
C onfin em ent en ergy (m eV)
LH 1
HH 2
CH 2
( 9 )1 HH 1
( 9 )2 HH 2
HH 1
150
HH
LH
CH
200
0
50
Well width (Å)
Figure 2.29 Valence band confinement energies versus the
thickness of the well in GaN/Al0.2Ga0.8N quantum wells. Courtesy
of Professor Bernard Gil.
100
(
)
71 1
LH 1
(
71 )2
LH 2
(
(
) CH 1
72 1
)
72 2 CH 2
2.11 Confined States
j229
40
Exciton binding energy (meV)
35
B Exciton
A Exciton
30
25
C Exciton
20
15
0
20
40
60
80
GaN Q Well thickness (Å)
Figure 2.30 A, B, and C exciton binding energies
as a function of the well width in a GaN/
Al0.2Ga0.8N system. Note that the exciton
binding energies reported for GaN range from
about 20 to 30 meV. The likely value of A exciton
binding energy in GaN is 21 meV. If so, the
absolute values of the binding energies shown
should be treated with caution. However, the
trend with quantum well thickness holds, which
is the reason for inclusion of this figure [431].
probability densities. The A, B, and C exciton binding energies computed as a
function of GaN well thickness are shown in Figure 2.30. Two essential points stand
out.
The first point is that the G9v and G17v states are confined in the well as is the
electron state. The largest binding energy corresponds to a well thickness of L 15 Å,
which is when the spreading of both electron and hole functions in the barrier area
minimal.
The second point is that the relatively light G27v hole state leaks out of the well (for
L < 100 Å) so that the electron–hole pair has a small binding energy and is almost
constant for 20 < L < 100 Å. Caution should be exercised in applying the calculations
of Ref. [432] for well thicknesses larger than about 100 Å, three times the Bohr radius.
In GaN-based systems, the terms quantum well and superlattices have been used
very liberally in that structures with well thicknesses well in access of the Bohr radius
are referred to as quantum wells. The term superlattice requires that barriers are
penetrable by the wave function, and further, the wave functions in adjoining wells
overlap and form the superlattice minibands. If these standards are strictly applied at
this point in time, there may not be much to discuss. Consequently, a conscious
decision was made to treat many heterostructures with reduced dimensions, in at
least one direction, as quantum-confined structures.
100
j 2 Electronic Band Structure and Polarization Effects
230
2.12
Polarization Effects
Solids are different from vacuum in that they respond to electric fields present or
applied. There are three forms of polarization that are present at the atomic level [2].
One is due to partial or complete alignment of dipole moments of polar molecules
with the electric field. When atoms forming the solid are different, as in binary,
ternary, and quaternary semiconductors, and they have different electronegativities,
any asymmetrical molecule has a permanent dipole moment, a process referred to as
dipole orientation or paraelectric response. This component cannot keep pace with
varying electric field above about 1010 Hz, causing a drop in the real part of the
dielectric constant and a jump in the imaginary part (the loss part – appreciable loss
factor). The loss is caused by dipoles attempting to respond to the field but seriously
lagging in phase. At higher frequencies, the dipoles cannot follow the field and the
effect is negligible including the loss factor. Electric field paves the way for rotation of
dipole to participate in the electric displacement and align the dipoles collectively,
barring thermal disturbances. In a completely or partially ionic solid, dipoles can be
induced by relative motion of positive and negative ions under the influence of
electric field, causing what is termed as the ionic polarization. Similar to the case of
dipole orientation, this process cannot respond to frequencies above 1013 Hz (Reststrahlen frequency), causing yet an additional drop in the dielectric constant and a
surge in the loss factor due to the phase lag. Again, as the frequency is increased
further, above the Reststrahlen frequency, ionic motion cannot respond to the field
and the loss factor due to this process diminishes. The third kind, which occurs in
every dielectric, is called the electronic polarization. This is caused by displacement of
electrons in an atom relative to the nucleus under the influence of electric field, in a
sense deforming the electron shells. The electronic polarization remains at frequencies above Reststrahlen frequency, making the real part of the dielectric constant
larger than unity. This process too cannot follow the field above 1015 Hz, above which
the dielectric constant of the solid becomes very close to unity.
Group III–V nitride semiconductors exhibit highly pronounced polarization
effects. Semiconductor nitrides lack center of inversion symmetry and exhibit
piezoelectric effects [209] when strained along h0 0 0 1i. Piezoelectric coefficients
in nitrides are almost an order of magnitude larger than in many of the traditional
group III–V semiconductors [209,433–437]. The strain-induced piezoelectric and
spontaneous polarization charges have profound effects on device structures. The
piezoelectric effect has two components. One is due to lattice mismatch (misfit)
strain while the other is due to thermal strain (ts) caused by the thermal expansion
coefficient difference between the substrate and the epitaxial layers. The low
symmetry in nitrides, specifically, the lack of center of inversion symmetry present
in zinc blende structure, may be interpreted as some sort of nonideality, which is not
the case. Nonvanishing spontaneous polarization is allowed in an ideal wurtzite
structure [212,438]. This spontaneous polarization is noteworthy, particularly when
heterointerfaces between two nitride semiconductors with varying electronegativity
are involved. This manifests itself as a polarization charge at heterointerfaces.
2.12 Polarization Effects
Spontaneous polarization was only understood fully not too long ago by King-Smith
and Vanderbilt [439] and Resta et al. [440].
In heterojunction devices such as modulation-doped field effect transistors (MODFETs) where strain and heterointerfaces are present, the polarization charge is present
and is inextricably connected to free carriers, which are indeed present. As such,
polarization charge affects device operation in all nitride-based devices, particularly
HFETs, and thus must be taken into consideration in device design unless nonpolar
surfaces such as the a-plane are used. The quality of films on nonpolar surface has not
kept pace with those on polar basal plane, which make the topic of discussion quite
relevant. As mentioned above, polarization charge arises from two sources: piezoelectric effects and the difference in spontaneous polarization between AlGaN and GaN,
even in the absence of strain. These charges exit in all compound semiconductors to
varying degrees unless self-cancelled by the symmetry of the particular orientation
under consideration such as the nonpolar surfaces/interfaces.
In relative terms, spontaneous polarization is larger than the piezoelectric polarization in AlGaN/GaN-based structures. In the case of InGaN/GaN structures,
spontaneous polarization is relatively small but not as small as the earlier predictions
called for, but still noteworthy, as spontaneous polarizations in GaN and InN are not
as different from one another. However, the strain-induced piezoelectric polarization
can be sizable. If and when defect-associated relaxation occurs reducing the strain in
the films, the strength of the piezoelectric polarization is lowered. Spontaneous
polarization and piezoelectric polarization affect the band diagram of heterostructures. The effects are very large and can easily obscure the engineered designs.
Polarization is dependent on the polarity of the crystal, namely, whether the bonds
along the c-direction are from cation sites to anion sites or visa versa. The convention
is that the [0 0 0 1] axis points from the face of the N-plane to the Ga-plane and
marks the positive z-direction. In other words, when the bonds along the c-direction
(single bonds) are from cation (Ga) to anion (N) atoms, the polarity is said to be
the Ga-polarity, and the direction of the bonds from Ga to N along the c-direction
marks the [0 0 0 1] direction, which is generally taken to be the þz-direction. By a
similar argument, when the bonds along the c-direction (single bonds) are from
anion (N) to cation (Ga) atoms, the polarity is said to be the N-polarity, and the
direction of the bonds from N to Ga along the c-direction marks the direction, which
is generally taken to be the z-direction. To shed further light, the Ga-polarity means
that if one were to cut the perfect solid along the c-plane where one breaks only a
single bond, one would end up with a Ga-terminated surface. The Ga- and N-polarity
of a model GaN crystal is shown in Figure 1.3. A schematic representation of
the spontaneous polarization in a model GaN/AlN/GaN wurtzitic crystal is shown in
Figure 2.31.
The spontaneous polarization Pspont (also commonly referred to as P0) in a solid
has not always been well defined, although much better understanding of it has been
emerging. Only those differences in P between two phases that can be linked by an
adiabatic transformation that maintains the insulating nature of the system throughout are well defined. For example, one phase can be considered unstrained and the
other strained. Vanderbilt proved that the polarization difference DP between the
j231
j 2 Electronic Band Structure and Polarization Effects
232
[0 0 0 1] Axis
P0AlN
P0GaN
N
N
Ga
Ga
Al
Al
N
N
N
Al
Al
N
N
Ga
Ga
N
P0
P0
GaN
P0GaN
P0AlN
AlN
Figure 2.31 Schematic depicting the convention
used for determining the polarity and crystalline
direction in wurtzitic nitride films. The diagram
shows the case for a Ga-polarity film with its
characteristic bonds parallel to the c-axis
(horizontal in the figure) going from the cation
(Ga or Al) to the anion (N). The spontaneous
polarization components P0Ga and P0AlN for a
periodic GaN/AlN structure are also indicated
with that for AlN having a larger magnitude. The
GaN
spontaneous polarization is negative and thus
points in the [0 0 0 1] direction. Caution must be
exercised here as there is no long-range
polarization field, just that it is limited to the
interface. The polarization in AlN is larger in
magnitude than in GaN. There exists a difference
in polarization at the interface, DP0 pointing in
direction for both GaN/AlN
the [0 0 0 1]
interfaces. The Born factor is defined in
Equation 2.82 [433].
wurtzite and zinc blende phases could be calculated by considering an interface
between the two phases and by defining Pspont to be zero in the zinc blende phase. In
short, by calculating the integral of a quantum mechanical Berry phase along a line in
the Brillouin zone from one end to the other in the bulk wurtzite symmetry leads to
polarization P with respect to that in zinc blende (which is zero by definition because
zinc blende is cubic and cannot have a spontaneous polarization in an infinite bulk
periodic crystal). The Berry phase actually represents an overlap integral between the
periodic part of the Bloch function at k and a neighboring k-point, k0 . Zorroddu
et al. [212] and Bernardini et al. [433] showed that the charges accumulating at each
interface in a self-consistent calculation can be obtained from the DP of the two bulk
layers forming the heterointerface. The relation between the charge and P follows
basically from Gausss law. The bound charge density rb ¼ ÏP. This means that
across an abrupt interface with P1 on one side and P2 on the other side, one gets
P
P2 P 1 ¼ i rs (surface charge density at the interface with the appropriate signs).
Even though it is overly simplistic, a graphical picture of polarization due to strain
(piezo component) and heterointerfaces (spontaneous component), the latter is in the
case of different ionicity, can be obtained, which is helpful. Shown in Figure 2.32 is a
ball-and-stick diagram of a tetrahedral bond between Ga and N in Ga- and N-polarity
configurations, showing the polarization vector due to the electron cloud being closer
to the N atoms. Actually, the cumulative polarization due to the triply bonded atoms is
along the direction of the single bond. The in-plane and vertical components of
polarization due to pairs of atoms cancel one another if the tetrahedron is ideal.
2.12 Polarization Effects
Ga Polarity
[0 0 0 1]
N Polarity
N
[0 0 0 1]
Ga
P0
P0
P0
P0zr
P0
P0
Ga
P0
P0zr
P0
P0
N
Ga
P0
N
j233
Ga
P0
N
Ga
Figure 2.32 Ball-and-stick configuration of an ideal GaN
tetrahedron with proper c/a ratio and internal parameter u for both
Ga and N polarity in a relaxed state.
However, when a Ga-polarity film is under homogeneous in-plane tensile strain,
the cumulative z-component, [0 0 0 1] direction, of the polarization associated with
the triple bonds decreases, causing a net polarization that would be along the ½0 0 0 1
direction, as shown Figure 2.33. In a nitrogen-polarity film, the same occurs except
that the net polarization would be in the opposite, [0 0 0 1], direction. When an inplane and homogeneous compressive strain is present, the net polarization would be
in the [0 0 0 1] direction in the Ga-polarity case and ½0 0 0 1 direction in the N-polarity
case, as shown in Figure 2.34.
Ga Polarity
[0 0 0 1]
N Polarity
N
P0
P0
P0
P0
P0zr
P0
P0
Ga
N
[0 0 0 1]
F
P0
P0
N
N
Ga
Ga
P0
Ga
Figure 2.33 Ball-and-stick configuration of a GaN tetrahedron for
both Ga and N polarity with a homogeneous in-plane tensile strain
showing a net polarization in the [0 0 0 1] direction for Ga-polarity
and [0 0 0 1] polarization for N polarity.
P0
P0
P0zr
P0
Ga
F
j 2 Electronic Band Structure and Polarization Effects
234
Ga Polarity
[0 0 0 1]
N Polarity
N
[0 0 0 1]
Ga
P0
P0
P0
P0zr
P0
Ga
P0
N
N
P0
P0
P0zr
P0
P0
N
Ga
Ga
P0
Ga
Figure 2.34 Ball-and-stick configuration of a GaN tetrahedron for
both Ga and N polarity with a homogeneous in-plane compressive
direction for Ga
strain showing a net polarization in the [0 0 0 1]
polarity and [0 0 0 1] polarization for N polarity.
The same graphical argument can be used to attain a mental image of spontaneous
polarization at heterointerfaces as well. For example, if we were to construct two
tetrahedra, one representing a GaN bilayer and another on top of it representing an
AlN bilayer, the top N atom shown in Figure 2.32 for the Ga-polarity configuration
would make triple bonds with it. Because AlN is more electronegative than GaN, the
net component of the polarization vector in the [0 0 0 1] direction in triply bonded N
with Al is larger in amplitude than in the GaN tetrahedron, and there would be a net
interfacial polarization in the ½0 0 0 1 direction even without strain. In short, the
source of piezoelectric polarization is strain in an electronegative binary. That for
spontaneous polarization is the change in electronegativity across an interface such
as the AlN and GaN interface.
Substrates upon which nitride films are grown lack the wurtzitic symmetry of
nitrides. Consequently, the polarity of the films may not be uniform, as schematically
depicted in Figure 4.21 where the section on the left is of Ga-polarity and the section
on the right is of N-polarity, representing a Holt-type inversion domain. In this type of
inversion domain, the wrong type of bonds, for example, GaGa and NN, are
formed at the boundary projected on (1 1 2 0) plane. The structural and electrical
details of the inversion domains observed and investigated in GaN can be found in
Section 4.1.3. Inversion domains combined with any strain in nitride-based films
lead to flipping piezoelectric fields with untold adverse effects on the characterization
of nitride films in general and the polarization effect in particular, and on the
exploitation of nitride semiconductors for devices. Such flipping fields would also
cause much increased scattering of carriers, as they traverse in the c-plane. Having
made the case, it should be mentioned that if proper measures are taken, the Gapolarity films grown by OMVPE and to a lesser extent by MBE are nearly or
completely inversion domain boundary free even on sapphire substrates. However,
2.12 Polarization Effects
in the case of MBE, unless optimum AlN buffer layers are employed or N-polarity
films are grown by incorporation of GaN initiation layers, inversion domain
boundaries do occur, sometimes in high concentrations, see Section 3.5.6.
The magnitude of the polarization charge converted into number of electrons can
be in the mid-1013 cm2 level for AlN/GaN heterointerfaces, which is huge by any
standard. For comparison, the interface charge in the GaAs/AlGaAs system is used
for MODFETs in less than 10% of this figure. An excellent review of the polarization
effects can be found in Ref. [440]. The magnitude of the polarization charge is
tabulated in Table 2.25 along with elastic coefficients taken from a series of
publications by Bernardini, Fiorentini, and Vanderbilt. The data in bold are those
reported in an earlier publication [433] and the remaining data points are taken from a
later publication [212]. Following the initial reports of piezoelectric and spontaneous
polarization [441], the authors returned to the topic [212] as the values of the initial
parameters were not consistent with other reports [181]. Bernardini et al. [442]
reanalyzed the polarization as obtained using the Berry phase method within two
different DFT exchange correlation schemes. Specifically, the authors used the
Vienna ab initio simulation package (VASP) and the pseudopotentials provided
therewith, as in Ref. [180]. The newer calculations were carried out using both the
generalized gradient corrected local density approximation (GGA) to density
Table 2.25 Elastic constants and spontaneous polarization charge
in nitride semiconductors.
2
e33 (C m )
LDA
GGA
e31 (C m2)
LDA
GGA
p
e31 LDA
GGA
C33 (GPa) GGA
C31 (GPa) GGA
P0 (C m2)
LDA
GGA
P0 (C m2), ideal wurtzite structure
R ¼ C31/C33 LDA
GGA
e31 (C13/C33)e33 (C m2)
AlN
GaN
InN
1.46
1.8
1.5
0.60
0.64
0.53
0.74
0.62
377
94
0.081
0.10
0.090
0.032
0.578
0.499
0.86
0.73
0.86
0.67
0.49
0.44
0.34
0.47
0.37
354
68
0.029
0.032
0.034
0.018
0.40
0.384
0.68
0.97
1.09
0.81
0.57
0.52
0.41
0.56
0.45
205
70
0.032
0.041
0.042
0.017
0.755
0.783
0.90
The data in bold are associated with DFT in the generalized gradient approximation (GGA) that
are more accurate than others reported prior. Moreover, the resultant predictions are in relatively
better agreement with experimental data as well as the bowing parameters observed in
polarization charge in alloys [212,442]. e31 and e33 are piezoelectric constants. C m2 is Coulomb
p
per square meter. C31 and C33 are elastic stiffness coefficient or elastic constants. e31 is the proper
piezoelectric constant. The data in bold, which are from Refs [212,442], are recommended. The
remaining data are from Refs [212,433,442].
j235
j 2 Electronic Band Structure and Polarization Effects
236
functional theory in the Perdew–Wang PW91 version, and the LDA in the Ceperley–
Alder–Perdew–Zunger form. For a glossary of these approximations within the DFT
method, the reader is referred to Section 2.15. Ultrasoft potentials were used treating
Ga and In d-electrons as valence at a conservative cutoff of 325 eV. Finally, the
reciprocal space summation was done on a (8 8 8) Monkhorst–Pack mesh. The results
of the refined GGA calculations in terms of spontaneous polarization, piezoelectric,
and elastic constants so calculated are tabulated in Table 2.25 along with earlier
calculations. The two sets of data, the earlier and refined, are within 10% agreement.
The results of more refined LDA calculations are also provided. For deference to
earlier work, the piezoelectric constants, albeit an incomplete list dealing with e14, for
GaN were estimated theoretically for cubic GaN [443] and used in early investigation
of piezoelectric effects in GaN [444] and deduced from the mobility data [445], which
is indirect particularly in samples containing many scatterers.
For comparison, the interface charge in the GaAs/AlGaAs system is used for
MODFETs (or HFET) in less than 10% of this figure. An excellent review of the
polarization effects can be found in Ref. [446].
2.12.1
Piezoelectric Polarization
In a polarizable medium, the displacement vector can be expressed in terms of two
components due to the dielectric nature of the medium and the polarizability nature
of the medium as [447]
!
!
!
!
!
!
D ¼ eE þ 4pP in cgs and D ¼ eE þ P in mks units;
!
!
!
$ !
$
!
ð2:60Þ
where E and P represent the electric field and polarization vectors. Considering only
the piezoelectric component, the piezoelectric polarization vector is given by [448]
PPE ¼ e e ;
ð2:61Þ
where e and e are the piezoelectric and the stress tensors. To gain a quantitative
understanding of the piezoelectric polarization, the piezoelectric tensor, which is
defined as the derivative of the polarization with respect to strain, must be considered.
In hexagonal P63mc symmetry, piezoelectric polarization is related to strain
through the piezoelectric tensor (ei,j) as [449]
3
2
exx
2 3 2
36 eyy 7
7
Px
0
0
0
0 e15 0 6
6 ezz 7
7:
6
4 Py 5 ¼ 4 0
5
ð2:62Þ
0
0 e24 0 0 6
eyz 7
7
e31 e31 e33 0
Pz
0 0 6
4 exz 5
exy
Note that e24 ¼ e15 for hexagonal symmetry that reduces to
2 3 2
3
e15 exz
Px
4 Py 5 ¼ 4 ðe24 ¼ e15 Þeyz
5;
e31 ðexx þ eyy Þ þ e33 ezz
Pz
ð2:63Þ
2.12 Polarization Effects
where Pi, eij, and eij represent the electric polarization, electric piezoelectric coefficient, and strain, respectively.
It is clear from Equation 2.63 that the piezoelectric polarization along the ½1
100mdirection Px ¼ e15exz ¼ 0, along the ½1120a-direction Py ¼ e15eyz ¼ 0, and along the
[0 0 0 1] c-direction Pz ¼ e31(exx þ eyy) þ e33ezz.
The strain components given in Equation 2.7 are repeated here for convenience,
without shear and eyz ¼ ezx ¼ exy ¼ 0. If only a biaxial strain is present, the in-plain
strain can be calculated by relative difference in the in-plane lattice constants of the
epitaxial layer and template (buffer or substrate) through
a a0
:
ð2:64Þ
exx eyy e11 e22 ¼
a0
Here, abuffer a0 and aepi a represent the relaxed (equilibrium) in-plane lattice
constants of the buffer layer or the substrate, depending on layers and their
thicknesses, and of the epitaxial layer of interest, the strained epitaxial layer,
respectively. The nomenclature as or a0 for the substrate (buffer) and ae or a for
the epitaxial layer is also used. The expression for the out-of-plane strain is
e33
ezz ¼
c c0
:
c0
ð2:65Þ
Similarly, c0 and c represent the relaxed and the out-of-plane lattice parameters,
which would correspond to the buffer layer and epitaxial layer, respectively. In case
the in-plane strain is anisotropic, e11 6¼ e22.
As it may have already become obvious, the nomenclature used in the literatures for
parameters surrounding strain and piezoelectric and elastic constants vary in that, for
example, exx and e11 are interchangeably used. The two-way transformation of x 1,
y 2, z 3 can be used to convert from one nomenclature to the other. Likewise, PPE,
pz
pz
PPE, PPE
are commonly used in literature interchangeably to depict
3 , P 3 , and P
piezoelectric polarization. If the subscript 3 is also used as in P PE
3 , it specifically
indicates that for biaxial in-plane strain, the only nonvanishing polarization is along the
c-axis. Even in cases where the subscript is not employed, the underlying assumption is
that the polarization is along the c-direction, as growth of nitride semiconductor
structures is performed predominantly on the basal plane. In the same vein, P0, Psp,
and Psp indicate spontaneous polarization along the c-axis. DPsp and DP0 both
represent differential spontaneous polarization at a heterointerface such as an
AlN/GaN interface, a topic which follows the discussion of piezoelectric polarization.
The components of the piezoelectric polarization tensor given by Equation 2.62
pz
can be expressed in terms of a summation, using Pi instead of PPE, as
X
pz
eij ej with i ¼ 1; 2; 3 and j ¼ 1; . . . ; 6;
ð2:66Þ
Pi ¼
j
pz
where P i is the ith component of the piezoelectric polarization.
The wurtzite symmetry reduces the number of independent components of the
elastic tensor e to three, namely, e15, e31, and e33. The third independent component of
the piezoelectric tensor, e15, is related to the polarization induced by a shear strain that
is not applicable to the epitaxial growth schemes employed. The index 3 corresponds
j237
j 2 Electronic Band Structure and Polarization Effects
238
to the direction of the c-axis. It is clear that the piezoelectric properties of the Wz
structures are somewhat more complicated. If we restrict ourselves to structures with
growth along the [0 0 0 1] direction or z-direction, or along the c-axis, only the e31 and
e33 components need to be considered. The piezoelectric polarization in the [0 0 0 1]
direction can be obtained by setting i ¼ 3. The electric polarization component in the
c-direction, which is designated by z- in the above nomenclature, is given by
Pz ¼ e31 exx þ e31 exx þ e33 ezz ¼ 2e31 exx þ e33 ezz :
For isotropic basal plane strain, the strain components exx
Equation 2.67 can be written as
Pz
ð2:67Þ
e?/2 and thus
pe
P 3 ¼ e31 e? þ e33 ezz :
ð2:68Þ
In hexagonal symmetry, strain in the z-direction can be expressed in terms of the
basal plane strain e? through the use of Poissons ratio, which is expressed in terms of
the elastic coefficients Cij as ezz ¼ 2(C13/C33)exx ¼ (C13/C33)e? (see Equations 2.7,
2.14–2.16 for specifics). In the case of externally applied pressure in addition to
mismatch strain, the out-of-plane strain can be related to the in-plane strain through
ezz ¼ [(p þ 2C13exx)]/C33, where p is the magnitude of compressive pressure (in the
same unit as the elastic coefficients). In terms of the nomenclature again, it should
also be noted that e1 e11 exx and e3 e33 ezz in the other notation used in the
literature and also in this text.
C13
e? :
ð2:69Þ
Pz ¼ e31 e33
C33
pz
3
The z-component of the electric polarization is also referred to as P PE
3 , P 3 , and P pz .
Piezoelectric polarization is also described in terms of piezoelectric moduli in the
literature, which is treated here for completeness. In terms of piezoelectric moduli,
dij, which are related to the piezoelectric constants by
X
eij ¼
dik Ckj with i ¼ 1; 2; 3 and j ¼ 1; . . . ; 6 and k ¼ 1; . . . ; 6: ð2:70Þ
k
Using Equation 2.70 in Equation 2.66 and strain–stress relationship (stress is equal to
the product of elastic constant and strain in a tensor form), the piezoelectric
polarization can be expressed in terms of piezoelectric moduli as
X
pz
Pi ¼
dij sj with i ¼ 1; 2; 3 and j ¼ 1; . . . ; 6:
ð2:71Þ
j
Symmetry considerations lead to d31 ¼ d32, d15 ¼ d24 and all other components dij ¼ 0,
and thus Equation 2.71 reduces to a set of three equations:
1
1
pz
pz
P1 ¼ d15 s5 ; P2 ¼ d15 s4 ;
2
2
pz
and P 3 ¼ d31 ðs1 þ s2 Þ þ d33 s3 :
ð2:72Þ
For biaxial strain, which is the case with epitaxial layers, additional conditions are
imposed in that s1 ¼ s2, s3 ¼ 0. Moreover, the shear stresses are negligible, which
leads to s4 ¼ s5 ¼ 0. Consequently, in cases primarily applicable to epitaxial layers
grown along the c-direction, the piezoelectric polarization is left with only one
2.12 Polarization Effects
nonvanishing component, which is in the growth direction and is given by, using
Equation 2.72,
pz
P3 ¼ 2d31 s1 :
ð2:73Þ
Utilizing stress–strain relationship
C2
s1 ¼ e1 C 11 þ C12 2 13 ;
C33
ð2:74Þ
we obtain
C2
pz
P3 ¼ 2d31 e1 C 11 þ C12 2 13 ;
C33
ð2:75Þ
pz
where P 3 represents the piezoelectric polarization along the c-direction and similar
to Equation 2.69, which expresses the same in terms of piezoelectric constants as
opposed to piezoelectric moduli. For hexagonal crystals, the relations between
piezoelectric constants and piezoelectric moduli expressed in Equation 2.70 can be
reduced to
e31 ¼ e32 ¼ C11 d31 þ C12 d32 þ C13 d33 ¼ ðC11 þ C 12 Þd31 þ C13 d33 ;
e33 ¼ 2C13 d31 þ C33 d33 ;
e15 ¼ e24 ¼ C 44 d15 ;
eij ¼ 0 for all other components:
ð2:76Þ
The nonvanishing component of the piezoelectric polarization of Equation 2.66 due
to only the biaxial strain is for i ¼ 3 and is given by, which recovers Equation 2.67,
pz
P3 ¼ e1 e31 þ e2 e32 þ e3 e33 ¼ 2e1 e31 þ e3 e33 ¼ e? e31 þ e3 e33 because e31 ¼ e32 :
ð2:77Þ
Steps can be taken to express Equation 2.77 in the form of Equations 2.68 and 2.69,
which are straightforward and therefore not presented.
We have so far focused on lattice mismatch induced strain. However, the thermal
expansion coefficients of the layers used to compose many of the heterostructures are
different, which upon cooling from growth temperature could lead to thermalinduced strain. A larger effect in this vein, however, is that caused by differences in
the thermal expansion coefficient between the substrate and epitaxial stack used. In
that case, the piezo component would have two parts, namely, the lm or misfit strain
and by the thermal strain leading to Ppe ¼ Plm þ Pts. Another issue that must be
considered is that the electric field induced due to strain (piezoelectric field) in
adjacent layers of a heterostructure comprised of A and B (i.e., A for AlGaN, B for
GaN, and A,B for AlGaN/GaN) is
sp
pe
E A;B ¼ E A;B þ E A;B :
ð2:78Þ
If, for example, A is composed of a ternary, then the linear interpolation for both sp
and pe polarizations can be used to a first extent. Again, the nonlinearities are
discussed later on in Section 2.12.3.
j239
j 2 Electronic Band Structure and Polarization Effects
240
Another relevant but not discussed nearly as much topic is that the properties that
cause the piezoelectric polarization can also lead to pyroelectric effects. Such
phenomena can be rather important in nitride-based devices, as the junction
temperature is high by the nature of the applications such as lasers and high-power
amplifiers. Consequently, the thermally induced electric field, pyroelectric effect,
would most likely be present [450] with consequences similar to those ascribed to
polarization effects.
If one considers the distortion of the u parameter as well, the piezoelectric
polarization can be expanded as
qP 3 ¼
qP3
qP3
qP3
ða a0 Þ þ
ðc c 0 Þ þ
ðu u0 Þ:
qa
qc
qu
ð2:79Þ
The internal u parameter is defined as the average value of the projection of the
connecting vector of a nitrogen atom with its first neighbor in the ð0 0 0 1Þ direction
along this same direction. The three parameters, a, c, and u, are not independent of
each other. If the partial derivatives in Equation 2.79 are known, following Ref. [209],
one can write for the two piezoelectric constants
qP3
4qc 0
qu
þ pffiffiffi 2 Z ;
qc
qc
3a0
ð2:80Þ
e31 ¼
a0 qP 3
2q
qu
þ pffiffiffi Z ;
2 qa
qa
3a0
ð2:81Þ
Z ¼
pffiffiffi 2
3a0 qP3
¼ ZT3 ;
4q qu
ð2:82Þ
e33 ¼ c 0
where
is the axial component of the Born, or the transverse component of the charge tensor
ZT3 . Various structural parameters that are useful in treating the polarization issue in
nitride semiconductors are tabulated in Table 2.26.
Table 2.26 Structural parameters of wurtzitic AlN, GaN, and InN
reported in Ref. [209] and updated with DFT in the GGA
approximation in Ref. [212].
a0 (Å)
c0/a0
Ref. [209] GGA [212] Exp.
AlN 5.814
GaN 6.040
InN 6.660
3.1095
3.1986
3.5848
u0
Ref. [209] GGA [212] Exp.
3.1106 1.6190
3.1890 1.6336
3.538 1.6270
1.6060
1.632
1.6180
Ref. [209] GGA [212] Exp.
1.6008 0.380
1.6263 0.376
1.6119 0.377
0.3798
0.3762
0.377
0.3821
0.377
It should be mentioned that GGA produced data for structural as well as polarization-related
parameters, see Table 2.25, are in better agreement with refined experimental data.
2.12 Polarization Effects
In Equation 2.80, it is implicit that the vector connecting the cation with the anion
has a modulus uc associated with the internal cell parameter and points in the
direction of the c-axis. The first term in Equations 2.80 and 2.81 signifies the term
called the clamped-ion term, and represents the effect of the strain on the electronic
structure. The second term represents the effect of internal strain on the polarization.
The derivatives of u with respect to c and a in Equations 2.80 and 2.81 are related to the
strain derivatives of u through c0du/dc ¼ du/de3 and a0du/da ¼ 2du/de1.
In addition to binaries, the nitride heterojunction system utilizes ternary and to a
lesser extent quaternary alloys as well. Knowing the piezoelectric parameters of the
end binary points is generally sufficient, to a first order, to discern parameters for
more complex alloys. For example, in the case of AlxGa1xN, the piezoelectric
polarization vector expression, using linear interpolation within the framework of
Vegards law, can be described as [448]
$
$
!
Ppe ¼ ½x e AlN þ ð1 xÞ e GaN e ðxÞ:
ð2:83Þ
The same argument can be extended to piezoelectric polarization in quaternary
alloys such as AlxInyGa1xyN in a similar fashion as
$
$
$
$
!
Ppe ¼ ½x e AlN þ ð1 xÞ e InN þ ð1 x yÞ e GaN e ðxÞ:
ð2:84Þ
The linear interpolation is very convenient and does give reasonably accurate
values. However, as will be discussed later in this section, while the Vegards law
applies to the alloys, the polarization charge itself is not a linear function of
composition [220,221].
2.12.2
Spontaneous Polarization
Spontaneous polarization calculated for the binary nitride semiconductors are
tabulated in Table 2.25. For ternary and quaternary alloys, the simplest approach
is to use a linear combination of the binary end points, taking into account that the
mole fraction can be used under the auspices of the Vegards law. However, this linear
interpolation falls short of agreeing with the experimental variation of spontaneous
polarization with respect to the mole fraction. Consequently, nonlinear models have
been developed that are discussed later in Section 2.12.3. For now, the linear
interpolation is applied for simplicity in which the spontaneous polarization in
quaternary alloys such as AlxInyGa1xyN can be expressed as
sp ðx; yÞ ¼ x P
sp þ yP
sp
sp
P
InN þ ð1 x yÞP GaN :
AlN
ð2:85Þ
The ternary cases can be obtained by simply setting either x or y to zero. This is again
predicated on the assumption that polarization charge obeys Vegards law, as shown
below. Later in this section, the nonlinearity involved is discussed. Using the GGA
calculation results for the spontaneous polarization and linear interpolation, as in
Equation 2.95, for ternaries one gets
j241
j 2 Electronic Band Structure and Polarization Effects
242
sp
PAlx Ga1 x N ¼ 0:09x 0:034ð1 xÞ;
sp
PInx Ga1 x N ¼ 0:042x 0:034ð1 xÞ;
sp
PAlx In1 x N ¼ 0:09x 0:042ð1 xÞ:
ð2:86Þ
The total polarization charge inclusive of spontaneous and piezoelectric must be
considered in dealing with heterojunctions such as quantum wells, quantum wires,
and quantum dots. The case of the quantum wells and dots are treated in Volume 2,
Chapter 5. The case of the modulation-doped structures is treated in Volume 3,
Chapter 3. Therefore, the total polarization involving layers A (AlxGa1xN) and B
(GaN) in contact is
Ptotal ¼ P sp þ Ppe :
ð2:87Þ
2
Spontaneous polarization P (C m ), Born Z or the transverse component of the
charge tensor ZT3, piezoelectric constants (C m 2), elastic constants (GPa), and the
ratio R ¼ 2C31/C33 of wurtzitic nitrides, as obtained in the LDA and GGA approximation [212] are tabulated in Table 2.27. Also, tabulated following the elastic
ðpÞ
constants e33 and e31 is e31 , which is the applicable piezoelectric constant in the
context of experiments dealing with current flow across the sample. The constant e31
is relevant to systems in depolarizing fields such as nitride nanostructures [448]. A
comprehensive table including experimental and calculated values of elastic compliance, elastic constants, and piezoelectric constants as well as the Poisson number of
wurtzite binary group III nitrides at room temperature are shown in Table 2.28.
Table 2.27 Spontaneous polarization, Born effective charges, Z*
(in units of e), piezoelectric constants, dynamical charges, elastic
constants (GPa), and the ratio R ¼ 2C31/C33 of wurtzitic nitrides,
as obtained with DFT calculations in the LDA and GGA
approximation.
AlN
LDA
LDA
[179]
GGA
GaN
LDA
[179]
LDA
GGA
InN
LDA
LDA
[179]
GGA
P
(C m2)
Z
e33
(C m2)
e31
(C m2)
«(p)
31
(C m 2)
C33
(GPa)
0.100
2.652
1.80
0.64
0.74
384
373
111
108
0.578
0.579
0.090
2.653
1.50
0.53
0.62
377
94
0.499
0.032
2.51
0.86
0.44
0.47
415
83
0.400
0.034
2.67
0.67
0.34
0.37
405
354
103
68
0.508
0.384
0.041
3.045
1.09
0.52
0.56
233
224
88
92
0.755
0.821
0.042
3.105
0.81
0.41
0.45
205
70
0.683
The last column reports the proper e31 piezoelectric constant [212].
C31
(GPa)
R¼
2C31/C33
0.22c
367
135
103
405
95
0.52
3.267
1.043
0.566
2.757
10.53
Theory [179]
1.253
2.291
1.579
0.34
0.67
0.38
68
354
Theory [209,212]
GaN
396
137
108
373
116
0.58
2.993
0.868
0.615
3.037
8.621
370a
145a
110a
390a
90a
0.56
3.326
1.118
0.623
2.915
11.11
0.30d
Theory [179]
Experiment
2.298
5.352
2.069
0.53
1.50
0.50
94
377
Theory [212,433]
AlN
a
For an expanded list of elastic constants, see tables in Chapter 1 under mechanical properties [84].
Ref. [200].
b
Ref. [198].
c
Shur, M.S., Bykhovski, A.D. and Gaska, R. (1999) MRS Internet Journal of Nitride Semiconductor Research, S41, G16.
d
Ref. [437].
C11 (GPa)
C12 (GPa)
C13 (GPa)
C33 (GPa)
C44 (GPa)
n(0001)
S11 (1012 N m2)
S12 (1012 N m2)
S13 (1012 N m2)
S33 (1012 N m2)
S44 (1012 N m2)
e31 (C m2)
e33 (C m2)
e24 ¼ e15 (C m2)
d31 (1012 C m2 Pa)
d33 (1012 C m2 Pa)
d15 (1012 C m2 Pa)
Parameter
410a
140a
100a
390a
120a
0.51
2.854
0.849
0.514
2.828
8.333
0.58b
1.55b
0.48b
2.65
5.53
4.08
Experiment
223
115
92
224
48
0.82
6.535
2.724
1.565
5.750
20.83
Theory [179]
Table 2.28 Experimental and predicted elastic compliance, elastic constants, and piezoelectric constants as well as the Poisson number
of wurtzite binary group III nitrides at room temperature (theory [179,212,433]).
3.147
6.201
2.292
0.41
0.81
0.68
70
205
Theory [212,433]
InN
2.12 Polarization Effects
j243
j 2 Electronic Band Structure and Polarization Effects
244
In heterojunctions containing donors and acceptors and shallow defects, the
associated free carriers within the Fermi statistics diffuse to the semiconductor with
the smaller bandgap where they are confined, due to potential barriers, to potential
minima. The resulting charge separation due to free carriers causes an internal
electric field, screening field, which is represented by the first term in Equation 2.60. In
addition, an electric field can also be induced by the application of an external voltage
such as done through the use of Schottky barriers, metal oxide semiconductor
structures, and p–n junctions. Spontaneous and strain-induced piezoelectric polarization can influence the final status of the interfacial free-charge density in these
heterostructures. Any shallow defects (induced fields would change the ionization
ratio), free carriers, and surface contacts must be included for a complete treatment.
Let us now calculate the polarization charge for model heterojunctions using the
linear interpolation method. For AlxGa1xN coherently strained on a relaxed GaN
substrate, the strain e? is expected to be proportional to x and given by e? ¼ 2
(aGaN aAlGaN)/aAlGaN, which is 0.051x and is tensile. The piezoelectric polarization
1 direction. The correspondis then Ppiezo ¼ 0.0464x, that is, pointing in the ½0 0 0 ing difference in spontaneous polarization between AlxGa1xN and GaN is also
expected to be proportional to x, the AlN mole fraction, and is given by DPspon ¼
0.056x. Consequently, the two are in the same direction for this particular
orientation and are comparable in magnitude. This treatment assumes that the
polarization charge scales linearly with alloy composition, which does not necessarily
hold but is used for simplicity. The matter is discussed below in more detail. The total
polarization for AlN–GaN interface, which is defined in this case as the sum of the
piezoelectric polarization and the differential polarization charge is 0.102x. Note
that these are all in C m2 and that 1 C m2 ¼ 0.624 · 1015 electrons cm2. Thus,
for x on the order of 0.1, we are dealing with total polarization charge of the order of
mid-1012 cm2.
In case the ternary AlxIn1xN is used for the barrier, the composition of
Al0.82In0.18N can be grown lattice matched to GaN [451] and the piezoelectric
polarization vanishes. For lower Al concentrations, that is, x < 0.82, the piezoelectric
polarization increases due to the increase in biaxial compressive strain. For higher Al
concentrations, that is, x > 0.82, the layer is under tensile strain and the piezoelectric
polarization becomes negative.
For a coherently strained InxGa1xN layer on relaxed GaN, the difference in
spontaneous polarization is much smaller, DPspon ¼ 0.012x. Furthermore, the
InxGa1xN layer on GaN would be under compressive strain e? ¼ 0.203x and
Ppiezo ¼ þ0.139x. Here, the piezoelectric polarization dominates and is opposite in
direction to the spontaneous polarization charge but even larger in absolute
magnitude.
Unlike for tensile-strained AlxGa1xN or AlxIn1xN (for large x-values) on GaN
layer, wherein the piezoelectric and the spontaneous polarization are negative and
point in the same direction, thus they add up, the spontaneous and piezoelectric
polarizations oppose one another for compressively strained InxGa1xN or
AlxIn1xN (for small x-values) layers. To calculate the differential spontaneous and
piezoelectric polarization associated with alloys, one can to a first order employ a
2.12 Polarization Effects
linear interpolation for the spontaneous polarization and piezoelectric and elastic
constants from the binary compounds [452].
In addition to nonlinear behavior of polarization that is discussed in Section 3.14.3,
there are technological issues that must be considered. Some further words of
caution about the above estimates based on linear interpolation are needed. If the
AlGaN layers are not pseudomorphic but partially relaxed (by misfit dislocations for
example), then the piezoelectric effect would be reduced but the spontaneous
polarization would still be present. Finally, if domains with inverted polarity exist,
the overall polarization effects may be washed out. Also note that in an inverted
structure with nitrogen (N) polarity toward the surface, it may be possible to create a
two-dimensional hole gas (2DHG) at the AlGaN/substrate GaN interface, provided
that free holes are available. However, if an n-type GaN layer is placed on top, a 2DEG
may form on top of the AlGaN layer.
Polarization effects and devices are inextricable. In devices with a large concentration of free carriers, the polarization charge would be screened. In devices where
the modulation of charge is the basis of operation such as the MODFETs, a detailed
accounting of all polarization charge must be undertaken. In considering a Normal
MODFET (N-MODFET) structure where the larger bandgap AlGaN donor layer is
deposited on top of a GaN channel layer, both the spontaneous polarization and the
piezoelectric polarization must be accounted for. For an N-MODFET structure with
Ga-polarity, the potential will slope down from the surface of the AlGaN layer toward
the AlGaN–GaN interface. The topic is discussed in some detail for both AlGaN/GaN
and AlxIn1xN/GaN structures in Section 2.12.4.1 and for the InGaN/GaN heterojunction system in Section 2.12.4.2.
2.12.3
Nonlinearity of Polarization
In the treatment above and in the literature for quite sometime, a linear interpolation
from the binary end points were used to deal with both piezoelectric and spontaneous
components of polarization. Despite the linear interpolation method being reasonably successful in getting the figures to a first extent, as done in conjunction with
Equation 2.86, discrepancies with experiments were noted. Thus, efforts continued
to refine the polarization figures and, while in the process, entertain whether a
bowing parameter such as the case in bandgap of ternaries, while the lattice
parameter obeys the Vegards law, that is, linear interpolation, could also be considered for polarization. This forms the basis of the treatment of the problem by
Bernardini and Fiorentini [220,221,453] and others [454,455].
The nonlinearity is quite pronounced in AlInN and InGaN alloys for which the
binary constituents are very largely lattice mismatched. Spontaneous polarization
bowing strongly depends on the microscopic nature of the alloy. What is more is that
chemical ordering in the form of short period superlattices may increase the bowing
up to a factor of five. Similarly, the piezoelectric polarization is also nonlinear.
However, in random alloys, this nonlinearity is entirely due to a nonlinear strain
dependence on piezoelectric polarization in pure end binary compounds while the
j245
j 2 Electronic Band Structure and Polarization Effects
246
piezoelectric coefficients follow Vegards law. On the contrary, in chemically ordered
InGaN and AlInN alloys piezoelectric coefficients deviate from Vegards law, this
effect reduces the strength of the piezoelectric polarization up to 38% of its value in
AlInN alloy. However, linear extrapolation is simple and gets reasonably accurate
figures. The details of this nonlinearity are discussed below.
To reiterate, discrepancies between the experimental data and the theory using
linear interpolation led Bernardini and Fiorentini to consider a bowing parameter for
polarization in conjunction with ternaries, and it follows that this applies to quaternaries as well. For freestanding alloys, meaning relaxed for this purpose, and assuming
that the ternary nitride alloys have random microscopic structure with no strain, the
values of the lattice constants within the realm of Vegards law are given by
aAlx Ga1 x N ¼ xaAlN þ ð1 xÞaGaN ;
c Alx Ga1 x N ¼ xc AlN þ ð1 xÞcGaN :
ð2:88Þ
The spontaneous polarization Psp of an alloy, for example, AlxGa1xN, can be
expressed in terms of the polarization values of the binary constituents, AlN and GaN,
in the realm of nonlinearities (i.e., non-Vegard behavior). The polarization in an alloy
can be described in a generic measurable quantity to a first approximation by a
parabolic model involving a bowing parameter, similar to that used for the bandgap of
alloys.
In conjunction with nonlinear polarization effects in alloys, Bernardini and
Fiorentini [453] considered ordered structures to get at the spontaneous polarization
across the entire composition range by calculating it for compositions of 0.25, 0.5,
and 0.75 in addition to the binary end points. The chalcopyrite-like (CH) structure,
used for the 0.5 composition, is formed by each anion site being surrounded by two
cations of one species and two of the other, with the overall condition of conforming to
periodical (2 · 2 · 2) wurtzite supercell. This structure is highly symmetric, as there
are only two kinds of inequivalent anion sites, differing in the orientation of the
neighbors but not in their chemical identity. Among the possible ordered structures,
CH is in a sense the most homogeneous for a given composition. A further useful
ordered structure considered is a luzonite-like (LZ) structure, used for the 0.25 and
0.75 points, resembling zinc blende based alloys. In this structure, each nitrogen
atom is surrounded by three cations of one species and one of the other: in a sense,
this is the analog to the CH structure for molar fractions x ¼ 0.25 and 0.75. In the
comparison between CH and random structures, the latter used for the 0.5 composition could provide insight into the effect of randomness versus ordering without the
biases due to specific superlattice ordering, as in the CuPt (CP) structure used for the
0.5 composition point. Luzonite-like structure can provide values of the polarization
in the intermediate molar fractions.
Shown in Figure 2.35 is a comparison of calculated equilibrium basal and axial
lattice parameters a and c for three binaries and their alloys with those determined in
the realm of Vegards law (dashed lines). The agreement between the equilibrium a
and c lattice parameters so calculated and those determined using Vegards law is
quite good. The dependence of the polarization on composition is then the same as
that on the lattice parameter(s), modulo a multiplicative factor.
2.12 Polarization Effects
5.9
InN
c-Lattice constant (Å)
5.7
In x G
a
1–x
N
5.5
In x
A l 1 –x
N
GaN
5.3
5.1
A lx G a
AlN
1 –x N
AlN
4.9
0
0.2
0.4
0.6
1.0
0.8
Molar fraction, x
InN
a-Lattice constant (Å)
3.6
In x G
a 1 –x
N
In x
3.4
A l 1 –x
N
GaN
A lx G a
3.2
1 –x
N
AlN
AlN
3.0
0
0.2
0.4
0.6
0.8
1.0
Molar fraction, x
Random alloy
CH-like
CP-like
Figure 2.35 The basal plane lattice parameter a and axial lattice
parameter c of wurtzite nitride binaries and alloys directly
calculated versus those determined by Vegards law. The open
circles denote the random alloy with 0.5 molar fraction. The
dashed lines are Vegards law. Courtesy of F. Bernaridini and
V. Fiorentini.
Shown in Figure 2.36 are again the spontaneous polarization values for the
aforementioned ternaries in the freestanding strain-free form. The solid lines
represent interpolations utilizing Equation 2.95 for AlInN, the binary points of
which were determined using 32-supercell calculations. The dashed lines represent
the simple Vegards law based interpolations and the numbers indicate the bowing
parameters in terms of C m2.
j247
j 2 Electronic Band Structure and Polarization Effects
248
–0.02
In x Ga 1–x N
GaN
Spontaneous polarization (C m–2)
+0.038
InN
–0.04
+0.071
+0.019
–0.06
In x Al1–x N
Alx Ga1–xN
–0.08
AlN
AlN
–0.10
0
0.2
0.4
0.6
0.8
1
Molar fraction, x
Figure 2.36 Spontaneous polarization versus molar fraction in
freestanding (strain free) random nitride alloys (solid circles). The
solid lines represent the results of the bowing model of
Equation 2.95. The dashed lines are determined by Vegards law.
The numbers shown in the figure are the bowing parameters
expressed in units of C m2. Courtesy of F. Bernaridini and
V. Fiorentini.
Depicted in Figure 2.37 are the spontaneous polarization values versus the molar
fraction. The solid circles, squares, and triangles represent the values for the random
alloy, CH-/LZ-, and CP-like alloys, respectively. The dashed lines represent the values
calculated using Vegards law. The numbers shown in the figure represent the
bowing parameters for the CH-/LZ- and CP-like ordered alloys.
The CH and LZ calculations can be used to verify efficacy of the interpolation
model inclusive of the bowing parameter. The ordered LZ structure is analogous to
CH for molar fractions of 0.25 and 0.75. The extent to which the LZ values deviate
from those calculated by Equation 2.95 and the CH (x ¼ 0.5) value indicates whether
or not nonparabolicity occurs in the P(x) relation for CH-like order. Because the
polarization of the CH structure behaves qualitatively as that of the random structure
(Figure 2.37), the conclusions drawn for CH are applicable to the random phase.
Fortuitously, the values of the polarization calculated for the LZ structures at
molar fractions 0.25 and 0.75, shown in Figure 2.37, are very close to the parabolic
curve for InGaN and AlGaN calculated using the quadratic expression depicted in
Equation 2.95, paving the way for the use of the analytical (quadratic) expression for
polarization calculations. However, for AlInN, the calculated values are somewhat
2.12 Polarization Effects
Spontaneous polarization (C m–2)
0.02
+0.333
InxAl1–xN
InxGa1–xN
0.00
+0.193
–0.02
InxGa1–xN
–0.04
GaN
+0.095
–0.06
InxAl1–xN
InN
AIxGa1–xN
+0.037
–0.08
Random alloy
CH-like
CP-like
AIN
AIN
–0.10
0
0.2
0.4
0.6
1
Molar fraction, x
Figure 2.37 Spontaneous polarization versus
the molar fraction in all three ternary nitride
alloys. Circles, squares, and triangles represent
random alloy, CH-/LZ-, and CP-like structures,
respectively. The dashed/dotted lines (blue) with
solid triangles are for the CP-like alloys, the
dashed lines (green) with solid squares are for
CH-like alloys, and solid lines (black) with filled
circles are for random alloys. The black dashed
lines represent the data calculated using
Vegards law. Numbers indicated in the figure are
for CP and CH-/LZ-like ordered alloy bowing
parameters in terms of C m2. Courtesy of F.
Bernaridini and V. Fiorentini. (Please find a color
version of this figure on the color tables.)
above the quadratic relation for x ¼ 0.25 and below it for x ¼ 0.75, indicating some
nonparabolicity in polarization. Specifically, the bowing is higher for low In concentration in AlInN. This nonparabolicity is relatively modest, of order 10%, as compared
to the quadratic nonlinearity Equation 2.95 for AlInN. One can then conclude that the
analytical (quadratic) expression would predict polarization in AlGaN and InGaN
fairly accurately and also for AlInN but with about 10% accuracy.
To understand the physical origin of the spontaneous polarization bowing, Bernardini and Fiorentini [453] decomposed the spontaneous polarization into
three distinct components on the basis of their genesis, namely, the internal structural
and bond alternation, volume deformation, and disorder. The internal structural and
bond alternation (strain) can be caused by varying cation–anion bond lengths. The
volume deformation can be due to compression or dilation of the bulk binaries from
their original equilibrium lattice constants to the alloy values. The disorder effect
is due to the random distribution of the chemical elements on the cation
sites. Bernardini and Fiorentini [453] showed that in ordered alloys the structural
j249
j 2 Electronic Band Structure and Polarization Effects
250
contribution is dominant, the volume deformation accounts for one-third of the
bowing found in random alloys, and the effect of disorder appears insignificant in
terms of its effect on the bowing of spontaneous polarization.
2.12.3.1 Origin of the Nonlinearity
To understand the origin of the structural contribution to the nonlinear behavior,
Bernardini and Fiorentini [453] considered that while the bandgap is a scalar, the
polarization is a vector of defined direction. The basal plane normal in wurtzite
structures is the [0 0 0 1] direction. Thus, the bond length and angle alternation will
affect the polarization bowing only if it changes the projection of the bond length
along the c-axis, which is the [0 0 0 1] axis. This is consistent with the notion that the
polarization in pure binaries is strongly affected by the relative displacement of the
cation and anion sublattice sites in the [0 0 0 1] direction [441]. Also consistent is that
there is a clear correlation between the u parameter of the wurtzite structure, which is
the bond length along the singular polar (or pyroelectric) axis, and the value of the
polarization. Shown in Figure 2.38 is the calculated spontaneous polarization of
freestanding x ¼ 0.5 alloys of AlGaN, InGaN, and AlInN versus the average internal
0.02
Random
CH – like
CP – like
Spontaneous polarization (C m–2)
0.00
InGaN
AlInN
– 0.02
– 0.04
AlGaN
– 0.06
0.380
0.378
0.376
0.374
Average u lattice parameter
Figure 2.38 Spontaneous polarization versus the average internal
parameter u in AlGaN, InGaN, and InAlN ternary alloys. Open
circles, squares, and triangles, refer to random, CH-like, and CPlike structures, respectively. Courtesy of F. Bernaridini and V.
Fiorentini.
0.372
0.370
2.12 Polarization Effects
parameter u. The internal u parameter is defined as the average value of the
projection of the connecting vector of a nitrogen atom with its first neighbor in the
ð0 0 0 1Þ direction along this same direction. This is in the realm of the wurtzite
structure convention in which each anion is situated at the (0, 0, u) Cartesian point
from the cations and all the vertical bonds between Ga and N point along [0 0 0 1]
direction. This definition can also be used for random phase alloys in spite of the
displacement from the ideal sites. Figure 2.38 shows that for a given alloy composition, the spontaneous polarization of relaxed (freestanding) nitride alloys of different
microscopic structure depends linearly on the average internal u parameter of the
alloy structure. This indicates that spontaneous polarization differences between
alloys of the same composition are primarily due to structural and bond alternation
effects; disorder appears to have a negligible influence.
The structural and bond alternation effects discussed above can also shed light on
the random and CP phases in AlGaN having almost the same average u, hence, nearly
the same polarization. In InGaN, the random alloy has a larger u than the CH phase,
while the opposite is true for AlInN and AlGaN accounting for the CH versus random
bowing behavior in InGaN being opposite of that in AlInN and InGaN. Moreover, the
large bowing of CP-ordered AlInN and InGaN is consistent with the very large
deviation of the average u as compared to the random and CH-like structures.
If the internal strain were the only source of polarization bowing, all of the points in
Figure 2.38 would fall on the same straight line. This not being the case suggests that
another factor related to the chemical identity of the constituents plays a role of some
importance and brings to the role of volume deformation. To investigate this role,
Bernardini and Fiorentini [453] set up a model based on the polarization in a
constrained ideal wurtzite structure in which only the a parameter is a variable,
whereas c and u are fixed atp
the
determined by maximal sphere packing,
ffiffiffiffiffiffiffivalues
ffi
namely, u ¼ 0.375 and c=a ¼ 8=3. Each nitrogen atom is then surrounded by four
equidistant cations, meaning all bonds have the same length for a given lattice
constant a. By design, the bond alternation caused by perturbation in the internal
parameter u would not play any role, and the effects of chemical identity of the
constituents can be easily distinguished. To continue to tackle the aspect of volume
deformation, Bernardini and Fiorentini [453] assumed that Vegards law holds for
the lattice constant a. Naturally, this establishes a linear relationship between the
composition and the lattice constant. This segues into the calculation of the
polarization in each of the binary nitrides in their ideal structure as a function of
the lattice constant a(x). Finally, they could express the alloy polarization as a
composition-weighted Vegard-like average of the polarizations of the binary end
points (through a reduction of Equation 2.85) as
aðxÞ
Psp ðAlx Ga1 x NÞ ¼ xPaðxÞ
sp ðAlNÞ þ ð1 xÞP sp ðGaNÞ:
ð2:89Þ
In this approach, any nonlinearity must have its origin in the different response of
polarization to perturbations in a(x), hence, to hydrostatic compression. To illustrate
this point, shown in Figure 2.39 are calculated polarizations in the ideal wurtzite
structure for the three ternary alloys, polarization of the binaries in the ideal
j251
j 2 Electronic Band Structure and Polarization Effects
252
InGaN
Spontaneous polarization (C m–2)
–0.015
GaN
InN
–0.020
AlInN
–0.025
AlGaN
AlN
GaN
InN
–0.030
AlN
–0.035
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
Equilibrium lattice constant (Å)
Figure 2.39 Spontaneous polarization versus
the lattice constant in ideal wurtzite structures.
Solid circles depict the values of binary
compounds and random ternary alloys. Open
circles, squares, and triangles represent the
polarization calculated as a function of the lattice
constant in bulk AlN, GaN, and InN. Solid lines
correspond to Vegard interpolations based on
the ideal binaries under hydrostatic pressure.
The dashed lines represent the Vegard
interpolation of the polarization using the values
for the binaries at equilibrium. The data show
that the polarization in the alloys is a direct result
of the hydrostatic pressure and thus volume
deformation. Courtesy of F. Bernaridini and V.
Fiorentini.
structure, and polarization interpolated by the Vegard interpolation of Equation 2.89.
It can be seen that the calculated values of polarization and those interpolated by
Vegards prediction agree well. Because the Vegard interpolation intrinsically account for the volume deformation, the origin of the volume deformation component
of the nonlinearity and its large values in In-containing alloys become clear.
Essentially, this has its genesis in the fact that polarization decreases with hydrostatic
pressure in AlN and GaN, while it increases in InN. Also to be noted, polarizations in
the ideal structure are between 35 and 50% of their values in freestanding (relaxed)
alloys, and despite the absence of bond alternation, which is designed for the purpose
separating the components in effect, the bowing is still very large.
The above model dealing with the effect of strain on polarization provides the basis
for developing an expression of the bowing parameter bmodel for an ideal wurtzite
structure alloy as a function of the polarization response to hydrostatic pressure (for
the model case of AlGaN):
0
1
qP
qP
@ GaN AlN A
bAlGaN
model ¼ ðaGaN aAlN Þ
qa
qa
a¼að1=2Þ
0
1
ð2:90Þ
2
2
1
q
P
q
P
GaN
AlN A
2@
þ ðaGaN aAlN Þ
:
qa2
qa2
4
a¼að1=2Þ
2.12 Polarization Effects
The agreement of the latter expression with the b resulting from a fit to the calculated
values is very good (e.g., for the extreme case of AlInN, bmodel 20.0225 C m2, while
from direct calculation, we get 20.0208 C m2). On the basis of the model, it is now
understandable that the AlGaN bowing is pretty moderate because the region of
interest is small (3.1–3.2 Å) and the responses to hydrostatic pressure of AlN and GaN
are similar. On the contrary, in the large range 3.1–3.6 Å, AlN and InN have opposite
behavior, when the huge bowing is found in AlInN alloys. The same goes, although to
a lesser extent, for the InGaN alloys.
2.12.3.2 Nonlinearities in Spontaneous Polarization
Any nonlinearity in the spontaneous polarization can be treated by using a bowing
parameter as commonly employed in interpolating the bandgap of an alloy from the
binary point with the help of a bowing parameter. In this vein, the spontaneous
polarization for a ternary Psp (AxB1xN) with A and B representing the metal
components and N representing nitrogen is given by [84,220,221]
sp
sp
sp
PAx B1 x N ¼ xP AN þ ð1 xÞP BN bAB xð1 xÞ:
sp
ð2:91Þ
sp
PAN and PBN are the spontaneous polarization terms for the end binaries forming the
alloy. The bowing parameter is as defined
bAB ¼ 2PAN þ 2P BN 4P A0:5 B0:5 N ;
ð2:92Þ
which requires only the knowledge of the polarization of the ternary alloy at the
midpoint, that is, molar fraction x ¼ 0.5. Knowledge of the bowing parameter from
Equation 2.97 would lead to the determination of the spontaneous polarization at any
composition. For AlxGa1xN, Equations 2.91 and 2.92 take the form
sp
sp
sp
PAlx Ga1 x N ¼ xPAlN þ ð1 xÞPGaN bAlx Ga1 x N xð1 xÞ;
ð2:93Þ
bAlx Ga1 x N ¼ 2P AlN þ 2P GaN 4P Al0:5 Ga0:5 N :
ð2:94Þ
with
The first two terms in Equation 2.93 are the usual linear interpolation terms between
the binary constituents. The third term, quadratic, represents the nonlinearity.
Higher order terms are neglected because their contribution is estimated to be less
than 10%. Using the numerical GGA values in Table 2.25 for the spontaneous
polarization in AlN and GaN and the bowing parameter for random alloy AlGaN
given in Refs [220,221] leads to
sp
PAlx Ga1 x N ¼ 0:09x 0:034ð1 xÞ þ 0:0191xð1 xÞ;
and
sp
sp
sp
PInx Ga1 x N ¼ xPInN þ ð1 xÞPGaN bInx Ga1 x N xð1 xÞ;
j253
j 2 Electronic Band Structure and Polarization Effects
254
with
bInx Ga1 x N ¼ 2P InN þ 2PGaN 4PIn0:5 Ga0:5 N :
Again, using the numerical GGA values in Table 2.25 and the bowing parameter for
random alloy InxGa1xN given in Refs [220,221] leads to
sp
PInx Ga1 x N ¼ 0:042x 0:034ð1 xÞ þ 0:0378xð1 xÞ;
and
sp
sp
sp
PAlx In1 x N ¼ xP AlN þ ð1 xÞP InN bAlx In1 x N xð1 xÞ;
with
bAlx In1 x N ¼ 2PAlN þ 2PInN 4P Al0:5 In0:5 N:
Using the numerical GGA values in Table 2.25 and the bowing parameter for random
alloy InxAl1xN given in Refs [220,221] leads to
sp
PAlx In1 x N ¼ 0:090x 0:042ð1 xÞ þ 0:0709xð1 xÞ:
ð2:95Þ
In calculating the bowing parameters for the three ternaries mentioned above,
Bernardini and Fiorentini [453] used a 32-atom supercell for both alloys and binary
nitrides for spontaneous polarization for the end binaries and ternaries. These
calculations, while being more efficient in terms of computer time, are not as
accurate as those reported in Refs [220,221,441]. In the above treatment, the bowing
parameters are taken from Ref. [453], while the spontaneous polarization for the end
binaries is taken from Ref. [441]. Relying simply on the 32-atom supercell calculations, the bowing parameters and related spontaneous polarization figures for
ordered alloys such as CuPt-like ordered alloy (CP-like), chalcopyrite-like ordered
alloy (CH-like), luzonite-like ordered alloy (LZ-like) have also been obtained by
Bernardini and Fiorentini [453]. In the series of tables below, the results of such
calculations for the aforementioned ordered alloys are given for completeness. The
binary figures in terms of spontaneous polarization and the lattice parameter
resulting from the 32-atom supercell calculations are also given for consistency.
Again, more accurate binary data exist in Refs [441].
For convenience, the lattice parameter (a), spontaneous polarization (Psp), and
bowing parameter (bAB) for the three ternary nitride alloys in the form of random,
ordered chalcopyrite (CH-like), ordered luzonite (LZ-like), and CuPt-ordered alloy
(CP-like) are tabulated in Tables 2.29–2.35, in addition to the data shown in
Figures 2.35–2.37.
Table 2.29 The lattice parameter (a) and spontaneous polarization
(Psp) for AlN, GaN, and InN determined by 32-atom supercell
calculations by Bernardini and Fiorentini.
AlN
a (Å)
Psp (C m2)
3.1058
0.0897
GaN
3.1956
0.0336
InN
3.5802
0.0434
2.12 Polarization Effects
Table 2.30 The lattice parameter (a), spontaneous polarization
(Psp), and the bowing parameter (bAB) for random alloy ternaries
with a molar fraction of x ¼ 0.5 determined by 32-atom supercell
calculations by Bernardini and Fiorentini [453].
R 50%
Al0.5Ga0.5N
In0.5Ga0.5N
Al0.5In0.5N
a (Å)
Psp (C m2)
bAB (C m2)
3.1500
0.0569
þ0.0191
3.3872
0.0290
þ0.0378
3.3352
0.0488
þ0.0709
Table 2.31 The lattice parameter (a), spontaneous polarization
(Psp), and the bowing parameter (bAB) for CuPt ordered alloy (CPlike) with a molar fraction of x ¼ 0.5 determined by 32-atom
supercell calculations by Bernardini and Fiorentini [453].
CP 50%
Al0.5Ga0.5N
In0.5Ga0.5N
Al0.5In0.5N
a (Å)
Psp (C m2)
bAB (C m2)
3.1489
0.0573
þ0.0176
3.3884
þ0.0098
þ0.1934
3.3222
þ0.0168
þ0.3336
Table 2.32 The lattice parameter (a) and spontaneous polarization
(Psp) for the luzonite (LZ-like) and chalcopyrite (CH-like) alloy with
a molar fraction of x ¼ 0.25 determined by 32-atom supercell
calculations by Bernardini and Fiorentini [453].
LZ 25%
Al0.25Ga0.75N
In0.25Ga0.75N
Al0.25In0.75N
a (Å)
Psp (C m2)
3.1724
0.0413
3.2920
0.0323
3.4510
0.0385
Table 2.33 The lattice parameter (a) and spontaneous polarization
(Psp) for the luzonite (LZ-like) and chalcopyrite (CH-like) alloy with
a molar fraction of x ¼ 0.5 determined by 32-atom supercell
calculations by Bernardini and Fiorentini [453].
CH 50%
Al0.5Ga0.5N
In0.5Ga0.5N
Al0.5In0.5N
a (Å)
Psp (C m2)
bAB (C m2)
3.1474
0.0523
þ0.0374
3.3949
0.0328
þ0.0226
3.3369
0.0427
þ0.0952
j255
j 2 Electronic Band Structure and Polarization Effects
256
Table 2.34 The lattice parameter (a) and spontaneous polarization
(Psp) for the luzonite (LZ-like) and chalcopyrite (CH-like) alloy with
a molar fraction of x ¼ 0.75 determined by 32-atom supercell
calculations by Bernardini and Fiorentini [453].
LZ 75%
Al0.75Ga0.25N
In0.75GaN0.25
Al0.75In0.25N
a (Å)
Psp (C m2)
3.1276
0.0690
3.4828
0.0366
3.2146
0.0564
Table 2.35 The bowing parameter (a) for the random alloy and
luzonite (LZ-like), chalcopyrite (CH-like), and CuPt ordered alloys
(CP-like) calculated by Bernardini and Fiorentini [453].
Random
CH and LZ
CP
AlGaN
InGaN
AlInN
þ0.019
þ0.037
þ0.018
þ0.038
þ0.023
þ0.193
þ0.071
þ0.095
þ0.333
2.12.3.3 Nonlinearities in Piezoelectric Polarization
The total polarization at heterointerfaces is the sum of spontaneous and piezoelectric
polarization. Having treated the nonlinearity in spontaneous polarization, attention
must be turned to piezoelectric polarization, specifically, its nonlinear dependence on
composition in alloys. Some if not all of the components of the heterojunctions are
grown pseudomorphically and are, therefore, under strain on the (0 0 0 1) axis. The
ensuing symmetry-conserving strain causes a change in polarization that amounts to a
piezoelectric polarization. The aim here is to show that piezoelectricity in nitride alloys
is nonlinear, and that this nonlinearity is due to a pure bulk effect with its nonlinear
behavior of bulk binary piezoelectric constants versus symmetry-conserving strain.
The model heterostructure considered by Bernardini and Fiorentini [453] is a
coherently strained alloy grown on a relaxed binary buffer layer (bulk for this
purpose) in which the in-plane lattice parameter is aalloy ¼ aGaN. The piezoelectric
component is the difference between the total polarization to be obtained and the
spontaneous polarization discussed above. Shown in Figure 2.40 is the piezoelectric polarization as a function of the alloy composition. Symbols, which have
similar designation as in the spontaneous polarization, represent the calculated
polarizations for AlGaN, InGaN, and InAlN alloys as a function of compositions.
After the publication of Ref. [453], to reconcile a discrepancy with a paper by
Al-Yacoub and Bellaiche [454] who showed that CuPt-like ordering in In0.5Ga0.5N
wurtzite-structure alloys causes a sizable deviation of the piezoelectric constants
from Vegards like behavior. Bernardini and Fiorentini [220,221] revisited the
piezoelectric polarization in nitride alloys. In fact, Figure 2.40 contained here
represents the updated calculations. The error in question stems from the fact that
the polarization values for the strained and unstrained alloys were subtracted
Piezoelectric polarization (C m–2)
2.12 Polarization Effects
InN
Vegard's
Random alloy
CH,LZ
CP
0.2
0.1
InGaN
AlInN
GaN
0.0
AlGaN
AlN
AlN
– 0.1
0
0.2
0.4
0.6
0.8
1
Alloy molar fraction
Figure 2.40 Piezoelectric component of the
macroscopic polarization in ternary nitride alloys
epitaxially strained on a relaxed GaN layer
(template). Open symbols represent the directly
calculated values for random alloy (circles), CHlike and LZ-like (squares), and CP-like (triangles)
structures, respectively. Dashed lines represent
the prediction of linear piezoelectricity, while the
solid lines are the prediction of Equation 2.96
using the nonlinear bulk polarization as shown in
Figure 2.41. Courtesy of F. Bernaridini and V.
Fiorentini.
correctly, but values calculated with different k-point meshes were used instead of
those for the same k-point [454,455].
It is clear that, contrary to the spontaneous component of polarization discussed
above, the piezoelectric polarization component hardly depends on the microscopic
structure of the alloy. One might then ask whether the piezoelectric polarization of
the alloy can be reproduced by a Vegard-like model interpolated from the binaries in
the form
pe
pe
pe
PAlGaN ðxÞ ¼ xPAlN ½eðxÞ þ ð1 xÞP GaN ½eðxÞ;
pe
P AlN ½eðxÞ
ð2:96Þ
pe
PGaN ½eðxÞ
where
and
represent the strain-dependent bulk piezoelectric
polarization of the binary end points. With obvious permutations, this expression can
be constructed for InGaN and InAlN ternaries as well. To a first approximation, one
may calculate the piezoelectric polarization of the binary compounds for symmetry
conserving in-plane and axial strains as
pe
PAlN ¼ e33 e3 þ 2e31 e1 :
ð2:97Þ
The piezoelectric constants e can be calculated for the equilibrium state of the binary,
AN, and as such they do not depend on strain. The dashed lines in Figure 2.40
represent the piezoelectric term as computed from the above relations using the
piezoelectric constants computed for the binaries [433]. The Vegards law of Equation 2.96 when combined with Equation 2.97 clearly fails to reproduce the calculated
polarization and misses the strong nonlinearity of the piezoelectric term evident in
j257
j 2 Electronic Band Structure and Polarization Effects
258
Figure 2.40. This is due to a valid nonlinearity of the bulk piezoelectricity of the binary
constituents, which is of nonstructural origin. It should be stated that bowing due to the
microscopic structure of the alloys is negligible. The argument forwarded by Bernardini and Fiorentini [453] is that they calculate the piezoelectric polarization as a
function of the basal strain for AlN, GaN, and InN while optimizing all structural
parameters. The results depicted in Figure 2.41 clearly indicate that the piezoelectric
polarization of the binaries is an appreciably nonlinear function of the lattice parameter
a, which is related to basal strain. Because all lattice parameters closely follow Vegards
law, the nonlinearity cannot be related to deviations from linearity in the structure.
Bernardini and Fiorentini [453] substitute the nonlinear piezoelectric polarization
computed for the binaries into the Vegard interpolation, Equation 2.96 In doing so,
they obtain excellent agreement with the polarization calculated directly for the alloys as
shown with solid lines in Figure 2.40. This led them to conclude that the nonlinearity in
bulk piezoelectricity dominates over any effects related to disorder, structure, bond
length and angle alternation, and so on. Importantly, they concluded that Vegards law
still holds in calculating the piezoelectric polarization of the III–V nitrides alloys,
provided that the nonlinearity of the bulk piezoelectric of the constituents is accounted
for. Serendipitously, this means that the piezoelectric polarization of any nitride alloy at
any strain can be found by noting the value for x (the composition), followed by
calculating the basal plane strain, e(x) from Vegards law, and Ppe from Equation 2.96
using the nonlinear piezoelectric polarization of the binaries (Figure 2.41). This
approach is of paramount value in the modeling of nitride heterostructures, especially
those with high In content, and AlInN alloys.
It should be noted that nonlinearities in the calculated piezoelectric constants of
AlN and GaN have also been reported by Shimada et al. [456] but in the realm of
Piezoelectric polarization (C m–2)
0.4
AlN
GaN
InN
0.3
AlN
InN
0.2
GaN
0.1
0.0
0.00
– 0.05
– 0.10
– 0.15
Basal strain
Figure 2.41 Piezoelectric polarization in binary nitrides as a
function of basal strain (symbols and solid lines) compared
to linear piezoelectricity prediction (dashed lines). The c- and
u-lattice parameters are optimized for each strain. Courtesy of
F. Bernaridini and V. Fiorentini.
2.12 Polarization Effects
volume-conserving strain. A direct quantitative comparison with the results of
Shimada et al. and Bernardini and Fiorentini is not possible, because the latter
seeks to optimize the volume of the cell so that the stress component along [0 0 0 1]
vanishes, which is more appropriate for epitaxial structures. In spite of this
substantial difference, there is a common trend between the derivative of the
piezoelectric polarization shown in Figure 2.41 (i.e., an effective piezoconstant) and
the values of the piezoelectric constants reported in Ref. [456].
The nonlinear piezoelectricity of the binaries can be described by the relations (in
C m2) [84]
pz
PAlN ¼ 1:808e þ 5:624e2 for e < 0;
pz
P AlN ¼ 1:808e 7:888e2 for e>0;
pz
PGaN ¼ 0:918e þ 9:541e2 ;
pz
PInN ¼ 1:373e þ 7:559e2 :
ð2:98Þ
It is important again to note that the nonlinearity in the bulk piezoelectricity exceeds
any effects related to disorder or bond alternation, which have been taken into
account [220,221]. The calculation of the piezoelectric polarization of an AxB1xN
alloy for any level of strain would proceed with first calculating the strain e ¼ e(x) for a
given molar fraction, x, using the Vegards law, and the piezoelectric polarization by
pz
pz
pz
PAx B1 x N ¼ xP AN ðeÞ þ ð1 xÞPBN ðeÞ;
pz
ð2:99Þ
pz
where PAN ðeÞ and PBN ðeÞ are the end binary strain dependent piezoelectric
polarizations that can be calculated for a given strain, eðxÞ, using Equation 2.98.
Application of this process to each of the three ternaries for all the possible cases of
ternaries is as follows. Using Equation 2.96 and linear interpolation for the elastic
constants that are tabulated in Table 2.28 and strain determined from Equation 2.64,
the piezoelectric polarization between a given ternary and binary can be calculated, as
represented below (in C m 2) [84].
pz
PAlx Ga1 x N=GaN ¼ 0:0525x þ 0:0282xð1 xÞ;
pz
PAlx Ga1 x N=AlN ¼ 0:026x þ 0:0282ð1 xÞ;
pz
PAlx Ga1 x N=InN ¼ 0:28x 0:113ð1 xÞ þ 0:042xð1 xÞ:
ð2:100Þ
pz
PInx Ga1 x N=GaN ¼ 0:148x þ 0:0424xð1 xÞ;
pz
PInx Ga1 x N=AlN ¼ 0:182x þ 0:026ð1 xÞ 0:0456xð1 xÞ;
pz
PInx Ga1 x N=InN ¼ 0:113ð1 xÞ þ 0:0276xð1 xÞ:
ð2:101Þ
pz
PAlx In1 x N=GaN ¼ 0:0525x þ 0:148ð1 xÞ þ 0:0938xð1 xÞ;
pz
PAlx In1 x N=AlN ¼ 0:182ð1 xÞ þ 0:092xð1 xÞ;
pz
PAlx In1 x N=InN ¼ 0:028x þ 0:104xð1 xÞ:
ð2:102Þ
The calculated values are a function of molar fraction for the ternaries for the
epitaxial layer and template combinations represented in Figure 2.42, assuming that
the template is fully relaxed and the epitaxial layer is completely coherently strained
j259
j 2 Electronic Band Structure and Polarization Effects
260
with strain relaxation. For partial relaxation, unless the degree to which the relaxation
that occurs is known, the calculations cannot be made. The extent of relaxation just
depends on whether strain-relieving defects propagate from the template to the
epitaxial layers. In addition, the effect of cooldown-induced thermal mismatch strain
due to cooling from the growth temperature down to the operating temperature of the
structure must also be considered. As stated, the results shown are for a perfect
system with fully relaxed template and fully strained epitaxial layer on top of it. We
should point out that for a particular pair with a particular composition, Al0.82In0.18N/
GaN heterostructure, there is a perfect lattice match; thus, the misfit-induced
piezoelectric polarization is equal to zero. It should also be pointed out that other
0.2
Al x In1–x N/AlN
In x Ga1-x N/AlN
b = 0.092
Piezoelectric polarization (C m–2)
b = – 0.046
0.1
A1 x Ga 1–x N/AlN
b = 0.025
0.0
–0.1
0
(a)
0.2
0.4
0.6
0.8
1.0
Molar fraction, x
Figure 2.42 (a) Piezoelectric polarization of fully
and coherently strained ternary alloys on fully
relaxed AlN templates in the case of which the
ensuing positive piezoelectric polarization and
the negative spontaneous polarization are
antiparallel. The bowing parameters describing
the nonlinearity in the compositional
dependence of Ppz are also indicated. (b) The
piezoelectric polarization of coherently strained
ternary alloys on fully relaxed GaN template.
Note that Al0.82In0.18N/GaN heterojunction
(indicated with an arrow) is lattice matched, thus
strain and piezoelectric polarization vanishes.
(c) Also note that other experiments indicate the
lattice matching composition for AlInN on GaN
where the piezoelectric polarization charge
vanishes to be different. The piezoelectric
polarization of coherently strained ternary alloys
on fully relaxed InN template. For ternary alloys
grown on InN, the negative piezoelectric
polarization and the spontaneous polarization
are parallel and oriented to along the c-axis.
Courtesy of O. Ambacher.
2.12 Polarization Effects
0.2
Piezoelectric polarization (C m–2)
Al x In1–x N/AlN
b = 0.094
In x Ga 1–x N/GaN
b = – 0.042
0.1
0.0
A1 x Ga 1–x N/GaN
b = 0.028
–0.1
0
0.2
(b)
0.8
0.6
0.4
1.0
Molar fraction, x
0.0
Al x In 1–x N/InN
b = 0.104
Piezoelectric polarization (C m–2)
In x Ga 1–x N/InN
b = – 0.028
–0.1
–0.2
Al x Ga1–xN/InN
b = 0.042
–0.3
0
(c)
Figure 2.42 (Continued )
0.2
0.4
0.6
Molar fraction, x
0.8
1.0
j261
j 2 Electronic Band Structure and Polarization Effects
262
Nonlinear
InN
0.4
Piezoelectric polarization, P3 (C m–2)
AlN
GaN
0.2
0.0
AlN
–0.2
–0.4
–0.2
–0.1
0.0
0.1
0.2
Strain, ε
pz
Figure 2.43 The piezoelectric polarization (P 3 ) of wurtzitic binary
group III nitrides under biaxial tensile or compressive strain
calculated using Equations 2.98 and 2.99, the premise of which
relies on nonlinearity as depicted in the figure. Only the AlN case is
shown for positive strain consistent with Equation 2.98. In
addition, the values of polarization exceed those of the linear
model expressed in Equation 2.69. Courtesy of O. Ambacher.
experiments indicate the lattice matching composition for AlInN on GaN, where the
piezoelectric polarization charge vanishes, to be different.
The piezoelectric polarization versus strain, ignoring nonlinearity, can be calculated using Equation 2.69 for both compressive and tensile biaxial strain for all the
epitaxial layer/template configurations. Use of Equations 2.98 and 2.99, however,
takes the nonlinearity discussed above into consideration, as shown in Figure 2.43.
As expected, the piezoelectric polarization for a given strain increases from GaN to
InN and AlN. Significantly, higher piezoelectric polarizations, especially in cases of
high strains (high In concentrations), result when nonlinearities in the piezoelectric
charge are considered.
The piezoelectric polarization associated with coherently strained random ternary
AlGaN, InGaN, and AlInN alloys grown on GaN templates is shown in Figure 2.44.
To reiterate, the piezoelectric polarization is nonlinear with respect to the alloy
2.12 Polarization Effects
Piezoelectric polarization, P3 (C m–2)
0.2
Al x In1–x N
In x Ga 1–x N
0.1
0.0
Al xGa1–x N/
0
0.2
0.4
0.6
0.8
1.0
Molar fraction, x
Figure 2.44 The piezoelectric polarization of
random coherent ternary alloys on relaxed GaN
templates. Those calculated using Equation 2.69
and linear interpolations of the physical
properties such as the piezoelectric constants
and elastic coefficients (exy , Cxy) for the relevant
binaries are shown in dashed lines. Calculations
considering the nonlinearity in the piezoelectric
polarization in terms of strain reflected in
Equations 2.98 and 2.99 are shown in solid lines.
For alloys under high biaxial strain such as those
containing high In concentration, the
piezoelectric polarization is underestimated by
the linear approach. It should be pointed out that
the lattice matching composition for AlInN on
GaN where the piezoelectric polarization charge
vanishes is converging on 16–18% In in the
InxAl1xN lattice. Courtesy of O. Ambacher.
composition, spanning a range of 0.461 to 0.315 C m2, the end points corresponding to coherently strained AlN on InN and InN on AlN, respectively. A point of side
interest, albeit not practical, is that in alloys with high indium concentration, leading
to high strain, the piezoelectric polarization is larger than that of the spontaneous
polarization. As mentioned earlier in conjunction with Figure 2.42 that Al0.82In0.18N
can be grown on GaN in a lattice-matched form making the piezoelectric polarization
for this heterostructure vanish. To underline the point, the nonlinear model
described by Equations 2.98 and 2.99 leads to larger piezoelectric polarization charge
than linear interpolation where strain, elastic constants, and piezoelectric constants
are assumed to vary linearly with composition.
j263
j 2 Electronic Band Structure and Polarization Effects
264
2.12.4
Polarization in Heterostructures
Investigation of heterostructures such as quantum well and single heterointerfaces
such as those present in modulation-doped structures are ideal platforms for putting to
test the knowledge base of spontaneous and piezoelectric polarization. Depending on
the polarity of the sample, that is, Ga or N, the order of growth, meaning GaN on AlGaN
or the other way around, and the buffer layer that determines to a first extent whether
the barrier, the well, or both are in strain, the piezoelectric and spontaneous polarization charge add or subtract. Regardless of this, the resultant band bending leads to a red
shift in energy due to the presence of electric field, commonly referred to as quantumconfined Stark effect (QCSE), and the ensuing deformation of the wave functions being
pushed to opposite ends of the interface leads to reduction of radiative efficiency,
increase in lifetime, and also blue shift with injection that is screening. In parallel, if the
well size is small enough, carrier confinement induces a blue shift. Consequently, the
red shift induced by polarization and the blue shift induced by localization compete in
determining the transition energy. The optical transitions both in CW and timeresolved forms are discussed in detail in Volume 2, Chapter 5. Here, the manifestation
of polarization charge in heterostructures is treated.
We should mention that the electric field resulting from the polarization in GaN
would cause a large Stark shift in optical measurements and reduce the effective
bandgap in, for example, quantum wells (see Volume 2, Chapter 5), which is of
paramount importance in optoelectronic devices. The field induced has been
calculated in the context of the GaAs-based superlattices, which show notable Stark
shift when grown along the [1 1 1] direction [457].
Stark shift can be screened on a length scale of the order of the Debye length
pThe
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
ekT=q2 n by injecting free carriers in the GaN layer(s), as is the case in LEDs, lasers,
and PL experiments with large excitation intensities. In such a case, the strain-induced
field causes carrier separation that, in turn, causes a field that opposes the strain field.
Strain-induced polarization in such a heterostructure can also lead to a net electric field
that can be measured as a voltage drop across the sample. The impact of the Stark shift
and concomitant screening has been considered for laser gain also [458].
Fiorentini et al. [448] and Bernardini and Fiorentini [459] demonstrated that for an
alternating sequence of wells (w) and barriers (b), the total electric field in the well can
be calculated by recognizing that the normal components of the displacement vector
D of Equation 2.60 are continuous (provided that there is no interface charge).
total
eW E W þ 4pPtotal
W ¼ DW ¼ DB ¼ eB E B þ 4pP B
ð2:103Þ
in cgs units (for MKS remove the 4p terms).
Utilizing Equation 2.103 together with the periodicity-imposed equality of voltage
drops being equal but with opposite sign
LW E W ¼ LB E B :
One arrives at
ð2:104Þ
2.12 Polarization Effects
total
E W ¼ 4pLB ðPtotal
W P B Þ=ðLW eB þ LB eW Þ;
or
total
E W ¼ 4pðPtotal
W P B Þ=ðLW =LB eB þ eW Þ;
total
E B ¼ 4pLW ðPtotal
B P W Þ=ðLB eW þ LW eB Þ;
0
1
L
B
total @
or E B ¼ 4pðPtotal
þ eB A:
B P W Þ=
LW eW
ð2:105Þ
where eW and eB are the dielectric constant of the well and barrier layers, respectively.
Likewise, LW and LB represent the well and barrier thicknesses. The same notation is
used for polarization also. Both W and B superscripts and subscripts relate to wells
and barriers, respectively. The total polarization term can be changed to piezoelectric
term and spontaneous polarization term for cases when only the former or the latter
is in effect, respectively.
total
Explicit in Equation 2.105 is that whenever Ptotal
W „P B there will be an electric field.
Due to strain or screening, the electric field will be present in both the well and the
barrier. If we limit ourselves to piezoelectric polarization and somehow achieve
relaxed heterostructures, the piezoelectric polarization charge and therefore the field
will be zero.
The electric field in wells and barriers in a quantum well has two components, one
from spontaneous and the other from piezoelectric polarization. If the thickness of
the wells and barrier are the same, the field in wells and barriers are related to each
other as
sp
pe
EW þ EW ¼ EW ¼ EB
sp
pe
ðE B þ E B Þ;
ð2:106Þ
where EW and EB represent the electric field in the well and barrier material. The
superscripts indicate the field due to spontaneous and the other from piezoelectric
polarization. Additional comments that can be made are that if lattice-matched
AlInGaN alloy is used, the piezoelectric component in that material is eliminated.
However, the spontaneous component would still be present. Another point to be
recognized is that the piezoelectric field induced in InGaN and AlGaN layers of the
same composition of In in the former and Al in latter, if grown on relaxed GaN, is
larger in the former because of the larger lattice mismatch between GaN and InN as
compared to that between GaN and AlN.
The spontaneous polarization in alloys can be found using Equations 2.85 and 2.86
together with values listed for the binary end points in Table 2.25. The spontaneous
polarization so formulated together with piezoelectric polarization of Equation 2.77
(and also Equation 2.83) would allow the computation of total polarization charge.
This must be done for both the barrier and well material. The electric field can then be
calculated using Equation 2.105.
Any free carrier present in the well and barrier regions as well as those injected by
optical and/or electrical means tend to screen the polarization-induced field. A
complete picture can be obtained by solving self-consistently a set of effective mass
theory or tight binding theory and simultaneously the Schr€
odinger–Poisson equation [460], which is discussed in Section 2.1.14. Combining the ab initio calculations
j265
j 2 Electronic Band Structure and Polarization Effects
266
of Fiorentini and Schr€odinger–Poisson solver of Di Carlo allowed the authors to
calculate the charge distribution and field profiles in quantum wells in the presence
of free carriers [448,461]. Poissons equation is solved using the boundary condition
that the electric field is zero at the ends of the simulated regions. This corresponds to
LW reaching the infinity limit in Equation 2.105. The potential thus obtained across
the structure is plugged into the tight binding Schr€
odinger equation, which is solved
to obtain energies and wave functions [462]. The new quasi-Fermi levels are then
calculated, which then lead to carrier concentrations, and the procedure is iterated
until self-consistency is obtained.
A case of interest for FETs and parts of quantum wells is one that features a ternary
on the surface where polarization charge would exist. The same is true for the
interface between an AxB1xN and a GaN heterostructure. The characteristic of this
charge is that it changes abruptly, leading to a fixed two-dimensional polarization
charge density s on the surface and at the interface, which is given by
sp
pz
sAx B1 x N ¼ P Ax B1 x N þ P Ax B1 x N
sp
pz
on the surface and
sp
pz
sAx B1 x N=GaN ¼ ðPGaN þ P GaN Þ ðP Ax B1 x N þ P Ax B1 x N Þ at the interface;
ð2:107Þ
respectively.
Figure 2.45 shows the polarization-induced surface and interface sheet density s/q
for relaxed and coherently strained binary nitrides and ternary–binary AxB1xN/GaN
interfaces, such as AlxGa1xN/GaN.
The spontaneous polarization surface charge density on relaxed InN, GaN, and AlN
layers is 2.62 · 1013 cm2, 2.12 · 1013 cm2, and 5.62 · 1013 cm2, respectively (there
is no piezoelectric charge in this case). If a biaxial compressive strain of e ¼ 0.002 is
present, the surface charge is reduced to 0.72 · 1013 cm2 · 1013, 0.74 · 1013, and
3.22 · 1013 cm2, as can be deduced from Figure 2.45a. For compressively strained
InN, GaN, and AlN at the level of e ¼ 0.025, 0.030, and 0.045, respectively,
the piezoelectric polarization fully compensates the spontaneous polarization and
thus no polarization-induced field should exist. As can be seen from Figure 2.45a,
polarization-induced surface charge is reduced by compressive and increased by
tensile strain. For fully relaxed layers grown on the c-axis on a substrate, the
spontaneous polarization-induced surface charge is negative for Ga-polarity and
positive for N-polarity samples. The bound surface charge can be screened by
oppositely charged surface defects and charges adsorbed on the surface. If this
screening were incomplete, the carrier concentration profiles in the crystals would
be different. Consequently, surface band bending, as well as that of interface, would be
affected by polarization, and further such band bending would be dependent of the
polarity of the sample. Experimental results are not sufficiently complete to draw a
definitive conclusion.
For coherently strained Ga-face AlxGa1xN/GaN (for 0 < x 1) and AlxIn1xN/
GaN (for 0.71 < x 1 interfaces), the polarization-induced interface charge is
calculated to be positive as shown in Figure 2.45b. For both ternary and binary
interfaces, the bound polarization charge increases nonlinearly with composition, x,
2.12 Polarization Effects
up to 7.06 · 1013 cm2. However, for Ga-face InxGa1xN/GaN (for the entire compositional range, 0 < x 1) and AlxIn1xN/GaN (or 0 < x 0.71), the polarization charge
is calculated to be negative and nonlinear with respect to composition. At the limit
for the former, InN/GaN structure, a bound sheet density of 14.4 · 1013 cm2
is calculated. Upon screening in n-type heterostructure, a positive polarization sheet
4
3
AlN
Bound surface density s /q (1014 cm–2)
2
1
0
AlN
–1
InN
–2
–3
GaN
–4
–0.2
–0.1
0.0
0.1
0.2
Strain, ε
(a)
Figure 2.45 (a) In-plane biaxial strain
dependence of surface polarization
(piezoelectric plus spontaneous) charge for
wurtzitic GaN, InN, and AlN. As in the case of
Figure 2.43, the AlN case is with positive strain.
(b) Bound interface charge density for coherent
AlxGa1xN, InxGa1xN, and AlxIn1xN grown on
relaxed Ga-polarity GaN. Polarization-induced
bound interface charge (positive in n-type
samples and negative in p-type samples) is
screened by free electrons in n-type and holes in
p-type samples, respectively, leading to twodimensional gas. Note that the interface charge
can be converted into carrier concentration by
dividing it with the electronic charge of
q ¼ 1.602 · 1019 C. Courtesy of O. Ambacher.
j267
j 2 Electronic Band Structure and Polarization Effects
268
2.0
Ga-Polarity on GaN
2DEG
Bound interface density s /q (1014cm–2)
1.5
1.0
AlN
0.5
N
A l xG a1 –x
GaN
0.0
–0.5
In
x Ga
N
I n 1 –x
Al x
–1.0
1–
x
N
InN
–1.5
InN
2DHG
–2.0
0
0.2
0.4
0.6
0.8
1.0
Molar fraction, x
(b)
Figure 2.45 (Continued )
charge, which is bound, leads to a 2DEG with a sheet carrier concentration close to the
concentration of the bound interface density þs/e.
Now that a conceptual issues related to polarization has been discussed, the
attention can now be turned to calculating the sheet charge distribution and band
structure at a single heterointerface typically used for MODFET. The available data
indicate that GaN channel devices are the only viable ones, even though In-containing
channels may someday work well as well. For the moment, the likely GaN channel
MODFET structures utilize AlxGa1xN, AlxIn1xN, or perhaps the quaternary. The
criterion is that the bandgap of AxB1xN ternary must be larger than that of GaN. As
discussed often, the AlxIn1xN ternary lattice matches GaN; however, the reported
values for lattice matching composition vary. For example, AlN molar fraction of 71%
(corresponds to InN molar fraction of 29%) and 82–83% (corresponds to InN molar
fraction of 18–17%, which is the more likely lattice matching composition) have been
reported to lattice match GaN [451]. To treat the MODFET interface charge problem,
2.12 Polarization Effects
the bandgap of nitride semiconductor alloys, which are discussed in Section 1.5 in
detail, must be known. For completeness, the expressions for the compositional
dependence of the alloy bandgap are repeated below. It should be pointed out that the
bandgap bowing parameter data bAx B1 x N for AlGaN are converging onto a value of
nearly 1 eV even though early figures spanned from 0.8 eV (upward bending) to
þ 2.6 eV (downward bending). The data for InGaN are fluidic also in part because of
reasons having to do with difficulties in determining the composition and bandgap,
detailed in Section 1.5.2, and also lack of high-quality samples with composition
midway in the alloy range. For example, optical reflectivity measurements together
with PL data corrected for Stokes shift led to a bInGaN ¼ 2.5 0.7 eV for 0.9 eV
bandgap of InN and bInGaN ¼ 3.5 0.7 eV for 1.9 eV InN bandgap [463]. PL data alone
in another report states the bowing parameter to be 4.5 eV [464]. The figure
determined by optical transmission measurements is 8.4 eV [465], while the theory
indicates 1.2 eV [466]. Other experimental values for lower InN molar fraction end of
the ternary are near null, meaning linear variation of the bandgap [467,468]. The
reported data in aggregate, taking into consideration the small bandgap of InN, leads
to a value of 2.53, as discussed in Section 1.5.2.
The value for the bowing parameter in AlInN is somewhat too controversial. For
example, one particular theoretical report points the bowing parameter to be
2.53 eV [469] while a value of 3.1 eV was determined by fitting the bandgap of this
alloy determined by photoreflection [470], 2.384 eV by absorption measurements but
by using 2.0 eV for the bandgap of InN, and 5.9 eV for AlN [471] and 5.4 eV in a review
article where 1.95 eV was used for InN bandgap [84]. However, when all the available
data are considered in aggregate with 0.7 eV bandgap for InN, a bowing parameter of
about 3 eV appears to be a very good value.
g
g
g
E Alx Ga1 x N ¼ xE AlN þ ð1 xÞE GaN bAlGaN xð1 xÞ
¼ 6:1x þ 3:42ð1 xÞ xð1 xÞ eV;
g
E Inx Ga1 x N
g
g
¼ xE InN þ ð1 xÞE GaN bInGaN xð1 xÞ
¼ 6:1x þ 0:7ð1 xÞ 1:43xð1 xÞ eV;
g
ð2:108Þ
g
E Alx InN ¼ xE AlN þ ð1 xÞE EInN bAlInN xð1 xÞ
¼ 6:1x þ 0:7ð1 xÞ bAlInN xð1 xÞ eV:
The presence of displacement or polarization gradient leads to a volume charge
density rv given by
!
!
!
r: D ¼ r:ðe E þ P total Þ ¼ rv
!
in MKS units: For cgs; add 4p before P :
ð2:109Þ
In a one-dimensional system, which is considered here along the c-axis, and utilizing
E ¼ dV/dz, Equation 2.109 can be rewritten as the Poissons equation in the form of
dD=dz ¼ d=dzð eðzÞdV=dz þ P total ðzÞÞ
¼ qrz ¼ q½N Dþ ðzÞ þ pðzÞ nðzÞ N A ðzÞ:
ð2:110Þ
j269
j 2 Electronic Band Structure and Polarization Effects
270
The term on the right-hand side of Equation 2.110 represents the volume density of net
charge, and the position-dependent quantities D, e, and V are the displacement field,
dielectric constant, and potential, respectively. The term Ptotal is the position-dependent
total transverse polarization (along the c-axis and perpendicular to the interfaces).
N Dþ ðzÞ and N A ðzÞ represent the ionized donor and acceptor concentrations, and p(z)
and n(z) represent the hole and electron concentrations, respectively. The effects of
composition, polarization, and free-carrier screening are thus fully included.
With the knowledge of the band profile for both the valence band and the
conduction band, one can determine the electronic states in the heterostructure by
solving the Schr€odinger (Equation 2.59) equation, as a function of the spatial
coordinate z. In the effective mass approximation, one needs to solve the following
eigenvalues problem:
2 d 1 dCi ðzÞ
h
þ V c ðzÞCi ðzÞ ¼ E i Ci ðzÞ;
2m0 dz mz dz
ð2:111Þ
for which the appropriate choice of the effective masses in the conduction band and
in the valence band is done by preliminary calculation using the tight binding
approximation. The solution of Equation 2.111 determines the eigenstates Ei in the
conduction band and in the valence band, and the corresponding eigenfunctions Ci.
It is imperative to note that free carriers cannot eradicate the polarization charge,
which is bound and invariable (unless structural changes are made), but one can
screen it to a degree determined by carrier concentration. Likewise, polarization
charge is bound charge and cannot by itself be the source of free carriers, but it
can cause a redistribution of free carriers that would tend to screen the polarization
charge.
In addition to the polarization charge related parameters that must be known to
solve the Schr€odinger and Poisson equations, one must also know the dielectric
constants, doping levels, effective masses, and band discontinuities, in addition to
compositions and the layer thicknesses used. The band discontinuity issue is
discussed in Volume 2, Chapter 1, but suffice it to say that the bandgap of the
ternaries is found from Equation 2.108 with the appropriate bowing parameter, and a
certain fraction of the total bandgap difference is assigned to the conduction band and
the rest to the valence band. To mitigate the process, the band alignment for all the
nitride heterostructures is of type I, which means that the larger bandgap straddles
the smaller bandgap one. Typically, 60–70% of the total band discontinuity is assigned
to the conduction band. A linear interpolation for the effective masses, given in
Chapter 1, in ternaries from binary end points can be used. While the tables provided
in Chapter 1 list the dielectric constants for binaries, knowledge of them leads to the
ternary dielectric constants via linear interpolation. Relative dielectric constants of
10.28, 10.31, and 14.61 (compares with 15.1 presented in Chapter 1) for dielectric
constants in GaN, AlN, and InN, respectively, have been calculated [472]. The GaN
dielectric constant is nominally larger than that of AlN, but in this particular
investigation the data are as listed above. Following the so calculated figures and
applying the linear interpolation leads to dielectric constant for ternaries.
2.12 Polarization Effects
eAlx Ga1 x N =e0 ¼ 10:28 þ 0:03x;
eInx Ga1 x N =e0 ¼ 10:28 þ 4:33x;
eAlx In1 x N =e0 ¼ 14:61 4:33x:
Using the values for the binary dielectric constants tabulated in Chapter 1, the linear
interpolation scheme for the ternary dielectric constants lead to
eAlx Ga1 x N =e0 ¼ 10:4 1:9x;
eInx Ga1 x N =e0 ¼ 10:4 þ 4:9x;
eAlx In1 x N =e0 ¼ 15:3 4:9x:
ð2:112Þ
In dealing with devices such as MODFETs where a Schottky barrier is placed
on top, the Poissons equation, through boundary conditions, is affected by the
metal barrier height as well as the applied bias on the metal. The Schottky barrier
height for the ternaries can also be interpolated from the binary end points. It
should also be noted that the barrier height depends on the particular metal used, the
details of which is discussed in Volume 2, Chapter 1. Because the Fermi level on the
surface of GaN is not fully pinned in that heavier metals with large work functions
lead to larger barrier heights, it is plausible and natural to assume that the barrier
height would increase with an increase in AlN content and decreases with InN
content. However, for InN the Fermi level is most likely in the conduction band
already, as in InSb and InAs. Limited studies of metal contact potential on nitride
semiconductors make it difficult to state a molar fraction dependence of this
parameter. However, Ti and Ni contacts on Al0.15Ga0.85N have been studied and
were compared to those on GaN. The barriers heights for Ti and Ni increased on
Al0.15Ga0.85N [473]. Comparing the data deduced for Ni using I–V, C–V, and
photoemission methods, barrier heights of 0.95 (0.84 eV if not corrected for the
nonideal ideality factor), 0.96, and 0.91 eV, respectively, have been obtained. For
Al0.15Ga0.85N, the same figures are 1.25 (1.03 eV if not corrected for the nonideal
ideality factor), 1.26, and 1.28 in order, using the same methods. These figures
represent an increase of about 0.3 eV in barrier height for Al0.15Ga0.85N over GaN.
Assuming that Ni is used for Schottky barriers, the barrier height for GaN is 0.95 eV,
and more boldly reported figures for one mole fraction do actually represent a figure
consistent with a linear interpolation, one can express the molar fraction dependence
of the barrier height on AlxGa1xN as
fAlx Ga1 x N ¼ 0:95 þ 2xV:
ð2:113Þ
Following Ref. [84], the linear interpolation for the other two ternaries modified for
barrier height on GaN used here are
fInx Ga1 x N ¼ 0:95 0:36xV;
fAlx In1 x N ¼ 0:59 þ 1:36xV:
ð2:114Þ
These expressions lead to barrier heights (qf or ef) of 0.95, 2.95, and 0.59 eV on
GaN, AlN, and InN, respectively. With the exception of the value for GaN, the rest is
really speculative at this point.
j271
j 2 Electronic Band Structure and Polarization Effects
272
2.12.4.1 Ga-Polarity Single AlGaN/GaN Interface
Returning to single-interface structures, such as those used in MODFETs (or HFETs),
the total interface charge, for example, at a gated AlxGa1xN and GaN interface, rs, for
an n-type case would be sum of total polarization charge rp and free-carrier charge ns.
er e0
E Fi DE C
;
ð2:115Þ
rs ¼ rp ns ¼ rp V G fB V p2 þ
qdAlGaN
q
where VG is the applied gate bias in terms of V, fB is the Schottky barrier height in
terms of V, on AlxGa1xN, EFi is the Fermi level in GaN at the interface with respect to
the edge of conduction at the interface, DEC is the conduction band discontinuity
between the ternary AlxGa1xN and GaN, and Vp2 is the voltage drop across the doped
AlGaN, which in turn is given by V p2 ¼ qN d d2d =2er e0 where Nd is the donor
concentration in AlxGa1xN, all assumed to be ionized, dd is the thickness of the
doped AlGaN, and ere0 is the dielectric constant of AlxGa1xN. Vp2 is negative for
depleting voltage in the case of which it would add to the Schottky barrier height. We
should point out at this stage that the form of Equation 2.115 is good for determining
the sheet carrier concentration at the interface, but when used for FETs both the
polarization and the charge induced by doping must be lumped into the threshold or
off voltage. For details see Volume 3, Chapter 3.
For a p-type semiconductor, the semiconductor charge as well as the reference for
the Fermi level and the band discontinuity should be that at the valence band
edges [88,474]. The effect of any undoped AlxGa1xN layer designated as having a
thickness of di is small because its thickness is several nanometers and is neglected in
Equation 2.115. This setback layer was originally employed by the author in the GaAs
system to further reduce remote Coulomb scattering for increased mobility. A
detailed treatment including the effect of di can be found in Ref. [474]. For an
undoped AlGaN/GaN heterostructure, the Vp2 term can be set to zero. In this case
the boundary conditions for potential or the Fermi level are made consistent with
the Schottky barrier height on the Alx Ga1 x N surface. In the bulk of the structure,
due the special nature of the quasi-triangular barrier at the interface, shown in
Figure 2.46, the Fermi level is generally taken to be near the midgap of the smaller
bandgap material, which in this case is GaN.
As in the case of quantum wells, the one-dimensional Schr€
odinger–Poisson
solver can be iteratively used to determine the band and carrier profile (simultaneous
solution of Equations 2.110 and 2.111 in an iterative mode either in the tight binding
realm or the effective mass approximation, but in the current example it is in the
effective mass realm). The bound charge can be represented by a thin and heavily
doped interfacial layer, the thickness of which is about 1 nm or less, keeping in mind
that the total charge associated with such a fictitious layer must be equal to the
bound charge. In one investigation, a thickness for this fictitious layer of 0.6 nm was
used. To reiterate, to solve this pair of equations boundary conditions at the interface
and surface as well as the structural parameters must be known. In a typical undoped
AlGaN/GaN MODFET, the GaN buffer layer is unintentionally doped with a level
of ND 1016 cm3, and the thickness of this buffer layer spans 1–3 mm. The
barrier thickness spans 10–20 nm. Because the structure is not intentionally doped,
2.12 Polarization Effects
Ec
AlGaN
GaN
di
qφB
Ec
ΔEc
-qVG
E1
E0
EF
EFi
dd
x=0
Figure 2.46 Conduction band edge of what is
generically referred to as a modulation-doped
structure based on the AlGaN/GaN system. The
origin of the interface charge is aggregate due to
polarization and free carriers. In Ga-polarity
samples, AlGaN grown on GaN produces
polarization charge (due to spontaneous
polarization and piezoelectric polarization
x
because AlGaN is under tensile strain), causing
accumulation of electrons at the interface in
addition to any electron donated by the any donor
impurities, intentional or unintentional, in
AlGaN. The diagram is shown for a doped
AlGaN, the doped portion of which is indicated
by dd and the undoped part is indicated by di.
it is assumed that unintentional impurities in the form of contamination or native
defects, such as O, Si, VN, are responsible for supplying the free carriers, which is
measured with the accompanying assumption that it is sufficient to completely
screen the positive polarization charge, þr/q, at the interface [476]. The influence
of doping specifically in the case of AlGaN/GaN has been treated, for example, by
Chu et al. [475]. In short, the sheet charge at the interface of such an undoped
structure is dominated by the polarization-induced charge. On Ga-polarity surfaces,
this charge increases with increasing AlN molar fraction in the barrier because both
the piezo and spontaneous components of the polarization charge increase, assuming of course that the barrier is coherently strained. The conduction band and
electron concentration profiles for an undoped Ga-face Al0.3Ga0.7N/GaN (30 nm/
2 mm) heterostructure with an Ni Schottky contact on top are shown in Figure 2.47a.
An electric field strength of about 0.4 MV cm1 in the barrier and a sheet electron
concentration of 1.2 · 1013 cm2 are induced by polarization. The bound sheet
density and 2DEG sheet carrier density induced by polarization in heterostructures
(identical to that shown in Figure 2.47a with the exception that the alloy compositions
in the barrier has been changed) are shown in Figure 2.47b and c with solid lines for
both the linear and nonlinear polarization cases. The underlying assumptions
are that the GaN buffer layer is relaxed, the barriers are coherently strained, and
the physical parameters of importance (Cij, eij, and Psp) linearly scale from binaries
to ternaries.
j273
j 2 Electronic Band Structure and Polarization Effects
274
The estimated error depicted by the gray area in Figure 2.47c is primarily due to
uncertainties in the barrier thickness and the conduction band offsets. The sheet
carrier concentrations of 2DEGs confined in an AlxGa1xN/GaN heterostructure for
x ¼ 0.5 have been measured by C–V profiling using Ti/Al ohmic and Ni Schottky
contacts [84]. The highest measured and calculated sheet carrier concentration for
2.0
+s
E = 0.41 MV cm–1
1.5
2DEG
1.0
Energy (eV)
13
Ns = 1.2 10 cm –2
eΦ
1.23 eV
0.5
CB
0
Δ
0.30 eV
–0.5
(a)
0
10
ΔE
EF
c
0.38 eV
E0 = 0.17 eV
30
40
20
Depth (nm)
50
Figure 2.47 (a) Self-consistent calculation of the
Schr€
odinger–Poisson equations for the
conduction band edge and the electron density
profile for an undoped Ga-polarity Al0.3Ga0.7N
(30 nm)/GaN (2 mm) single-interface
heterostructure. Also shown are the Schottky
metal on the surface and the polarizationinduced surface and interface charges. (b) The
polarization-induced bound interface charge
density as a function of the alloy composition in
the barrier for Ga-polarity AlxGa1xN/GaN
heterostructures for the case of relaxed buffer
layer and coherently strained barrier layer. The
upper solid line for sheet charge corresponds to
the case of linear interpolation of physical
parameters (Cij, eij, and Psp) from the binary
compounds. The lower solid line for the sheet
charge, on the contrary, corresponds to the case
of nonlinear extension of polarization from
binary end points. The dashed lines depict the
lagging sheet density with increasing molar
fraction, x, due to partial relaxation, which is
60
accounted for by the measured degree of barrier
relaxation into account. (c) 2DEG sheet carrier
concentrations ns in unintentionally doped Gaface AlxGa1xN/GaN heterostructures (with
30 nm AlGaN and 2000 nm GaN – the structure
referred to in (b)) as obtained by C–V profiling
versus alloy composition of the barrier (open
symbols), compared with the theoretical
predictions for the bound interface charge r/q
calculated using (i) a linear interpolation
between the macroscopic polarizations of the
binary compounds (upper solid line) and (ii) the
nonlinear piezoelectric and spontaneous
polarization (lower solid line). The sheet charge
of the 2DEGs is then calculated as in (ii),
considering in addition the depletion by a Ni
Schottky contact (dotted line). The dashed lines
account for the experimentally observed strain
relaxation of the barrier for x > about 0.4.
Courtesy of O. Ambacher. The related details can
be found in Refs [476,84,479].
2.12 Polarization Effects
1014
Bound interface density σ/q(cm–2)
Linear
interpolation
Coherently
strained
1013
Nonlinear
1012
0
0.4
0.2
(b)
0.6
0.8
1.0
Molar fraction, x
3.5
Sheet carrier concentration ns (1013 cm–2)
Relaxed
Ga-face
Alx Ga1–x N/GaN
(30/2000 nm)
3.0
Pseudomorphic
2.5
2.0
Partly
relaxed
Linear
interpolation
1.5
1.0
0.5
Nonlinear
0
(c)
0
0.1
0.3 0.4
0.2
Molar fraction, x
0.5
0.6
Figure 2.47 (Continued )
AlxGa1xN/GaN heterostructures is 2 · 1013 cm2 for x ¼ 0.37, as higher AlN molar
fractions in the alloy for 30 nm barrier causes a partial relaxation, lowering the piezo
contribution [476]. The extrapolation, indicated by dashed lines in Figure 2.47c,
represents the case when the mole fraction is sufficiently large to cause some degree
of relaxation. As expected, the sheet density decreases nonlinearly with reducing
mole fraction for the 30 nm barrier modeled. Among the two curve that bend over, the
upper one is the total calculated polarization charge and the lower one is the
j275
j 2 Electronic Band Structure and Polarization Effects
276
calculated screening charge using the one-dimensional Schr€
odinger–Poisson equations. When full or partial relaxation occurs, the piezo component of the polarization
charge is reduced while the spontaneous component remains unchanged. The
experimental data along with the error bars are shown in open circles. Overall,
there is then a reduction of the charge. Likewise, the screening sheet carrier
concentration is also reduced. The obvious conclusion that can be made is that a
much better agreement is attained between calculations and experiments when the
nonlinear polarization is used. The case of the nonlinear polarization is stronger in
the 2DEG case than it is in the quantum well case due to the direct nature of the
measurement.
Ridley et al. [477] presented an empirical expression relating the sheet carrier
concentration for a nominally undoped AlGaN/GaN single-interface heterostructure
having barrier thicknesses of greater than 15 nm and AlN molar fractions over 6%.
ns ðxÞ ¼ ½ 0:169 þ 2:61x þ 4:50x 2 1013 cm 2 :
ð2:116Þ
Naturally, the sheet charge becomes affected by parameters other than the AlN
molar fraction when the barriers are made much thinner, as can be deduced from
Equation 2.115. Using an expression similar to Equation 2.115, Ambacher et al. [84]
plotted the dependence of the sheet density of barrier thickness for several AlN molar
fractions, namely, x ¼ 0.15, 0.30, and 0.45. A priori, it is clear that beyond a certain
thickness of the barrier, the density should saturate even if coherent strain prevails, as
shown Figure 2.48. Ambacher et al. [84] also measured the sheet density for a set of
samples with x ¼ 0.3 by C–V profiling for barrier thicknesses spanning the range of
1–50 nm, and presented the data together with those from other reports.
2.12.4.2 Ga-Polarity Single AlxIn1xN/GaN Interface
This system, if for nothing else, is of importance because the entire structure can be
lattice matched, leading to vanishing piezoelectric polarization and thus allowing one
to investigate and probe only the spontaneous polarization in undoped structures.
Doing so would enhance our confidence in spontaneous polarization calculations, as
there are fewer parameters to be determined and thus less uncertainty. For high
concentrations of Al (for x > 0.6), the bandgap of AlxIn1xN is larger than that of
GaN, and if used in conjunction with Ga-polarity GaN, a 2DEG would result due to
polarization or doping in AlxIn1xN or both. For lattice-matched conditions where
the x-value is about 0.82, the bandgap of the AlxIn1xN alloy is about 4.7 eV to
produce sufficiently large band discontinuity with GaN, imperative for MODFET
structures. Ambacher et al. [84] prepared coherently strained AlxIn1xN (50 nm)/
GaN (540 nm) single-interface heterostructures with Al concentrations between 0.78
and 0.88, and applied X-ray diffraction to determine the structural state of GaN,
meaning relaxed or strained (GaN grown on sapphire is typically under compressive
strain due to thermal mismatch). Through the in-plane and out-of-plane strain
(which can both be measured using reciprocal space X-ray diffraction mapping) or
the lattice constants, the mole fraction of the barrier and, through repeated attempts,
the mole fraction giving rise to lattice matching conditions can be determined.
Ambacher et al. [84] determined the lattice matching composition of AlxIn1xN to
2.12 Polarization Effects
10
14
Sheet carrier concentration ns (cm–2)
x = 0.45
10
13
= 0.3
= 0.15
1012
Ga-face AlGaN/GaN
10
11
0
10
20
30
40
50
60
d AlGaN (nm)
Figure 2.48 Barrier thickness, d, dependence of sheet density in
nominally undoped and coherently strained AlxGa1xN/GaN
heterointerfaces for x ¼ 0.15, 0.30, and 0.45 (solid lines).
Experimental data available for x ¼ 0.3 measured by C–V profiling
for barrier thicknesses spanning 1 and 50 nm, representing an
aggregate from several reports, are shown [84].
GaN to be x ¼ (0.83 0.01), as shown in Figure 2.49, which is compared with other
experimental values of 0.82–0.83 [451]. The weak compressive strain in GaN was
determined to be e ¼ 1.9 · 103, which would result in a piezoelectric polarization
of 1.5 · 103 Cm2. The residual strain in GaN would lead to a bound sheet density
of only 1012 cm2, which is much smaller than the 1013 cm2 electron sheet
density. This implies explicitly that interface charge is dominated by the gradient
in the spontaneous polarization across the GaN/AlxIn1xN interface, as show in
Figure 2.50, which shows the calculated bound sheet density induced by a gradient in
spontaneous polarization (upper dashed line) and strain-induced piezoelectric (lower
solid line) for a range of compositions near the lattice matching conditions and the
resultant 2DEG sheet carrier density (upper solid line). Also shown are the experimental 2DEG densities obtained by C–V measurements. The polarization and thus
the sheet density have been obtained by a linear interpolation of the physical
parameters (Cij, eij, and PSP) of the binary compounds but by taking the nonlinearity
of Ppz and PSP into account. The calculations confirm the obvious in that the
measured high electron sheet density can be accounted for by the spontaneous
polarization charge.
j277
j 2 Electronic Band Structure and Polarization Effects
278
6
cAlInN (x)
4
aAlInN (x)
Strain, e (10–3)
2
Tensile strain
0
Compressive
strain
–2
–4
Lattice
matched
–6
0.75
0.85
0.80
Molar fraction, x
Figure 2.49 Strain in AlxIn1xN barrier along the
c-axis and on the basal plane as determined from
the measured lattice constants, cAlxIn1xN and
cAlxIn1xN by high-resolution X-ray diffraction
reciprocal mapping. AlxIn1xN for x ¼ 0.83 can be
0.90
grown lattice matched to GaN, leading to
vanishing piezoelectric polarization due to
misfit. However, residual strain, due to thermal
mismatch between GaN and the substrate, can
still induce piezoelectric polarization [84].
Note that for Ga-polarity samples and AlN compositions less than the lattice
match conditions, the AlxIn1xN barrier layer is under compressive strain in-plane
and therefore the piezo-induced polarization is opposite in sign to that for spontaneous polarization. When the AlN molar fraction exceeds that for matching condition, the in-plane strain is tensile in the case of which the two polarization charges are
additive. Near the lattice matching conditions, the calculated sheet density is about
2.95 · 1013 cm2 and falls above the measured values. The reason for this discrepancy
could be related to samples themselves, either in the form of defects near the
interface, surface adsorbates, and/or inhomogeneities caused by In, and underestimation of the bowing parameters associated with the polarization charge.
2.12.5
Polarization in Quantum Wells
For multiple interface heterostructures, the sheet carrier density and barrier thickness, as well as the width quantum wells, are of interest because the total potential
drop across the structure is directly proportional to the product of polarization field
and well width in constant field approximation if free-carrier screening is neglected.
2.12 Polarization Effects
4
s sp
e (P )
2
n (x)
S
1
0
–1
–2
0.75
s z
(P )
E
0.80
Lattice matched
Sheet carrier concentration (1013cm–2)
3
0.85
0.90
Molar fraction, x
Figure 2.50 Compositional dependence of the
spontaneous polarization (dashed line) and
piezoelectric polarization (lower solid line)
induced charge density reduced to interface
sheet charge in an AlxIn1xN/GaN
heterojunction using the nonlinear interpolation
for polarization discussed in the text. For AlN
molar fractions below the lattice matching
conditions, the two polarization charges oppose
and above the lattice matching value, they add.
The calculated sheet electron concentration
(upper solid line) and the measured sheet
electron density are also shown, the latter of
which has been obtained by both Hall effect
(solid symbols) and C–V profiling (open
symbols) [84].
This issue goes to the heart of lasers, particularly in the stages as the gain is built up, in
that the well widths greater than about 5 nm are not used. At higher injection levels,
the polarization-induced field is screened pretty much [458]. To illustrate the point,
the conduction band profile of a 10 nm GaN/In0.2Ga0.8N quantum well as calculated
by Della Salla et al. [461] is shown in Figure 2.51 for several sheet densities. Even at a
substantial sheet density of n2D ¼ 5 · 1012 cm2, a nearly uniform electrostatic field of
strength 2.5 MV cm1 is still present in the well. One needs to increase n2D to
5 · 1013 cm2 before recovering the quasi-field-free shape of the quantum well that is
needed for lasers. This is achieved much earlier in thinner quantum wells. In wells,
the electrons and holes are indeed spatially separated by the polarization field, but the
free carrier induced field acts to cancel the polarization field, which is efficient for
high sheet densities. This reestablishes the efficient electron–hole recombination.
Time-resolved PL (TRPL) is a wonderful way of seeing the effect of polarization.
Due to band bending induced by polarization, when the optical excitation pulse is
turned off, the free-carrier density goes down and so does the screening. As a result, a
j279
j 2 Electronic Band Structure and Polarization Effects
280
[0 0 0 1]
Conduction band edge energy (eV)
4.0
GaN/In0.2Ga0.8N
3.5
3.0
2.5
2.0
n2 D = 1013cm–2
0.5
1.0
2.0
5.0
1.5
1.0
0.5
0
10
20
30
Depth (nm)
40
50
Figure 2.51 Conduction band profile of a 10 nm GaN/In0.2Ga0.8N
quantum well for various levels of free carriers ranging from
5 · 1012 cm2 to 5 · 1013 cm2 either present by doping or
injection. The effect of polarization is nearly all but wiped out for
the largest sheet electron concentration [461].
red shift accompanied with increased carrier lifetime due to lowering of the overlap
between the electron and hole wave function occurs, as they are pushed to the
opposing end of each well [478]. For details, the reader is referred to Volume 2,
Chapter 5.
2.12.5.1 Nonlinear Polarization in Quantum Wells
Asinthecaseofbulk binariesand alloys, the polarization issue inheterostructuresneeds
a revisit to consider the nonlinearities discussed above in the context of binary and alloy
nitride bulk layers. Assuming that the ternary nitride alloys have random microscopic
structure, the spontaneous polarization of random ternary nitride alloys, in unit C m2,
has been expressed by Fiorentini et al. [479] to the second order in the composition
parameter x, as expressed in Equation 2.95 but repeated here for convenience.
sp
PAlx Ga1 x N ¼ 0:09x 0:034ð1 xÞ þ 0:019xð1 xÞ;
sp
PInx Ga1 x N ¼ 0:042x 0:034ð1 xÞ þ 0:038xð1 xÞ;
sp
PAlx In1 x N
ð2:117Þ
¼ 0:09x 0:042ð1 xÞ þ 0:071xð1 xÞ:
The first two terms in all three equations indicated in Equation 2.117 are the usual
linear interpolation between the binary compounds represented by Equation 2.86.
However, the third term is the so-called bowing term encompassing the quadratic
nonlinearity as in the case of the bandgap bowing parameter discussed in Section 1.5.
The coefficient of the third term is the bowing parameter discussed in conjunction
with Equations 2.95 and 2.92. Higher order terms are neglected, but their effect was
estimated to be smaller than 10% in the worst case being the AlInN alloy [453].
2.12 Polarization Effects
For piezoelectric polarization, it was shown [453] in conjunction with Equation 2.96
that Vegards law holds provided that the appreciable nonlinearity of the bulk
piezopolarization of the component binaries as a function of strain is accounted
for. Doing so has led, in general, to a good agreement with experimental results [480].
For a model AlxGa1xN alloy, the piezoelectric polarization can be related to the
binary end points using the Vegards law, but recognizing that the terms for the bulk
binaries must contain the nonlinear terms as indicated in Equation 2.96.
Such polarizations can be expressed accurately and compactly (in units of C m2) as
pe
PAlN ¼ 1:808e þ 5:624e2
pe
PAlN ¼ 1:808e 7:888e2
pe
PGaN ¼
pe
PInN ¼
for e < 0;
for e > 0;
0:918e þ 9:541e ;
2
ð2:118Þ
1:373e þ 7:559e2 ;
as a function of the basal strain of the alloy layer in question, with a(x) and asubs or a0 as
the lattice constants of the unstrained alloy at composition x and of the relaxed buffer
layer or the substrate.
eðxÞ ¼ ½asubst aðxÞ=aðxÞ:
ð2:119Þ
In the case of pseudomorphic growth on GaN buffer layers, basal strain e can be
calculated directly from the lattice constants, which are found to follow Vegards law as
a function of composition (depending on the lattice constants used):
aAlx Ga1 x N ðxÞ ¼ aGaN xðaGaN aAlN Þ ¼ 0:31986 0:00891x nm;
aInx Ga1 x N ðxÞ ¼ aGaN þ xðaInN aGaN Þ ¼ 0:31986 þ 0:03862x nm;
aAlx In1 x N ðxÞ ¼ aInN xðaInN aAlN Þ ¼ 0:35848 0:04753x nm:
ð2:120Þ
The combination of Equations 2.118–2.120 provide a convenient way of determining
the polarization dependence on basal strain. The coefficients in Equation 2.118 are
related (not equal) to piezoelectric constants and come about from the ab initio
calculations [453].
The polarization charge values calculated using Equations 2.118–2.120 for heterostructures can be used together with a self-consistent Schr€
odinger–Poisson solver
based, for example, on effective mass theory (not as accurate but efficient) or tight
binding (more accurate but computation intensive) to determine field and charge
distribution in the entire heterostructure as well as the effect of free carriers [448,460].
Typically, for a given structure or set of structures, two classes of observable interest
can be simulated and compared with experiment.
Experimental confirmation for the nonlinear theory can be garnered from quantum wells by probing the red shift for a given sized well and its dependence on
excitation, and measuring the magnitude and gate voltage dependence of the
interface sheet carrier concentration at an AlGaN–GaN interface with the aid of the
one-dimensional self-consistent simultaneous solution of Schr€
odinger and Poisson
equations in the effective mass approximation. Additional confidence can be
estimated by repeating these experiments for these structures where, for example,
j281
j 2 Electronic Band Structure and Polarization Effects
282
only the barrier mole fraction is changed [479]. Fortunately, the results are very
sensitive to the values of the polarization in the different layers, and therefore the
built-in field or the screening charge. The particular bowing parameters in the
polarization expression yielding the best agreement with experiment can be assumed
to be valid. The point should be made that the C–V measurements are probably the
most direct and optical shifts the least direct. The C–V data and the simulations based
on the theories described above agree very well [479]. The attempt here is to show that
inclusion of the nonlinear effects in calculations lead to a better agreement between
the experiments and theory in quantum wells and AlGaN/GaN heterointerfaces.
In terms of the quantum confined stark effect (QCSE), the photoluminescence
energy of AlxGa1xN/GaN QWs with well thicknesses of 1,1.5, 2.5, 4, 6, and 8 nm, and
with barrier alloy composition of x ¼ 0.08, 0.13, 0.17, and 0.27, respectively, reported
in Ref. [481] were used to determine the polarization-induced electric field. The field
determined by measuring red shift from the PL transition energies versus the mole
fraction in the barrier of quantum wells as well as calculated values from the
polarization charge determined using the linear and the nonlinear approach are
shown in Figure 2.52. In both types of calculations, the electric field in the GaN QWs
was obtained by solving self-consistently the coupled Schr€
odinger–Poisson equations
including polarization-induced interface charges. In the first approach the polarization of the AlGaN barriers that are assumed under tensile coherent strain, as the bulk
buffer layers were GaN, is determined by a linear interpolation between the elastic
2.5
Linear
interpolation
Electric field (MV cm–1)
2.0
Nonlinear
approach
1.5
1.0
Ga-polar
AlxGa1–xN/GaN
QWs
0.5
0
0
0.1 0.2 0.3 0.4 0.5 0.6
AIxGa1–x N molar fraction, x
Figure 2.52 Polarization-induced electric fields
in high resistivity and Ga-face AlxGa1xN/GaN
MQWs versus the alloy composition of the
barrier. The upper line represents the field
predicted by linear interpolation of binary
compound polarization. The lower line
represents the calculated field using the
nonlinear polarization concept. Open circles
are deduced, via self-consistent effective mass
calculations, from the polarization-induced Stark
shift of excitonic recombination reported in
Ref. [481] via a self-consistent effective mass
calculation. In addition, the experimental data
published by Langer et al. [482] and Kim
et al. [483] are also shown in filled
circles [479].
2.12 Polarization Effects
and piezoelectric constants and the spontaneous polarizations of the binary compounds AlN and GaN. In the second approach, the nonlinearity of the polarization of
AlGaN as described by Equations 2.117 and 2.118 was taken into consideration.
Clearly, the electric field calculated including the nonlinearity in polarization versus
barrier alloy composition does much better at reproducing the experimental data
taken from Ref. [481]. In addition, the experimental data published by Langer
et al. [482] and Kim et al. [483] are also shown in filled circles.
A more convincing arguments for nonlinearity in polarization in the context of
polarization charge and resultant red shift in the spectra is made by pressuredependent measurements of the transition energies as presented by Vaschenko
et al. [484]. They considered a quantum well system with background unintentional
dopants and excitation-induced carriers in the case of which the field deviates from
that indicated in Equation 2.105.
total
ðPtotal
Vs
W PB Þ þ r
þ
LW þ LB
LW eB þ LB eW
1 d LW
1 d LB
qN D
þ
;
2
2
eW
eB
E W ¼ LB
ð2:121Þ
where eW,B is the permittivity of the GaN wells and AlxGa1xN barriers, respectively,
(assumed to be independent of pressure in this work), LW,B are the cumulative
thicknesses of the wells and the barriers in the MQW structure, r is the total twodimensional photogenerated charge density in the wells, Vs is the surface barrier
potential determined as in Ref. [485], ND ¼ 1017 cm3 is the assumed background
doping concentration based on bulk GaN layers grown by MBE, which was used to
produce the structure, and d is the distance from the barrier–buffer interface to the
well where the field is calculated. The first term in Equation 2.121 is similar to
Equation 2.105 but the former having an additional charge r (the total two-dimensional photogenerated charge) in the first term in addition to second and third terms.
The second term represents the field due to the surface barrier potential which
neglects the effect of dielectric discontinuity between AlGaN and GaN, and the third
terms represents the effect of dopant on the field. If Vs, r, and ND were made to go to
zero, Equation 2.121 reduces to Equation 2.105.
To find PW PB as a function of pressure, the experimentally measured PL peak
energy variation, which was assumed to be representing the n ¼ 1 electron and heavyhole transitions, with well width was fitted to the calculated dependence of the same
transition, The nonlinear behavior in this case is revealed by determining the
PW PB (barrier–well polarization difference) as a function of applied hydrostatic
pressure.
Figure 2.53 shows the fit to the measured PL peak energies in samples in which the
AlN mole fraction varied as 0.2, 0.5, and 0.8 at a pressure of 5 GPa. The solid lines are
calculations with the polarization-induced field, whereas the dashed ones are without
such field. The experimental data are shown in open symbols. The good agreement
between the calculations where PW PB was treated as fitting parameter and the
experiment underscores the crucial nature of the built-in electric field in the
determination of the well width dependence of the ground-state energy.
j283
j 2 Electronic Band Structure and Polarization Effects
284
4.2
4.0
3.8
Photon energy (eV)
3.6
3.4
3.2
3.0
2.8
2.6
x = 0.2
x = 0.5
2.4
x = 0.8
2.2
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
Well width (nm)
Figure 2.53 Well width dependence of the PL peak energy at
5 GPa. Open symbols correspond to the experimental points and
the solid lines depict the fits to the experimental data obtained
with PW PB as an adjustable parameter. The dashed lines
correspond to the e1 hh1 transitions calculated assuming zero
field [484].
The polarization difference PW PB and resulting electric field in the 2.9 nm wells
for the x ¼ 0.5 MQW sample, as determined by the procedure described above, is
presented in Figure 2.54. For the x ¼ 05, and the others that are not shown here,
PW PB noticeably increases with pressure, resulting in an increase of the built-in
field in accordance with the mole fraction in the barrier reaching 0.76 MV cm1 in the
samples with x ¼ 0.5 at 8 GPa. Figure 2.54 also shows the calculated results of the
pressure dependence of PW PB in that the solid lines correspond to PW PB
calculated with the linear polarization [448]. This linear polarization model overestimates the values of PW PB at atmospheric pressure and underestimates the
pressure dependence as compared to experiments. The dashed-dotted lines represent calculations where only the volume-conserving strain dependence of the GaN
and AlN piezoelectric coefficients is taken into account [456]. Clearly, this model
agrees better with the experimental data than the linear polarization model that is
consistent with the conclusions of Perlin et al. [486], where the pressure dependence
of PL in GaN/Al0.13Ga0.87N QWs was found to be adequately described by the volumeconserving strain dependence of the piezoelectric coefficients. Lastly, the dashed line
2.12 Polarization Effects
0.058
3.6
0.056
0.052
3.2
0.050
3.0
0.048
2.8
0.046
0.044
Built in electric field, MV/cm
Polarization difference (Pw-PB), (cm–2)
3.4
0.054
2.6
0.042
2.4
0.040
T = 35 K
x = 0.5
0.038
0
2
4
6
8
10
2.2
Pressure (GPa)
Figure 2.54 Pressure dependence of PW PB
and corresponding electric field in 2.9 nm
AlxGa1xN/GaN MQWs with x ¼ 0.5. The open
circles represent the calculated points obtained
from the fit to the PL data as shown in
Figure 2.53. The solid line corresponds to
PW PB calculated with the linear polarization
that overestimates the data obtained from
experiments at zero pressures and
underestimates the slope of the pressure
dependence of polarization. The dashed-dotted
line shows the calculations, where only the
volume-conserving strain dependence of the
GaN and AlN piezoelectric coefficients is
considered, which overestimates the differential
polarization but does very well in terms of the
pressure dependence of the differential
polarization. Lastly, the dashed line shows the
result of calculations using the nonlinear
polarization behavior with the distinctly best
overall fit to the experimentally determined
values. The dotted line is a guide to eye [484].
shows the result of calculations where the nonlinear behavior of both the spontaneous and piezoelectric polarizations had been taken into account using the results of
Bernardini and Fiorentini [220,221]. Only the change in piezoelectric polarization
due to hydrostatic compression of the ideal crystal and that due to the increase in the
internal parameter u with pressure is considered here [86]. The spontaneous
polarization bowing was included at p ¼ 0 [220,221]. Although this is only an
approximation of the theory developed in Refs [220,221], this model also predicts
the slope of the pressure dependence of PW PB significantly larger than that of the
linear model.
In short, the added value of pressure dependence of differential polarization
causes this parameter to be very sensitive to which polarization picture is employed
for the transition energies in AlGaN/GaN MQWs to the point that one can clearly
state that the nonlinear dependence of the piezoelectric polarization in GaN and AlN
unequivocally predicts the experimental data best.
j285
j 2 Electronic Band Structure and Polarization Effects
286
2.12.5.2 InGaN/GaN Quantum Wells
Relatively better transport properties of InN as compared to GaN and possible
translation of the same to InxGa1xN are one of the draws for considering InxGa1xN
channels [229] for MODFETs, discussed briefly in Volume 3, Chapter 3. However,
experimental results have so far been disappointing. On the contrary, InxGa1xN
proved to be the magical material for its high radiative recombination efficiency
for optical emitters. As detailed in Volume 3, Chapters 1 and 2, all the highperformance optical emitters feature InxGa1xN in one form or another in their
active regions. Unlike the FETcases where GaN is used as the active layer and also the
buffer layer where the buffer layer is high resistivity, the conductivity of InxGa1xN is
high, which rules out its use as the buffer layer in an InxGa1xN channel FET.
Consequently, even if InxGa1xN were to be considered for the active layer, it must
be grown on GaN buffer layers. As for the barrier layer, it can be made of GaN or
some composition of AlxGa1xN. In any case, the InxGa1xN layer would be
straddled by large bandgap material on both sides with the resultant single quantum
well structure. Consequently, polarization in single-well InxGa1xN quantum
wells must be considered as discussed here. In a single quantum well structure,
in addition to interface sheet charge that contains information on polarization
and can be measured by electrical means, these structures also offer an additional
avenue to probe the polarization-induced charge through quantum-confined Stark
shift. While the optical properties of InxGa1xN quantum wells are discussed in
great detail in Volume 2, Chapter 5, including the associated QCSE and any Stokes
shift, a discussion of single quantum well for the sole purpose of polarization effects
is provided here for completeness. Ambacher et al. [84] reviewed the issue in
conjunction with nominally undoped, n-type GaN/In0.13Ga0.87N/GaN structures
with Ga-polarity, where the width of the quantum well dInx Ga1 x N varied between
sp
0.9 and 54 nm. The spontaneous polarization of the InGaN layer, P In0:13 Ga0:87 N ,
2
is 0.031 Cm and points in the ½0 0 0 1 direction and the piezoelectric polarization is calculated to be 0.016 Cm 2, which is antiparallel to the spontaneous
polarization because the InxGa1xN quantum well is under compressive strain.
The bound charge at the GaN–InGaN interface near the surface is positive and that
at the lower interface is negative for Ga-polarity sample due the compressive
strain that InxGa1xN is under. This implies that the electron accumulation
caused by screening would occur at the interface near the surface in n-type samples.
If p-type samples were considered, a hole accumulation would occur at the other
interface.
The total polarization-induced interface sheet density is then given by
sp
pz
sp
pz
ðPGaN þ P GaN Þ ðPInx Ga1 x N þ PInx Ga1 x N Þ:
ð2:122Þ
Recognizing that GaN is relaxed in this case, the samples are grown on GaN buffer
layers that are presumed to be relaxed and any residual strain is neglected. This leads
pz
to PGaN ¼ 0, and substituting the numerical values, one gets for the total polarization
ð 0:034 þ 0Þ ð 0:031 þ 0:016Þ Cm 2
¼ q1:18 1013 Ccm 2 ;
or 1:18 1013 electrons cm 2 :
2.12 Polarization Effects
InGaN SQW
x = 0.13
d = 54 nm
ns = 5 × 1012 cm–2
22
10
26 nm
Electron concentration (cm–3)
2.0 ×
× 10
1012
10
10 16
12 nm
2.6 × 1011
18
16
10
10
20
1018
2
20
10
10
10
22
1014
Electron concentration (cm-3)
10
10 12
4.3 nm
3.6 × 1011
+s –s
14
12
10
1
10
2
2
× 10
10
3
10 4
Depth (nm)
Figure 2.55 Electron concentration profiles unintentionally
doped, n-type GaN/In0.13Ga0.87N/GaN QWs having well widths of
4.3, 12, 26, and 54 nm, respectively, as deduced from C–V depth
profiling [84].
The GaN/In0.13Ga0.87N/GaN the heterostructures with GaN top layer and In0.13Ga0.87N quantum well with thicknesses of 130 and 20 nm, respectively, have been
examined by Ambacher et al. [84], and their electron profiles, as determined by C–V
measurements, are shown in Figure 2.55. For well widths less than 4 nm, any
electron accumulation was not observed, which implies failure to screen the bound
charge fully. In fact, as the quantum well thickness was increased from 4.3 to 54 nm,
the sheet carrier concentration increases from ns ¼ 3.6 · 1010 to 5 · 1012 cm2. The
increase in 2DEG sheet carrier concentration with well width, which follows a 2.5
power of well width, is much faster than the well width (the volume), which may be
attributed to the system not being in equilibrium in terms of screening for thinner
wells. Overall, the sheet density is about half of what is expected even in thick
quantum wells.
The discrepancy between predictions and experiments raises an interesting
question if the polarization-induced charge is fully screened by electrons and ionized
donors. It is clear that an electric field causes band bending, the total extent of which
scales with thickness, which in turn causes a red shift in radiative recombination
transition energy. Assuming that the polarization-induced charge at the free surface
and the GaN–substrate interface are fully screened, an electric field in the constant
j287
j 2 Electronic Band Structure and Polarization Effects
288
field approximation forms, which can be expressed as
E well ¼
ptotal
e0 ðeInGaN
r
1Þ
¼
total
P total
GaN P InGaN
:
InGaN
e0 ðer
1Þ
ð2:123Þ
The terms here have their usual meanings.
The electric field has been predicted to be 2.2 MV cm1 for In0.13Ga0.87N QWs,
which causes band bending to the extent of turning otherwise square quantum well
potential distribution to a triangular distribution in the constant field approximation.
The resultant red shift in transition energy, Stark shift, can be related to the
polarization charge through the field as
g
E InGaN E energy ¼ qE well W þ
9phqE W
pffiffiffi
8 2
2=3 1
mnInGaN
þ
1
p
mInGaN
1=3
;
ð2:124Þ
where mnInGaN
p
and mInGaN
are the electron and hole effective masses in InGaN. For an
InN mole fraction of 0.13, these values will not be much different from those for GaN.
W represents the InGaN well width. This would provide an additional means for
determining the polarization charge with an all-optical method such as photoluminescence. Extreme care must then be exercised to be certain about the InN composition and its bandgap as well as making sure that the optical transitions observed are
accurately related to the band edge, meaning their nature must be known. The
experiments must also be conducted at low injection levels as to not screen the charge,
and high injection levels to screen the charge. If screening is full and low injection
levels is truly low, then the difference in energy between the very low and very high
injection levels would represent the red shift due to the polarization charge, as was
done by Ambacher et al. [84] who also determined the bandgap of InGaN by reflection
measurements such as spectroscopic ellipsometry and room-temperature PL using
an excitation energy of 3.41 eV, only absorbed by the InGaN well. The QW region was
pumped with high-intensity light to generate a high density of electron–hole pairs to
fully screen the polarization-induced charge, thus the field. For In0.13Ga0.87N QWs
with widths of greater than 26 nm, a bandgap of 2.902 0.012 eV was measured,
which is consistent with the value for bulk (2.946 eV). The spectroscopic ellipsometry
and PL data show a monotonic increase in energy, reaching a value of 3.21 eV for a well
width of 0.9 nm, as the Stark shift diminishes with decrease in well width while the
quantum confinement increases, as depicted in Figure 2.56. The PL spectra measured
by 3.81 eVexcitation with a pump power density of 103 Wcm2 yield a PL peak position
that is increasingly shifted to lower energies if the QW width is increased from 0.9 to
5.3 nm (Stark effect). Knowledge of this red shift in the PL peak position together with
Equation 2.124 allows one to calculate the electric field strength as 0.83 MV cm1. This
field corresponds to a polarization-induced bound charge density of about 5 · 1012
cm2 and compares well with the data of 0.62 MV cm1 and charge of 3.7 · 1012 cm2
for In0.12Ga0.88N/GaN MQWs by Wetzel et al. [487].
As can be deduced from Figure 2.56, the polarization-induced red shift (Stark shift)
is not notable for QWs wider than 26 nm where the bandgap of InGaN and the PL
2.12 Polarization Effects
3.4
3.2
g
Energy (eV)
3.0
2.8
E InGaN
660 meV
E 0 e, 0 h
2.6
5 nm
2.4
2.2
10 –1
10 1
10 0
InGaN SQW thickness (nm)
Figure 2.56 The dependence of the effective
bandgap (without confinement effects) of
In0.13Ga0.87N used in QWs (solid line) and the
energy of the radiative recombination in QWs
assumed to be between electron ground state
(E0e) and hole ground state (E0h) on well width.
The effective bandgap is determined by
10 2
spectroscopic ellipsometry and RT PL with
3.41 eV excitation (high and low intensity for PL
to account for the Stark shift). Also noted in the
figure is that a change of 5 nm in the well
thickness leads to a change of 660 meV change in
the QW emission energy [84].
peak position agree within 40 meV. It also appears that polarization-induced charged
may either be fully screened, negating the constant field approximation throughout
the quantum well, or be the notorious In fluctuations discussed in details in Chapter 3
in terms of growth, Chapter 4 in terms of its effects on optical transitions, and
Volume 3, Chapters 1 and 2 in terms of its effects on optical emitters.
2.12.6
Effect of Dislocations on Piezoelectric Polarization
To fully consider the true nature of III-Nitride epilayers, the effect of dislocations on
piezoelectric polarization must be included. The case does hardly needs to be
made owing to the high density of dislocation (edge, screw, and mixed) that alters
the strain distribution and thus the piezoelectric polarization. In this vein, Shi
et al. [488] calculated the piezoelectric polarization around c-oriented screw and edge
dislocations in Wz GaN and found that polarization around screw dislocations
(having Burgers vector h0 0 0 1i) has no z-component, which is similar to a magnetic
field around a conducting line, so there is no charge induced either at the core
or around the screw dislocation. In the case of edge dislocations (having Burgers
vector 1/3h1 2 0i), in which the strain field is compressive on one side and tensile on
j289
j 2 Electronic Band Structure and Polarization Effects
290
(0 0 0 1) c-plane
-
Qs
+
Pe
Ps
bs
be
+
-
Figure 2.57 Schematic diagram illustrating the dislocation
geometry, associated polarizations and charge densities [488].
the other, calculations show that piezoelectric polarization has only the z-component
and its divergence vanishes at the core and around the dislocation. But at the
interface, it results in an effective surface charge for the difference in polarization
across the interface, as shown in Figure 2.57. It was estimated that for a c-oriented
edge dislocation the charge density could reach 1011 e cm2 within 0.1 mm of the core.
On the experimental side, electron holography was applied to investigate the built-in
field caused by polarization [489] and charge distribution at the dislocation [490]. In
undoped GaN, the holography results confirm that all dislocations are negatively
charged and the line charge densities are calculated as 1 and 0.3 for screw and edge
dislocations, respectively. Cai and Ponce [491] argued that screw dislocations always
have relatively higher charge density. The electrical activity associated with extended
defects is discussed in considerable detail in Section 4.1.6. We should be cognizant of
the fact that not only extended defects in general cause local strain and therefore
inhomogeneous strain but they also attract and often trap impurities, point defects,
and free charge. Moreover, the strain component affects the charge through the
piezoelectric component.
2.12.7
Thermal Mismatch Induced Strain
Having made the case that strain induced by both lattice mismatch and also by
thermal mismatch plays a profound role in polarization, let us discuss the thermal
mismatch case in some detail because the lattice mismatch component got good
deal of coverage already. Within the realm of thermal mismatch, the dominant
component is that introduced by the nonnative substrates used. The substrates
used are many in number and kind, but the dominant ones are sapphire and SiC
both of which introduce sizable thermal mismatch. An incomplete list of
2.12 Polarization Effects
substrates used, in addition to the ones cited, include other substrates, GaN, AlN,
g-LiAlO2, b-LiGaO2, NdGaO3, Si, GaAs, MgO, ZnO, ScAlMgO4, MgAl2O4, and (La,
Sr)(Al,Ta)O3. For a complete discussion refer to Chapter 4 and for a complete
compilation refer to Ref. [492]. In a nut shell, the thermal stress is relatively small
if GaN layers are grown on AlN, SiC, ScAlMgO4, Si, GaAs, and ZnO. However, it
is relatively larger when all other substrates are used. The stress in GaN is compressive
for all the substrates substrate except Si and SiC, which due to their small expansion
coefficients give rise to tensile strain, which is notorious for layer cracking. The overall
stress remains the same for nominally thick GaN layers when a sapphire substrate is
used with or without an AlN buffer layer but reduces by an order when a 6H-SiC
substrate is used with an AlN buffer layer. In these pages, the treatment reported in
Refs [492,493], which followed the model of Olsen and Ettenberg [494], for an arbitrary
stack of epitaxial layers on a substrate is followed.
The three-layer heterostructure stack layer model used to study the thermal-induced
strain is shown in Figure 2.58 with length L, width W, Youngs modulus Ei, layer
thickness ti, moments Mi, coefficient of thermal expansion a, forces Fi, strain e, and
curvature k. The index i ¼ 1 represents the substrate and i ¼ 2, 3, 4, . . . represents the
epitaxial layers.
Figure 2.58 (a) A set of unstrained platelets of
thicknesses ti (i ¼ 1, 2, 3, . . .) used to construct
the composite layer. (b) A case depicting the
films and the substrate to be in contact,
representing the epitaxial growth of two different
films on a substrate with a larger thermal
expansion coefficient (positive bending) as
compared to the films. The dimensions are
characterized by length L (into the page), width
W, and thickness ti. The terms Ei, Mi, and Fi
represent the Youngs modulus, moments
(bending forces), and forces, respectively.
j291
j 2 Electronic Band Structure and Polarization Effects
292
For one epitaxial layer with i ¼ 1 representing the substrate and i ¼ 2 representing
the epitaxial layer on it, one can surmise that from equilibrium of force, the total force
must vanish as
F 1 þ F 2 ¼ 0:
ð2:125Þ
Similarly, equilibrium of moments leads to
Wk
t1
t2
¼ 0:
ðE 1 t31 þ E 2 t32 Þ þ F 1 þ F 2 t1 þ
2
2
12
ð2:126Þ
The strain at the interface between the film and the substrate in terms of mechanical
parameters can be expressed as
e¼
F2
F1
ðt1 þ t2 Þk
:
2
E 2 t2 L E 1 t1 L
ð2:127Þ
The strain is also determined from the difference between the coefficients of thermal
expansion (CTE) of the substrate and the film multiplied by the difference between
growth and room temperature, DT.
e ¼ DTða1 a2 Þ:
ð2:128Þ
Considering a negligibly small bending stress, the one-dimensional stress in the ith
epitaxial layer is then taken as constant and given by
si ð1DÞ ¼
Fi
;
ti W
ð2:129Þ
where si(1D) is the one-dimensional stress.
By assuming a spherical bending for a square sample (meaning L W), the twodimensional stress can be deduced from the one-dimensional stress expression of
Equation 2.129 as follows:
si ð2DÞ ¼
si ð1DÞ
ð1 vÞ 1
;
ð2:130Þ
where si(2D) and n are the two-dimensional stress and Poissons ratio, respectively.
For two epitaxial layers the equilibrium of forces Equation 2.125, the equilibrium of moments Equation 2.126, strain in terms of mechanical parameters
Equation 2.127, strain in terms of the difference in CTE, and differential temperature
Equation 2.128 take the form of
F 1 þ F 2 þ F 3 ¼ 0;
ð2:131Þ
Wk
t1
t2
t3
ðE 1 t31 þ E 2 t32 þ E 3 t33 Þ þ F 1 þ F 2 t1 þ
þ F 3 t1 þ t2 ¼ 0;
2
2
2
12
ð2:132Þ
e1 ¼
F2
F1
ðt1 þ t2 Þk
¼ DTða1 a2 Þ;
2
E 2 t2 L E 1 t1 L
ð2:133Þ
2.12 Polarization Effects
e2 ¼
F3
F2
ðt2 þ t3 Þk
¼ DTða2 a3 Þ;
2
E 3 t3 L E 2 t2 L
ð2:134Þ
where e1 and e2 represent the strain between the first epilayer and substrate and the
second and first epilayers, respectively. Then si(2D), the two-dimensional stress in
the epilayer i, is given as
si ð2DÞ ¼
ðF i =ti WÞ
ð1 nÞ 1
:
ð2:135Þ
To calculate the misfit strain and to some extent CTE mismatch, the epitaxial
relationship between the epitaxial layer and the substrate must be known, which is
known simply as epitaxial relationship. That relationship between GaN and a variety
of other substrates are discussed in Section 3.3. In spite of this, a succinct description
is provided here as a part of the present topic on stress and strain for continuity and to
help support the discussion on polarization. For sapphire substrates, orientations of
(0 0 0 1), ð0 1 1 0Þ, ð2 1 1 0Þ, and ð0 1 1 2Þ (basically c-, m-, a-, and r-planes) have been
used, see Table 3.6. The largest lattice mismatch, 33%, is between (0 0 0 1) GaN and
ð2 1 1 0Þ sapphire along the ½0 1 1 0==½0 1 1 0 in-plane direction. The smallest
mismatch, 1.19%, is between ð2 1 1 0Þ GaN and ð0 1 1 2Þ sapphire (r-plane) along
the ½0 0 0 1==½0 1 1 1 in-plane direction. This orientation produces a-plane sapphire
on r-plane sapphire and is discussed at some length in Sections 3.3.1 and 3.5.11.
From the view point of lattice mismatch alone, the r-plane of sapphire is predicted to
be most suitable for GaN growth. A thermal strain of 0.18% exists in GaN (0 0 0 1)
when grown on sapphire (0 0 0 1), which is compressive.
The next common substrate used to grow GaN is various polytypes of SiC. The
epitaxial relationship on SiC substrates of hexagonal symmetry is tabulated in
Table 3.7. The lattice parameter misfits between GaN and 6H-SiC, 3C-SiC, and
4H-SiC are very close to each other, namely, 3.48%, 3.46%, and 3.50%, respectively,
and the corresponding thermal strains are 0.01, 0.09, and 0.03, respectively. The
thermal strain in GaN on SiC is tensile and notorious for causing cracks in the
epitaxial layers, beginning at about 2 mm thickness.
In case AlN substrates would become available, growth on that substrate is also
considered. Lattice misfit between GaN and AlN is only 2.41% and the thermal
strain is 0.06% and 4.08% for epitaxial relationships (0 0 0 1)//(0 0 0 1) and
ð1 1 2 0Þ==ð1 1 2 0Þ, respectively. From the thermal strain point of view, growth on
c-plane AlN is preferred.
GaN and ZnO share the stacking order and close lattice parameter, the epitaxial
relationship for which is provided in Section 3.3.4. GaN can be grown on (0 0 0 1) ZnO,
with the lattice parameter mismatch being only 1.97% and the misfit strain 0.21%.
Some oxides have also been explored because of the small lattice misfit with GaN
1 0 0Þ==ð1 0 0Þ
they provide. The epitaxial relationship between GaN and LiAlO2 is ð1 with a lattice misfit of 0.31% along the [0 0 0 1]//[0 1 0] in-plane direction and a misfit
strain of 0.41%. Some details can be found in Section 3.3.5. The structure of
LiGaO2 is similar to the wurtzite structure, but because Li and Ga atoms have
different ionic radii, the crystal has orthorhombic structure. Figure 3.36 shows the
j293
j 2 Electronic Band Structure and Polarization Effects
294
transformation of the hexagonal unit cell of GaN to an orthorhombic cell, which has
lattice parameters close to that of LiGaO2. Similarly, Table 3.10 shows the corresponding lattice parameters of GaN before and after the transformation to orthorhombic
cell as well as that of orthorhombic LiGaO2 unit cell. The misfit along [1 0 0] is 2.13%,
with a misfit strain of 0.54%.
Hexagonal (0 0 0 1) GaN has been prepared on (0 0 0 1) ScAlMgO4. The lattice
parameter mismatch between the ½1 0 1 0 GaN and ½1 0 1 0 ScAlMgO4 is 1.49%
and the misfit strain is 0.15%. Wurtzite (0 0 0 1) GaN films have been grown on
cubic (1 1 1) spinel (MgAl2O4) substrates, as detailed in Section 3.5.10. The lattice
parameter mismatch between the ½1 1 2 0 GaN and ½1 1 0 MgAl2O4 is 11.55% and
the misfit strain is 0.06%. Zinc blende GaN was grown on (0 0 1) MgO with an
epitaxial misfit of 6.99% and a misfit strain of 1.19%. Zinc blende GaN was grown
on (0 0 1) MgO with an epitaxial misfit of 6.99% and a misfit strain of 1.19%. Mixed
perovskite (La,Sr)(Al,Ta)O3 (LSAT) that is grown by Czochralski method could also be
a promising substrate for GaN epitaxial layers and are reported. The lattice parameter
25
AlN
Si
6HSiC
4HSiC
3CSiC
LiGaO2
Al2O3
MgO
GaAs
ZnO
MgAl2O4
LiAlO2
ScMgAlO4
NdGaO3
Curvature (1 m–1)
20
15
10
5
0.0
–5
0.0
20
40
60
Thickness (μm)
Figure 2.59 A compilation of the variation of thermal curvature, a
measure of strain, in epitaxial GaN layers grown on different
substrates with respect to layer thickness [492]. (Please find a
color version of this figure on the color tables.)
80
100
2.12 Polarization Effects
Figure 2.59 (Continued )
mismatch between ½1 1 2 0 GaN//LSAT ½1 1 0 is 16.64% and the corresponding
misfit strain is 0.04%.
Cubic GaN films can be epitaxially grown onto (0 0 1) Si and hexagonal polytype
on (1 1 1) Si with a lattice misfit of 16.93% and misfit strain of 0.19%. The epitaxial
relationship between GaN andSi is discussed in Section 3.3.3 and tabulated in Table 3.8.
As for the case on GaAs, GaN with (0 0 0 1) orientation can be grown on (1 1 1) GaAs
with an in-plane lattice arrangement of ½1 1 2 0==½1 1 0. The corresponding lattice
misfit is 20.19% and the misfit strain 0.07%.
Perovskite oxide substrates have also been considered as substrates for GaN and
related structures in an effort to perhaps find a better match and/or utilize the
j295
j 2 Electronic Band Structure and Polarization Effects
296
nonlinear optical properties of perovskite oxide along with what GaN has to offer. The
lattice mismatch of GaN to NdGaO3 has been calculated by assuming a perovskite cell
of NdGaO3 with lattice parameters a, b, and c each being equal to 3.86 Å. Next, a new
unit cell is constructed with a0 and b0 , where a0 and b0 are the diagonals of the old
perovskite cell, as shown in Figure 3.37. The c0 -axis of the new cell is parallel to the caxis of the perovskite cell, but its length is doubled. In a sense this transforms a
perovskite unit cell to a tetragonal unit cell. Accordingly, (1 0 0) plane becomes ð1 1 0Þ
plane and (0 0 1) becomes (0 0 1). The corresponding lattice misfit is 1.72% and the
misfit strain is 0.66%.
Owing to the difference in stress between the thin film and substrate, the
composite of film and substrate bends, which is the basis for many predigital
thermometers and temperature controllers with spiral elements. The strain can be
deduced from bending radius, for example, by optical means even during growth at
elevated temperature, to monitor the evolution of stress and in attempts to reduce
stress by epitaxial heterojunction layer design as performed on SiC substrates, see
Section 3.5.3. It is therefore imperative to establish the relationship between the
radius of curvature and the difference in the strain between the film and substrate, as
discussed in the model of Olsen and Ettenberg [494]. In the calculation of curvature
and stress instead of an average CTE over the entire range of temperature, the
variation CTE with temperature has been considered for accuracy [492]. The final
5108
Stress on various substrates (Pa)
0
–5108
–1109
AlN
Si
6HSiC
4HSiC
3CSiC
LiGaO2
Al2O3
MgO
GaAs
ZnO
MgAl2O4
LiAlO2
ScMgAlO4
NdGaO3
–1.5109
–2109
–2.5109
–3109
0100
210–5
410–5
610–5
810–5
Thickness (m)
Figure 2.60 A compilation of residual thermal stresses in
epitaxial GaN layer on different substrates with respect to layer
thickness [492]. (Please find a color version of this figure on the
color tables.)
110–4
2.12 Polarization Effects
j297
Stress versus thickness of GaN/potential substrates
5.00E+08
0.00E+00
0
0.00002
0.00004
0.00006
0.00008
0.0001
AlN
Si
MgO
–5.00E+08
3C-SiC
6H-SiC
4H-SiC
Stress (Pa)
–1.00E+09
ZnO
Al2O3
LiGaO2
–1.50E+09
MgAl2O4
GaAs
NdGaO3**
ScAlMgO**
–2.00E+09
LiAlO2
LSAT
–2.50E+09
–3.00E+09
Thickness (m)
Figure 2.60 (Continued )
curvature and stress is then the integrated values from growth temperature to room
temperature. Figures 2.59 and 2.60 show the curvature and the residual thermal
stresses in epitaxial GaN layers grown on various substrates with respect to layer
thickness, respectively. The parameters associated with each of the substrate used as
well as the growth temperatures are tabulated in Table 2.36. An inspection of
Figure 2.60 leads to the conclusion that the thermal stress is relatively small when
GaN is grown on AlN, SiC, ScAlMgO4, Si, GaAs, and ZnO and much higher when
NdGaO3 and MgO substrates are used. In all other cases (Al2O3, LiAlO2, LiGaO2,
MgAl2O4, LSAT) the values are intermediate. Of paramount importance, the thermal
stress is tensile in GaN when grown on Si and SiC, whereas in all other cases it is
j 2 Electronic Band Structure and Polarization Effects
298
Table 2.36 Properties and residual thermal stress of 1 mm epitaxial
GaN film with other III-N compounds and substrates [492].
Substrate
Melting point ( C)
GaN
AlN
a-Al2O3
6H-SiC
3C-SiC
4H-SiC
c-LiAlO2
b-LiGaO2
Si
GaAs
NdGaO3
MgO
ZnO
ScAlMgO4
MgAl2O4
>1700 at 2 kbar
2400 C at 30 bar
2030
2700 sublimes
1825 sublimes
2797
1700
1595
1415
1238
1600
2852
1975
2130
CTE (·106 C)
(room temperature)
Growth temperature
of GaN by grown
OMVPE ( C)
aa ¼ 4.997, ac ¼ 4.481, a ¼ 5.45
aa ¼ 5.411
aa ¼ 8.31, ac ¼ 8.5
aa ¼ 4.76, ac ¼ 4.46
4.5 [76]
4.75
aa ¼ 12.1
aa ¼ 10.1, ab ¼ 21.1, ac ¼ 13.6
3.9
6.7
aa ¼ 11.9, ab ¼ 6.6, ac ¼ 5.8
13.9
aa ¼ 6.9, ac ¼ 4.75
aa ¼ 6.2, ac ¼ 12.2
8.7
950–1050
450–1040
950–1100
1000
600
600–1000
600
700
810
450–800
700
650
1000
compressive in nature, which leads to serious cracking issues. This is caused by CTE
in SiC and Si being much smaller than that for GaN. As a result, during cooldown
GaN is not freely permitted to reduce its lattice constant to the extent GaN naturally
would like to, and consequently, the film remains under tensile strain, which causes
cracks when the film thickness is about 2 mm or larger.
Focusing on the most commonly used substrates for GaN and related epitaxy,
Figures 2.61 and 2.62 show variation of the residual thermal stress in GaN versus the
7 × 107
Stress (Pa)
6 × 107
5 × 107
4 × 107
3 × 107
2 × 107
0
20
40
60
80
Thickness (μm)
Figure 2.61 Residual thermal stress in GaN for a dual layer GaN/
AlN (0.1 mm) structure on 6H-SiC with respect to GaN
thickness [492].
100
References
–0.2
Stress (GPa)
–0.4
–0.6
–0.8
–1.0
–1.2
0
20
60
40
Thickness (μm)
80
100
Figure 2.62 Residual thermal stress in GaN for dual layer GaN/
AlN (0.1 mm) structure on sapphire (Al2O3) with respect to GaN
thickness [492].
thickness of GaN with an AlN buffer layer of thickness of 0.1 mm for dual-layer GaN/
AlN on 6H-SiC and GaN/AlN on Al2O3 heterostructures, respectively. There is very
little change in thermal stress when GaN is grown on Al2O3 with or without a buffer
layer of AlN. On the contrary, the stress decreases by an order by using a buffer layer
of AlN while growing GaN on 6H-SiC.
In summary, GaN layers grown on substrates such as AlN, SiC, ScAlMgO4, Si,
GaAs, and ZnO has a residual thermal stress that is smaller by a factor of two or more
as compared to the cases when the GaN layers are grown on other substrates.
Moreover and very pivotally, the thermal stress is tensile in nature when grown on Si
and SiC substrates whereas in all other cases it is compressive. The tensile residual
strain in GaN grown on SiC and Si is notorious for cracking, and multiheterolayer
buffer structures are used to deal with the problem, but not without limitations on
layers thicknesses for a given substrate temperature used.
References
1 Matsuoka, T., Tanka, H., Sasaki, T. and
Katsui, A. (1989) Wide-gap
semiconductor (In,Ga)N. Proceedings of
the Symposium on GaAs and Related
Compounds, Karuizawa, Japan, Institute
of Physics Conference Series 106,
Chapter 3, pp. 141–146.
2 Blakemore, J.S. (1985) Solid State Physics,
2nd edn, Cambridge University Press.
3 McKelvey, J.P. (1986) Solid State Physics,
4th edn, Krieger Publishing Company.
4 Ashcroft, N.W. and Mermin, N.D. (1976)
Solid State Physics, Holt-ReinhartWinston.
5 Bloch, F. (1928) Zeitschrift fur Physik, 52,
555.
6 Kronig, R.deL. and Penney, W.G. (1931)
Proceedings of the Royal Society A, 130, 499.
7 Herring, C. and Hill, A.C. (1940) Physical
Review, 58, 1332.
8 Woodruff, T.O. (1957) Solid State Physics,
4, 367.
9 Phillips, J.C. and Kleinman, L. (1959)
New method for calculating wave
functions in crystals and molecules.
Physical Review, 116 (2), 287–294.
10 Cohen, M.L. and Heine, V. (1970) The
fitting of pseudopotentials to experimental
j299
j 2 Electronic Band Structure and Polarization Effects
300
11
12
13
14
15
16
17
18
19
20
21
22
data and their subsequent application, in
Solid State Physics: Advances in Research
and Applications (eds H. Ehrenreich, F.
Seitz and D. Turnbull), Academic Press,
London, UK, pp. 37–248.
Harrison, W.A. (1980) Solid State Theory,
Dover.
Slater, J.C. (1937) Physical Review, 51, 846.
Hohenberg, P. and Kohn, W. (1964)
Inhomogeneous electron gas. Physical
Review, 136, B864–B871.
Dreizler, R.M. and Gross, E.K.U. (1996)
Density Functional Theory, Springer.
Kohn, W. and Sham, L.J. (1965) Selfconsistent equations including exchange
and correlation effects. Physical Review,
140, A1133–A1138.
Lambrecht, W.R.L. and Segall, B. (1998)
Band structure of the group-III nitrides,
in Gallium Nitride, Semiconductors and
Semimetals, vol. 50 (eds J.I. Pankove and
T.D. Moustakas), Academic Press, San
Diego, CA, pp. 369–402.
Hedin, L. (1965) New method for
calculating the one-particle Greens
function with application to the electrongas problem. Physical Review, 139,
A796–A823.
Hedin, L. and Lundqvist, S. (1969) Solid
State Physics: Advances in Research and
Applications, vol. 23 (eds F. Seitz, D.
Turnbull and H. Ehrenreich), Academic
Press, New York, p. 1.
Sham, L.J. and Schl€
uter, M. (1983)
Density-functional theory of the energy
gap. Physical Review Letters, 51 (20),
1888–1891.
Perdew, J.P. and Levy, M. (1983) Physical
content of the exact Kohn–Sham orbital
energies: band gaps and derivative
discontinuities. Physical Review Letters, 51
(20), 1884–1887.
Hybertsen, M.S. and Louie, S.G. (1986)
Electron correlation in semiconductors
and insulators: band gaps and
quasiparticle energies. Physical Review B:
Condensed Matter, 34 (8), 5390–5413.
Godby, R.W., Schl€
uter, M. and Sham, L.J.
(1988) Self-energy operators and
23
24
25
26
27
28
29
30
31
32
exchange-correlation potentials in
semiconductors. Physical Review B:
Condensed Matter, 37 (17), 10159–10175.
Aryasetiawan, F. and Gunnarsson, O.
(1994) Product-basis method for
calculating dielectric matrices. Physical
Review B: Condensed Matter, 49 (23),
16214–16222.
Aryasetiawan, F. and Gunnarsson, O.
(1994) Linear-muffin-tin-orbital method
with multiple orbitals per L channel.
Physical Review B: Condensed Matter, 49
(11), 7219–7232.
Rubio, A., Corkill, J.L., Cohen, M.L.,
Shirley, E.L. and Louie, S.G. (1993)
Quasiparticle band structure of AlN and
GaN. Physical Review B: Condensed Matter,
48 (16), 11810–11816.
Lambrecht, W.R.L., Segall, B., Rife, J.,
Hunter, W.R. and Wickenden, D.K. (1995)
Physical Review B: Condensed Matter, 51,
13516.
Jancu, J.-M., Bassani, F., Della Sala, F. and
Scholz, R. (2002) Transferable tightbinding parametrization for the group-III
nitrides. Applied Physics Letters, 81 (25),
4838–4841.
Andersen, O.K. (1975) Linear methods in
band theory. Physical Review B: Condensed
Matter, 12, 3060.
Wimmer, E., Krakauer, H., Weinert, M.
and Freeman, A. (1981) Physical Review B:
Condensed Matter, 24, 864.
Christensen, N.E. and Gorczyca, I. (1993)
Calculated structural phase transitions of
aluminum nitride under pressure.
Physical Review B: Condensed Matter, 47
(8), 4307–4314.
Christensen, N.E. and Gorczyca, I. (1994)
Optical and structural properties of III–V
nitrides under pressure. Physical Review
B: Condensed Matter, 50 (7), 4397–
4415.
Lambrecht, W.R.L. and Segall, B. (1995)
General remarks and notations on the
band structure of pure group III nitrides,
in Group III Nitrides (ed. J.H. Edgar),
EMIS Data Review Series, No. 11, IEE,
London, p. 125.
References
33 Van Camp, P.E., Van Doren, V.E. and
Devreese, J.T. (1991) High-pressure
properties of wurtzite- and rocksalt-type
aluminum nitride. Physical Review B:
Condensed Matter, 44 (16), 9056–9059.
34 Min, B.J., Chan, C.T. and Ho, K.M. (1992)
First-principles total-energy calculation of
gallium nitride. Physical Review B:
Condensed Matter, 45 (3), 1159–1162.
35 Yeh, C.-Y., Lu, Z.W., Froyen, S. and
Zunger, A. (1992) Zinc-blende–wurtzite
polytypism in semiconductors. Physical
Review B: Condensed Matter, 46 (16),
10086–10097.
36 Wright, A.F. and Nelson, J.S. (1995) Firstprinciples calculations for zinc-blende
AlInN alloys. Applied Physics Letters, 66
(25), 3465–3467.
37 Van Camp, P.E., Van Doren, V.E. and
Devreese, J.T. (1992) Solid State
Communications, 81, 23.
38 Fiorentini, V., Methfessel, M. and
Scheffler, M. (1993) Electronic and
structural properties of GaN by the fullpotential linear muffin-tin orbitals
method: the role of the d electrons.
Physical Review B: Condensed Matter, 47
(20), 13353–13362.
39 Lambrecht, W.R., Segall, B., Strite, S.,
Martin, G., Agarwal, A., Morkoç, H. and
Rockett, A. (1994) X-ray photoelectron
spectroscopy and theory of the valence
band and semicore Ga 3d states in GaN.
Physical Review B: Condensed Matter, 50
(19), 14155–14160.
40 Morkoç, H., Strite, S., Gao, G.B., Lin,
M.E., Sverdlov, B. and Burns, M. (1994)
Journal of Applied Physics, 76, 1363–1398.
41 Boguslawski, P., Briggs, E.L. and
Bernholc, J. (1995) Native defects in
gallium nitride. Physical Review B:
Condensed Matter, 51 (23), 17255–17258.
42 Northrup, J.E. and Neugebauer, J. (1996)
Theory of GaN ð1010Þ and ð1120Þ
surfaces. Physical Review B: Condensed
Matter, 53 (16), R10477–R10480.
43 Chen, J., Levine, Z.H. and Wilkins, J.W.
(1995) Calculated second-harmonic
susceptibilities of BN, AlN, and
44
45
46
47
48
49
50
51
52
53
GaN. Applied Physics Letters, 66 (9),
1129–1131.
Miwa, K. and Fukumoto, A. (1993) Firstprinciples calculation of the structural,
electronic, and vibrational properties of
gallium nitride and aluminum nitride.
Physical Review B: Condensed Matter, 48
(11), 7897–7902.
Pandey, R., Jaffe, J.E. and Harrison, N.M.
(1994) Ab initio study of high pressure
phase transition in GaN. Journal of Physics
and Chemistry of Solids, 55 (11),
1357–13561.
Pandey, R., Sutjianto, A., Seel, M. and
Jaffe, J.E. (1993) Electronic structure of
high pressure phase of AlN. Journal of
Materials Research, 8 (8), 1922–1927.
Ruiz, E., Alvarez, S. and Alemany, P.
(1994) Electronic structure and properties
of AlN. Physical Review B: Condensed
Matter, 49 (11), 7115–7123.
Palummo, M., Del Sole, R., reining, L.,
Bechstedt, F. and Cappellini, G. (1995)
Screening models and simplified GW
approaches: Si & GaN as test cases.
Solid State Communications, 95 (6),
393–398.
Bechstedt, F. and Del Sole, R. (1988)
Analytical treatment of band-gap
underestimates in the local-density
approximation. Physical Review B:
Condensed Matter, 38 (11), 7710–7716.
Bechstedt, F., Del Sole, R., Cappellini, G.
and Reining, L. (1992) An efficient
method for calculating quasiparticle
energies in semiconductors. Solid State
Communications, 84 (7), 765–770.
Palummo, M., Reining, L., Godby, R.W.,
Bertoni, C.M. and Bornsen, N. (1994)
Electronic structure of cubic GaN with
self-energy corrections. Europhysics
Letters, 26 (8), 607–612.
Della Sala, F., Di Carlo, A., Lugli, P.,
Bernardini, F., Scholz, R. and Jancu,
J.M. (1999) Applied Physics Letters, 74,
2002.
Yang, T., Nakajima, S. and Sakai, S. (1995)
Japanese Journal of Applied Physics, 34,
5912.
j301
j 2 Electronic Band Structure and Polarization Effects
302
54 Jogai, B. (1998) Effect of in-plane biaxial
strains on the band structure of wurtzite
GaN. Physical Review B: Condensed Matter,
57, 2382.
55 Fritsch, D., Schmidt, H. and Grundmann,
M. (2003) Band-structure pseudopotential
calculation of zinc-blende and wurtzite
AlN, GaN, and InN. Physical Review B:
Condensed Matter, 67, 235205.
56 Suzuki, M., Uenoyama, T. and Yanase, A.
(1995) First-principles calculations of
effective-mass parameters of AlN and
GaN. Physical Review B: Condensed Matter,
52 (11), 8132–8139.
57 Suzuki, M. and Uenoyama, T. (1995) Firstprinciples calculation of effective mass
parameters of gallium nitride. Japanese
Journal of Applied Physics, Part 1: Regular
Papers, Short Notes & Review Papers, 34
(7A), 3442–3446.
58 Chuang, S.L. and Chang, C.S. (1996) kp
method for strained wurtzite
semiconductors. Physical Review B:
Condensed Matter, 54 (4), 2491–2504.
59 Suzuki, M. and Uenoyama, T. (1998)
Electronic and optical properties of GaN
based quantum wells, in Group III Nitride
Semiconductor Compounds: Physics and
Applications (ed. B. Gil), Clarendon,
Oxford.
60 Suzuki, M. and Uenoyama, T. (1996)
Japanese Journal of Applied Physics, 35,
1420.
61 Kamiyama, S., Ohnaka, K., Suzuki, M.
and Uenoyama, T. (1995) Japanese Journal
of Applied Physics, 34, L821.
62 Sirenko, Yu.M., Jeon, J.B., Kim, K.W.,
Littlejohn, M.A. and Stroscio, M.A. (1996)
Physical Review B: Condensed Matter, 53,
1997.
63 Vainshtein, B.K. (1994) Fundamentals of
Crystals: Modern Crystallography, 2nd edn,
vol. 1, Springer, Berlin.
64 Zhao, G.L., Bagayoko, D. and Williams,
T.D. (1999) Physical Review B: Condensed
Matter, 60, 1563.
65 Madelung, O., Schulz, M. and Weiss, H.
(eds) (1982) Numerical Data and
Functional Relationships in Science and
66
67
68
69
70
71
72
73
74
75
76
77
78
Technology, Landolt-B€ornstein New Series,
Group III, vol. 17, part a, Springer,
New York.
Yeo, Y.C., Chong, T.C. and Li, M.F. (1998)
Electronic band structures and effectivemass parameters of wurtzite GaN and
InN. Journal of Applied Physics, 83 (3),
1429–1436.
Loughin, S., French, R.H., Ching, W.Y.,
Xu, Y.N. and Slack, G.A. (1993) Applied
Physics Letters, 63, 1182.
Foley, C.P. and Tansley, T.L. (1986)
Physical Review B: Condensed Matter, 33,
1430.
Fan, W.J., Li, M.F., Chong, T.C. and Xia,
J.B. (1996) Journal of Applied Physics, 79,
188.
Tadjer, A., Abbar, B., Rezki, M., Aourag, H.
and Certier, M. (1999) Journal of
Physics and Chemistry of Solids, 60, 419.
Pikus, G. (1962) Soviet Physics: JETP, 14,
1075.
Bir, G.L. and Pikus, G.E. (1974) Symmetry
and Strain-Induced Effects in
Semiconductors, John Wiley & Sons, Inc.,
New York.
Monemar, B., Bergman, J.P. and
Buyanova, I.A. (1997) Optical
characterization of GaN and related
material, in GaN and Related Material (ed.
S.J. Pearton), Gordon and Breach, New
York, pp. 85–140.
Ludwig, W. and Falter, C. (1996)
Symmetries in Physics. Group Theory
Applied to Physical Problems, Springer
Series in Solid-State Sciences, 2nd edn,
vol. 64, Springer, Berlin.
Inui, T., Tanabe, Y. and Onodera, Y. (1996)
Group Theory and Its Applications in Physics,
Springer Series Solid-State Sciences, 2nd
edn, vol. 78, Springer, Berlin.
Jogai, B. (1998) Solid State
Communications, 107, 345.
Jones, H. (1960) The Theory of Brillouin
Zones and Electronic States in Crystals,
North-Holland, Amsterdam.
Yu, P.Y. and Cardona, M. (1999)
Fundamentals of Semiconductors, 2nd edn,
Springer, Berlin.
References
79 Bassani, F. and Parravicini Pastori, G.
(1975) Electronic States and Optical
Transitions in Solids, Pergamon, New York.
80 Nye, J.F. (1964) Physical Properties of
Crystals, Oxford University Press; Nye,
J.F. (1985) Physical Properties of Crystals:
Their Representation by Tensors and
Matrices, Clarendon, Oxford; Nye, J.F.
(1998) Physical Properties of Crystals: Their
Representation by Tensors and Matrices,
Oxford University Press, New York.
81 Sheleg, A.U. and Savastenko, V.A. (1979)
Izvestiya Akademii Nauk USSR,
Neorganicheskie Materialy, 15, 1598.
82 Nye, J.F. (1985) Physical Properties of
Crystals: Their Representation by Tensors
and Matrices, Clarendon, Oxford.
83 Jogai, B. (2001) Three-dimensional strain
field calculations in multiple InN/AlN
wurtzite quantum dots. Journal of Applied
Physics, 90 (2), 699–704.
84 Ambacher, O., Majewski, J., Miskys, C.,
Link, A., Hermann, M., Eickhoff, M.,
Stutzmann, M., Bernardini, F.,
Fiorentini, V., Tilak, V., Schaff, B. and
Eastman, L.F. (2002) Pyroelectric
properties of Al(In)GaN/GaN hetero- and
quantum well structures. Journal of
Physics: Condensed Matter, 14, 3399–3434.
85 Wagner, J.-M. and Bechstedt, F. (2002)
Properties of strained wurtzite GaN and
AlN: ab initio studies. Physical Review B:
Condensed Matter, 66, 115202–115222.
86 Wagner, J.-M. and Bechstedt, F. (2000)
Physical Review B: Condensed Matter, 62,
4526.
87 Vinet, P., Ferrante, J., Smith, J.R. and
Rose, J.H. (1986) The Journal of Physical
Chemistry, 19, L467.
88 Morkoç, H., Sverdlov, B. and Gao, G.B.
(1993) Strained layer heterostructures
and their applications to MODFETs,
HBTs and lasers. Proceeding of IEEE, 81
(4), 492–556.
89 Suzuki, M. and Uenoyama, T. (1996)
Strain effect on electronic and optical
properties of GaN/AlGaN quantum-well
lasers. Journal of Applied Physics, 80 (12),
6868–6874.
90 Ren, G.B., Liu, Y.M. and Blood, P. (1999)
Valence-band structure of wurtzite GaN
including the spin–orbit interaction.
Applied Physics Letters, 74 (8), 117–119.
91 Albrecht, J.D., Ruden, P.P. and Reinecke,
T.L. (2002) Hole scattering near the
valence band edge in wurtzite gallium
nitride. Journal of Applied Physics, 92 (7),
3803–3814.
92 Vurgaftman, I., Meyer, J.R. and RamMohan, L.-R. (2001) Band parameters for
III–V compound semiconductors and
their alloys. Journal of Applied Physics, 89
(11), 5815–5875.
93 Sirenko, Yu.M., Jeon, J.B., Lee, B.C., Kim,
K.W., Littlejohn, M.A., Stroscio, M.A. and
Iafrate, G.J. (1997) Hole scattering and
optical transitions in wide-band-gap
nitrides: wurtzite and zinc-blende
structures. Physical Review B: Condensed
Matter, 55 (7), 4360–4375.
94 Yamaguchi, A.A., Mochizuki, Y.,
Sunakawa, H. and Usui, A. (1998)
Determination of valence band splitting
parameters in GaN. Journal of Applied
Physics, 83 (8), 4542–4544.
95 Rodina, V. and Meyer, B.K. (2001)
Anisotropy of conduction band g values
and interband momentum matrix
elements in wurtzite GaN. Physical Review
B: Condensed Matter, 64, 245209.
96 Hopfield, J.J. (1960) Journal of Physics and
Chemistry of Solids, 15, 97.
97 Dingle, R., Sell, D.D., Stokowski, S.E. and
Ilegems, M. (1971) Absorption,
reflectance, and luminescence of GaN
epitaxial layers. Physical Review B:
Condensed Matter, 4 (4), 1211–1218.
98 Gil, B., Briot, O. and Aulombard, R.-L.
(1995) Valence-band physics and the
optical properties of GaN epilayers grown
onto sapphire with wurtzite symmetry.
Physical Review B: Condensed Matter, 52
(24), R17028–R17031.
99 Reynolds, D.C., Look, D.C., Kim, W.,
Aktas, Ö., Botchkarev, A., Salvador, A.,
Morkoç, H. and Talwar, D.N. (1996)
Ground and excited state exciton spectra
from GaN grown by molecular beam
j303
j 2 Electronic Band Structure and Polarization Effects
304
100
101
102
103
104
105
106
107
epitaxy. Journal of Applied Physics, 80 (1),
594–596.
Shikanai, A., Azuhata, T., Sota, T.,
Chichibu, S., Kuramata, A., Horino, K.
and Nakamura, S. (1997) Biaxial strain
dependence of exciton resonance
energies in wurtzite GaN. Journal of
Applied Physics, 81 (1), 417–424.
Chen, G.D., Smith, M., Lin, J.Y., Jiang,
H.X., Wei, S.-H., Khan, M.A. and Sun,
C.J. (1996) Fundamental optical
transitions in GaN. Applied Physics Letters,
68 (20), 2784–2786.
Korona, K.P., Wysmolek, A., Paluka, K.,
Stepniewski, R., Baranowski, J.M.,
Grzegory, I., Lucznik, B., Wroblewski, M.
and Porowski, S. (1996) Exciton region
reflectance of homoepitaxial GaN layers.
Applied Physics Letters, 69 (6), 788–790.
Campo, J., Julier, M., Coquillat, D.,
Lascaray, J.P., Scalbert, D. and Briot, O.
(1997) Zeeman splittings of excitonic
transitions at the Gamma point in
wurtzite GaN: a magnetoreflectance
investigation. Physical Review B:
Condensed Matter, 56 (12), R7108–R7111.
Julier, M., Campo, J., Gil, B., Lascaray, J.P.
and Nakamura, S. (1998) Determination
of the spin-exchange interaction constant
in wurtzite GaN. Physical Review B:
Condensed Matter, 57 (12), R6791–R6794.
Edwards, N.V., Yoo, S.D., Bremser, M.D.,
Weeks, T.W., Jr, Nam, O.H., Davis, R.F.,
Liu, H., Stall, R.A., Horton, M.N.,
Perkins, N.R., Kuech, T.F. and Aspnes,
D.E. (1997) Variation of GaN valence
bands with biaxial stress and
quantification of residual stress. Applied
Physics Letters, 70 (15), 2001–2003.
Rodina, A.V., Dietrich, M., G€oldner, A.,
Eckey, L., Hoffmann, A., Efros, Al.L.,
Rosen, M. and Meyer, B.K. (2002)
Free excitons in wurtzite GaN.
Physical Review B: Condensed Matter,
64, 115204.
Wei, S.-H. and Zunger, A. (1996) Valence
band splittings and band offsets of AlN,
GaN, and InN. Applied Physics Letters, 69
(18), 2719–2721.
108 Yeo, Y.C., Chong, T.C. and Li, M.F. (1998)
Electronic band structures and effectivemass parameters of wurtzite GaN and
InN. Journal of Applied Physics, 83 (3),
1429–1436.
109 Kim, K., Lambrecht, W.R.L. and Segall, B.
(1996) Elastic constants and related
properties of tetrahedrally bonded BN,
AlN, GaN, and InN. Physical Review B:
Condensed Matter, 53 (24), 16310–16326.
110 Hsu, L. and Walukiewicz, W. (1997)
Electron mobility in AlxGa1xN/GaN
heterostructures. Physical Review B:
Condensed Matter, 56 (3), 1520–1528.
111 Knap, W., Borovitskaya, E., Shur, M.S.,
Hsu, L., Walukiewicz, W., Frayssinet, E.,
Lorenzini, P., Grandjean, N.,
Skierbiszewski, C., Prystawko, P.,
Leszczynski, M. and Grzegory, I. (2002)
Acoustic phonon scattering of twodimensional electrons in GaN/AlGaN
heterostructures. Applied Physics Letters,
80 (7), 1228–1230.
112 Shan, W., Hauenstein, R.J., Fischer, A.J.,
Song, J.J., Perry, W.G., Bremser, M.D.,
Davis, R.F. and Goldenberg, B. (1996)
Strain effects on excitonic transitions in
GaN: deformation potentials. Physical
Review B: Condensed Matter, 54 (19),
13460–13463.
113 Ghosh, S., Waltereit, P., Brandt, O.,
Grahn, H.T. and Ploog, K.H. (2002)
Electronic band structure of wurtzite GaN
under biaxial strain in the M plane
investigated with photoreflectance
spectroscopy. Physical Review B:
Condensed Matter, 65, 075202.
114 Gil, B. and Alemu, A. (1997) Optical
anisotropy of excitons in strained GaN
epilayers grown along the h1010i
direction. Physical Review B: Condensed
Matter, 56 (19), 12446–12453.
115 Ohtoshi, T., Niwa, A. and Kuroda, T.
(1997) Dependence of optical gain on
crystal orientation in wurtzite–GaN
strained quantum-well lasers. Journal of
Applied Physics, 82, (4), 1518–1520.
116 Suzuki, M. and Uenoyama, T. (1996)
Strain effect on electronic and optical
References
117
118
119
120
121
122
123
124
125
126
properties of GaN/AlGaN quantum-well
lasers. Journal of Applied Physics, 80,
6868–6874.
Alemu, A., Gil, B., Julier, M. and
Nakamura, S. (1998) Optical properties of
wurtzite GaN epilayers grown on A-plane
sapphire. Physical Review B: Condensed
Matter, 57 (7), 3761–3764.
Lambrecht, W.R.L. and Segall, B. (1995)
General remarks and notations on the
band structure of pure group III nitrides,
in Group III Nitrides (ed. J.H. Edgar),
EMIS Data Review Series, No. 11, IEE,
London, p. 125.
Huang, Y.S., Pollak, F.H., Park, S.S., Lee,
K.Y. and Morkoç, H. (2003) Contactless
electroreflectance investigation, in the
range of 20 K < T < 300 K of freestanding
wurtzite GaN prepared by hydride-vaporphase-epitaxy. Journal of Applied Physics,
94 (2), 899–903.
Logothetidis, S., Petalas, J., Cardona, M.
and Moustakas, T.D. (1994) Optical
properties and temperature dependence
of the interband transitions of cubic and
hexagonal GaN. Physical Review B:
Condensed Matter, 50 (24), 18017–18029.
Yang, Z. and Xu, Z. (1996) Electronic and
optical properties of unstrained and
strained wurtzite GaN. Physical
Review B: Condensed Matter, 54 (24),
17577–17584.
Bouhafs, B. and Lakdja, A. and Ruterana,
P. (2003) First-principles calculations of
the structural and electronic properties of
the III-nitrides-based superlattices.
Physica A, 17 (1–4), 235–237.
Barker, A.S., Jr and Illegems, M. (1973)
Infrared lattice vibrations and freeelectron dispersion in GaN. Physical
Review B: Condensed Matter, 7 (2), 743–750.
Rheinlander, B. and Neumann, H. (1974)
Physica Status Solidi b: Basic Research,
64, K123.
Sidorov, V.G., Sveshkova, L.S., Shagalov,
M.D. and Shalabutov, Yu.K. (1976) Soviet
Physics: Semiconductors, 10, 1309.
Pankove, J.I., Bloom, S. and Harbeke, G.
(1975) RCA Review, 36, 163.
127 Kesamanly, F.P. (1974) Soviet Physics:
Semiconductors, 8, 147.
128 Meyer, B.K., Volm, D., Graber, A., Alt,
H.C., Detchprohm, T., Amano, A. and
Akasaki, I. (1995) Shallow donors in GaN
– the binding energy and the electron
effective mass. Solid State
Communications, 95, 597–600.
129 Witowski, A.M., Pakula, K., Baranowski,
J.M., Sadowski, M.L. and Wyder, P. (1999)
Electron effective mass in hexagonal GaN.
Applied Physics Letters, 75 (26), 4154–4155.
130 Drechsler, M., Hoffman, D.M., Meyer,
B.K., Detchprohm, T., Amano, H. and
Akasaki, I. (1995) Determination of the
conduction band electron effective mass
in hexagonal GaN. Japanese Journal of
Applied Physics, Part 2: Letters, 34 (9B),
L1178–L1179.
131 Perlin, P., Litwin-Staszewska, E.,
Suchanek, B., Knap, W., Camassel, J.,
Suski, T., Piotrzkowski, R., Grzegory, I.,
Porowski, S., Kaminska, E. and Chervin,
J.C. (1996) Determination of the effective
mass of GaN from infrared reflectivity and
Hall effect. Applied Physics Letters, 68 (8),
1114–1116.
132 Wang, Y.J., Kaplan, R., Ng, H.K.,
Doverspike, K., Gaskill, D.K., Ikedo, T.,
Akasaki, I. and Amono, H. (1996)
Magneto-optical studies of GaN and GaN/
AlxGa1xN: donor Zeeman spectroscopy
and two-dimensional electron gas
cyclotron resonance. Journal of Applied
Physics, 79 (10), 8007–8010.
133 Knap, W., Contreras, S., Alause, H.,
Skierbiszewski, C., Camassel, J.,
Dyakonov, M., Robert, J.L., Yang, J., Chen,
Q., Asif Khan, M., Sadowski, M.L., Huant,
S., Yang, F.H., Goiran, M., Leotin, J. and
Shur, M.S. (1997) Cyclotron resonance
and quantum Hall effect studies of the
two-dimensional electron gas confined at
the GaN/AlGaN interface. Applied Physics
Letters, 70 (16), 2123–2125.
134 Kasic, A., Schubert, M., Einfeldt, S.,
Hommel, D. and Tiwald, T.E. (2000)
Free-carrier and phonon properties of
n- and p-type hexagonal GaN films
j305
j 2 Electronic Band Structure and Polarization Effects
306
135
136
137
138
139
140
141
measured by infrared ellipsometry.
Physical Review B: Condensed Matter, 62
(11), 7365–7377.
Elhamri, S., Newrock, R.S., Mast, D.B.,
Ahoujja, M., Mitchel, W.C., Redwing,
J.M., Tischler, M.A. and Flynn, J.S. (1998)
Al0.15Ga0.85N/GaN heterostructures:
effective mass and scattering times.
Physical Review B: Condensed Matter, 57
(3), 1374–1377.
Saxler, A., Debray, P., Perrin, R., Elhamri,
S., Mitchel, W.C., Elsass, C.R.,
Smorchkova, I.P., Heying, B., Haus, E.,
Fini, P., Ibbetson, J.P., Keller, S., Petroff,
P.M., DenBaars, S.P., Mishra, U.K. and
Speck, J.S. (2000) Characterization of an
AlGaN/GaN two-dimensional electron
gas structure. Journal of Applied Physics, 87
(1), 369–374.
Wong, L.W., Cai, S.J., Li, R., Wang, K.,
Jiang, H.W. and Chen, M. (1998)
Magnetotransport study on the twodimensional electron gas in AlGaN/GaN
heterostructures. Applied Physics Letters,
73 (10), 1391–1393.
Wang, T., Bai, J., Sakai, S., Ohno, Y. and
Ohno, H. (2000) Magnetotransport
studies of AlGaN/GaN heterostructures
grown on sapphire substrates: effective
mass and scattering time. Applied Physics
Letters, 76 (19), 2737–2739.
Hang, D.R., Liang, C.-T., Huang, C.F.,
Chang, Y.H., Chen, Y.F., Jiang, H.X. and
Lin, J.Y. (2001) Effective mass of twodimensional electron gas in an
Al0.2Ga0.8N/GaN heterojunction. Applied
Physics Letters, 79 (1), 66–68.
Goano, M., Bellotti, E., Ghillino, E.,
Ghione, G. and Brennan, K.F. (2000)
Band structure nonlocal pseudopotential
calculation of the III-nitride wurtzite
phase materials system. Part I. Binary
compounds GaN, AlN, and InN. Journal of
Applied Physics, 88 (11), 6467–6475.
Eliseev, P.G., Perlin, P., Lee, J. and
Osinski, M. (1997) Blue, temperatureinduced shift and band-tail emission in
InGaN-based light sources. Applied
Physics Letters, 71 (5), 569–571.
142 Gurioli, M., Vinattieri, A., Pastor, J.M. and
Colocci, M. (1994) Exciton thermalization
in quantum-well structures. Physical
Review B: Condensed Matter, 50,
11817–11826.
143 Monemar, B. (1974) Fundamental energy
gap of GaN from photoluminescence
excitation spectra. Physical Review B:
Condensed Matter, 10 (2), 676–681.
144 Teisseyre, H., Perlin, P., Suski, T.,
Grzegory, I., Porowski, S., Jun, J.,
Pietraszko, A. and Moustakas, T.D. (1994)
Temperature dependence of the energy
gap in GaN bulk single crystals and
epitaxial layer. Journal of Applied Physics,
76 (4), 2429–2434.
145 Shan, W., Schmidt, T.J., Yang, X.H.,
Hwang, S.J., Song, J.J. and Goldenberg,
B. (1995) Temperature dependence of
interband transitions in GaN grown
by metalorganic chemical vapor
deposition. Applied Physics Letters, 66 (8),
985–987.
146 Petalas, J., Logothetidis, S., Boultadakis,
S., Alouani, M. and Wills, J.M. (1995)
Optical and electronic-structure study of
cubic and hexagonal GaN thin films.
Physical Review B: Condensed Matter, 52
(11), 8082–8091.
147 Salvador, A., Liu, G., Kim, W., Aktas, O.,
Botchkarev, A. and Morkoç, H. (1995)
Properties of a Si doped GaN/AlGaN
single quantum well. Applied Physics
Letters, 67, 3322–3324.
148 Manasreh, M.O. (1996) Optical
absorption near the band edge in GaN
grown by metalorganic chemical-vapor
deposition. Physical Review B: Condensed
Matter, 53 (24), 16425–16428.
149 Li, C.F., Huang, Y.S., Malikova, L. and
Pollak, F.H. (1997) Temperature
dependence of the energies and
broadening parameters of the interband
excitonic transitions in wurtzite GaN.
Physical Review B: Condensed Matter, 55
(15), 9251–9254.
150 Zubrilov, A.S., Melnik, Yu.V., Nikolaev,
A.E., Jacobson, M.A., Nelson, D.K. and
Dmitriev, V.A. (1999) Optical properties of
References
151
152
153
154
155
156
157
158
159
160
161
162
gallium nitride bulk crystals grown by
chloride vapor phase epitaxy.
Semiconductors, 33 (10), 1067–1071.
Reynolds, D.C., Hoelscher, J., Litton, C.W.
and Collins, T.C. (2002) Temperature
dependence of free excitons in GaN.
Journal of Applied Physics, 92 (9),
5596–5598.
Vurgaftman, I. and Meyer, J.R. (2003)
Band parameters for nitrogen-containing
semiconductors. Journal of Applied
Physics, 94 (6), 3675–3696.
Dugdale, D.J., Brand, S. and Abram, R.A.
(2000) Direct calculation of kp
parameters for wurtzite AlN, GaN, and
InN. Physical Review B: Condensed Matter,
61 (19), 12933–12938.
Kim, K., Lambrecht, W.R.L., Segall, B. and
van Schilfgaarde, M. (1997) Effective
masses and valence-band splittings in
GaN and AlN. Physical Review B:
Condensed Matter, 56 (12), 7363–7375.
Kawabe, K., Tredgold, R.H. and Inuishi, Y.
(1967) Electrical Engineering in Japan,
87, 62.
Balkanski, M. and de Cloizeaux, J. (1960)
Journal de Physique et le Radium, 21, 825.
Hopfield, J.J. and Thomas, D.G. (1963)
Physical Review, 132, 563.
Kane, E.O. (1958) Journal of Physics and
Chemistry of Solids, 6, 236; Kane, E.O.
(1966) Semiconductors and Semimetals, vol.
1 (eds R.K. Willardson and A.C., Beer),
Academic Press; Kane, E.O. (1982)
Handbook on Semiconductors, vol. 1 (ed. W.
Paul), North-Holland, New York, p. 193.
Bloom, S., Harbeke, G., Meier, E. and
Ortenburger, I.B. (1974) Physica Status
Solidi b: Basic Research, 66, 161.
Strite, S., Ran, J., Li, Z., Salvador, A.,
Chen, H., Smith, D.J., Choyke, W.J. and
Morkoç, H. (1991) Journal of Vacuum
Science & Technology B: Microelectronics
and Nanometer Structures, 9, 1924.
Sitar, Z., Paisley, M.J., Ruan, J., Choyke,
J.W. and Davis, R.F. (1992) Journal of
Materials Science Letters, 11, 261.
Okumura, H., Yoshida, S. and Okahisa, T.
(1994) Applied Physics Letters, 64, 2997.
163 Pugh, S.K., Dugdale, D.J., Brand, S. and
Abram, R.A. (1999) Electronic structure
calculations on nitride semiconductors.
Semiconductor Science and Technology, 14,
23–31.
164 Menniger, J., Jahn, U., Brandt, O., Yang,
H. and Ploog, K. (1996) Physical Review B:
Condensed Matter, 53, 1881.
165 Wu, J., Yaguchi, H., Onable, K. and Ito, R.
(1997) Applied Physics Letters, 71, 2067.
166 Xu, D., Yang, H., Li, J.B., Zhao, D.G., Li,
S.F., Zhuang, S.M., Wu, R.H., Chen, Y.
and Li, G.H. (2000) Applied Physics Letters,
76, 3025.
167 Ramirez-Flores, G., Navarro-Contreras,
H., Lastras-Martinez, A., Powell, R.C. and
Greene, J.E. (1994) Physical Review B:
Condensed Matter, 50, 8433.
168 Fanciulli, M., Lei, T. and Moustakas, T.D.
(1993) Conduction-electron spin
resonance in zinc-blende GaN thin films.
Physical Review B: Condensed Matter, 48
(20), 15144–15147.
169 Chow, W.W., Wright, A.F. and Nelson, J.S.
(1996) Theoretical study of room
temperature optical gain in GaN strained
quantum wells. Applied Physics Letters, 68
(3), 296–298.
170 Suzuki, M. and Uenoyama, T. (1996)
Optical gain and crystal symmetry in
III–V nitride lasers. Applied Physics Letters,
69 (22), 3378–3380.
171 Ramos, L.E., Teles, L.K., Scolfaro, L.M.R.,
Castineira, J.L.P., Rosa, A.L. and Leite,
J.R. (2001) Structural, electronic, and
effective-mass properties of silicon
and zinc-blende group-III nitride
semiconductor compounds.
Physical Review B: Condensed Matter,
63, 165210.
172 Pfeffer, P. and Zawadzki, W. (1988)
Resonant and nonresonant polarons in
semiconductors. Physical Review B:
Condensed Matter, 37 (5), 2695–2698.
173 Meney, T., OReilly, E.P. and Adams, A.R.
(1996) Optical gain in wide bandgap
GaN quantum well lasers.
Semiconductor Science and Technology,
11, 897–903.
j307
j 2 Electronic Band Structure and Polarization Effects
308
174 Fan, W.J., Li, M.F., Chong, T.C. and Xia,
J.B. (1996) Solid State Communications,
97, 381.
175 Ohtoshi, T., Niwa, A. and Kuroda, T.
(1998) Orientation dependence of optical
gain in zincblende-GaN strainedquantum-well lasers. IEEE Journal of
Selected Topics in Quantum Electronics, 4
(3), 527–530.
176 Wei, S.-H. and Zunger, A. (1999)
Predicted band-gap pressure coefficients
of all diamond and zinc-blende
semiconductors: chemical trends.
Physical Review B: Condensed Matter, 60
(8), 5404–5411.
177 Van de Walle, C.G. and Neugebauer, J.
(1997) Small valence-band offsets at GaN/
InGaN heterojunctions. Applied Physics
Letters, 70 (19), 2577–2579.
178 Binggeli, N., Ferrara, P. and Baldereschi,
A. (2001) Band-offset trends in nitride
heterojunctions. Physical Review B:
Condensed Matter, 63, 245306.
179 Wright, A.F. (1997) Elastic properties of
zinc-blende and wurtzite AlN, GaN, and
InN. Journal of Applied Physics, 82 (6),
2833–2839.
180 Kim, K., Lambrecht, W.R.L. and Segall, B.
(1997) Erratum: Elastic constants and
related properties of tetrahedrally bonded
BN, AlN, GaN, and InN [Phys. Rev. B 53,
16310 (1996)]. Physical Review B:
Condensed Matter, 56 (11), 7018.
181 Bechstedt, F., Grossner, U. and
Furthm€
uller, J. (2000) Dynamics and
polarization of group-III nitride lattices:
a first-principles study. Physical Review B:
Condensed Matter, 62 (12), 8003–8011.
182 Yim, W.M., Stofko, E.J., Zanzucchi, P.J.,
Pankove, J.I., Ettenberg, M. and Gilbert,
S.L. (1973) Journal of Applied Physics,
44, 292.
183 Perry, P.B. and Rutz, R.F. (1978) The
optical absorption edge of single-crystal
AlN prepared by a close-spaced vapor
process. Applied Physics Letters, 33 (4),
319–321.
184 Guo, Q. and Yoshida, A. (1994) Japanese
Journal of Applied Physics, Part 1: Regular
185
186
187
188
189
190
191
192
193
Papers, Short Notes & Review Papers, 33,
2454.
Vispute, R.D., Wu, H. and Narayan, J.
(1995) High quality epitaxial aluminum
nitride layers on sapphire by pulsed laser
deposition. Applied Physics Letters, 67 (11),
1549–1551.
Tang, X., Hossain, F., Wongchotigul, K.
and Spencer, M.G. (1998) Near band-edge
transition in aluminum nitride thin films
grown by metal organic chemical vapor
deposition. Applied Physics Letters, 72 (12),
1501–1503.
Brunner, D., Angerer, H., Bustarret, E.,
Freudenberg, F., Hoepler, R., Dimitrov,
R., Ambacher, O. and Stutzmann, M.
(1997) Optical constants of epitaxial
AlGaN films and their temperature
dependence. Journal of Applied Physics, 82
(10), 5090–5096.
Wethkamp, T., Wilmers, K., Cobet, C.,
Esser, N., Richter, W., Ambacher, O.,
Stutzmann, M. and Cardona, M. (1999)
Physical Review B: Condensed Matter,
59, 1845.
Kuokstis, E., Zhang, J., Fareed, Q., Yang,
J.W., Simin, G., Asif Khan, M., Gaska, R.,
Shur, M., Rojo, C. and Schowalter, L.
(2002) Near-band-edge
photoluminescence of wurtzite-type AlN.
Applied Physics Letters, 81 (15), 2755–2757.
Guo, Q., Nishio, M., Ogawa, H. and
Yoshida, A. (2001) Temperature effect
on the electronic structure of AlN. Physical
Review B: Condensed Matter, 64, 113105.
Silveira, E., Freitas, J.A., Jr, Kneissl, M.,
Treat, D.W., Johnson, N.M., Slack, G.A.
and Schowalter, L.J. (2004) Nearbandedge cathodoluminescence of an
AlN homoepitaxial film. Applied Physics
Letters, 84 (18), 3501–3503.
Slack, G.A., Schowalter, L.J., Morelli, D.
and Freitas, J.A., Jr (2002) Journal of
Crystal Growth, 246, 287.
Shimada, K., Sota, T. and Suzuki, K.
(1998) First-principles study on electronic
and elastic properties of BN, AlN, and
GaN. Journal of Applied Physics, 84, (9),
4951–4958.
References
194 Silveira, E., Freitas, J.A., Jr, Glembocki,
O.J., Slack, G.A. and Schowalter, L.J.
(2005) Excitonic structure of bulk AlN
from optical reflectivity and
cathodoluminescence measurements.
Physical Review B: Condensed Matter, 71,
041201(R)-1–041201(R)-4.
195 Oshikiri, M., Aryasetiawan, F., Imanaka,
Y. and Kido, G. (2002) Physical Review B:
Condensed Matter, 66, 125204.
196 Persson, C., Sernelius, B.E., Ferreira da
Silva, A., Ahuja, R. and Johansson, B.
(2001) Journal of Physics: Condensed
Matter, 13, 8915.
197 Shan, W., Ager, J.W., III, Walukiewicz, W.,
Haller, E.E., Little, B.D., Song, J.J.,
Schurman, M., Feng, Z.C., Stall, R.A. and
Goldenberg, B. (1998) Near-band-edge
photoluminescence emission in
AlxGa1xN under high pressure. Applied
Physics Letters, 72 (18), 2274–2276.
198 Tsubouchi, K., Sugai, K. and Mikoshiba, N.
(1981) Ultrasonics Symposium, vol. 1 (ed.
B.R. McAvoy), IEEE, New York, p. 375.
199 McNeil, L.E., Grimditch, M. and French,
R.H. (1993) Journal of the American
Ceramic Society, 76, 1132.
200 Deger, C., Born, E., Angerer, H.,
Ambacher, O., Stutzmann, M.,
Hornsteiner, J., Riha, E. and Fischerauer,
G. (1998) Sound velocity of AlxGa1xN
thin films obtained by surface acousticwave measurements. Applied Physics
Letters, 72 (19), 2400–2402.
201 Davydov, S.Yu. and Davydov, S.Yu.
(2002) Evaluation of physical parameters
for the group III nitrates: BN, AlN,
GaN, and InN. Semiconductors, 36
(1), 41–44.
202 Davydov, V.Yu., Kitaev, Yu.E., Goncharuk,
I.N., Smirnov, A.N., Graul, J.,
Semchinova, O., Uffmann, D., Smirnov,
M.B., Mirgorodsky, A.P. and Evarestov,
R.A. (1998) Phonon dispersion and
Raman scattering in hexagonal GaN and
AlN. Physical Review B: Condensed Matter,
58 (19), 12899–12907.
203 Ruiz, E., Alvarez, S. and Alemany, P.
(1994) Electronic structure and properties
204
205
206
207
208
209
210
211
212
213
of AlN. Physical Review B: Condensed
Matter, 49 (11), 7115–7123.
Kato, R. and Hama, J. (1994) Firstprinciples calculation of the elastic
stiffness tensor of aluminium nitride
under high pressure. Journal of Physics:
Condensed Matter, 6, 7617–7632.
Hutson, A.R. (1963) US Patent No.
3,090,876, May 21.
Tsubouchi, K., Sugai, K. and Mikoshiba,
N. (1982) Proceedings of the IEEE
Ultrasonic Symposium, IEEE, New York,
p. 340.
Muensit, S., Goldys, E.M. and Guy, I.L.
(1999) Shear piezoelectric coefficients of
gallium nitride and aluminum nitride.
Applied Physics Letters, 75 (25), 3965–3967.
Lueng, C.M., Chan, H.W., Surya, C. and
Choy, C.L. (2000) Piezoelectric coefficient
of aluminum nitride and gallium nitride.
Journal of Applied Physics, 88 (9),
5360–5363.
Bernardini, F., Fiorentini, V. and
Vanderbilt, D. (1997) Spontaneous
polarization and piezoelectric constants
in III–V nitrides. Physical Review B:
Condensed Matter, 56, R10024.
Bernardini, F. and Fiorentini, V. (2002)
First-principles calculation
of the
$
piezoelectric tensor d of III–V nitrides.
Applied Physics Letters, 80 (22), 4145–4147.
Kamiya, T. (1996) Japanese Journal of
Applied Physics, Part 1: Regular Papers,
Short Notes & Review Papers, 35, 4421.
Zoroddu, A., Bernardini, F., Ruggerone,
P. and Fiorentini, V. (2001) Firstprinciples prediction of structure,
energetics, formation enthalpy, elastic
constants, polarization, and piezoelectric
constants of AlN, GaN, and InN:
comparison of local and gradientcorrected density-functional theory.
Physical Review B: Condensed Matter, 64,
45208.
Nardelli, M.B., Rapcewicz, K., Bernholc, J.
(1997) Strain effects on the interface
properties of nitride semiconductors.
Physical Review B: Condensed Matter, 55
(12), R7323–R7326.
j309
j 2 Electronic Band Structure and Polarization Effects
310
214 Leroux, M., Grandjean, N., Massies, J.,
Gil, B., Lefebvre, P. and Bigenwald, P.
(1999) Physical Review B: Condensed
Matter, 60, 1496.
215 Grandjean, N., Damilano, B., Dalmasso,
S., Leroux, M., Laugt, M. and Massies, J.
(1999) Journal of Applied Physics, 86, 3714.
216 Garrido, J.A., Sanchez-Rojas, J.L.,
Jimenez, A., Munoz, E., Omnes, F. and
Gibart, P. (1999) Applied Physics Letters, 75,
2407.
217 Hogg, R.A., Norman, C.E., Shields, A.J.,
Pepper, M. and Iizuka, M. (2000) Applied
Physics Letters, 76, 1428.
218 Park, S.-H. and Chuang, S.-L. (2000)
Applied Physics Letters, 76, 1981.
219 Cingolani, R., Botchkarev, A., Tang, H.,
Morkoç, H., Coliı, G., Lomascolo, M., Di
Carlo, A. and Lugli, P. (2000) Spontaneous
polarization and piezoelectric field in
GaN/Al0.15Ga0.85N quantum wells: impact
on the optical spectra. Physical Review B:
Condensed Matter, 61 (4), 2711–2715.
220 Bernardini, F. and Fiorentini, V. (2001)
Nonlinear macroscopic polarization in
III–V nitride alloys. Physical Review B:
Condensed Matter, 64, 085207.
221 Bernardini, F. and Fiorentini, V. (2002)
Erratum: Nonlinear macroscopic
polarization in III–V nitride alloys [Phys.
Rev. B 64, 085207 (2001)]. Physical Review
B: Condensed Matter, 65, 129903(E).
222 Fiorentini, V., Bernardini, F. and
Ambacher, O. (2002) Evidence for
nonlinear macroscopic polarization in
III–V nitride alloy heterostructures.
Applied Physics Letters, 80 (7), 1204–1206.
223 Petrov, I., Mojab, E., Powell, R.C., Greene,
J.E., Hultman, L. and Sundgren, J.-E.
(1992) Synthesis of metastable epitaxial
zinc-blende-structure AlN by solid-state
reaction. Applied Physics Letters, 60 (20),
2491–2493.
224 Gerthsen, D., Neubauer, B., Dieker, Ch.,
th, H. (1999)
Lantier, R., Rizzi, A. and Lu
Molecular beam epitaxy (MBE) growth
and structural properties of GaN and AlN
on 3C-SiC(0 0 1) substrates. Journal of
Crystal Growth, 200, 353–361.
225 Thompson, M.P., Auner, G.W., Zheleva,
T.S., Jones, K.A., Simko, S.J. and Hilfiker,
J.N. (2001) Deposition factors and band
gap of zinc-blende AlN. Journal of Applied
Physics, 89 (6), 3331–3336.
226 Lebedev, V., Cimalla, V., Kaiser, U.,
Foerster, Ch., Pezoldt, J., Biskupek, J. and
Ambacher, O. (2005) Effect of nanoscale
surface morphology on the phase stability
of 3C-AlN films on Si(1 1 1). Journal of
Applied Physics, 97, 114306–1.
227 Cardona, M. and Christensen, N.E. (2000)
Spin-orbit splittings in AlN, GaN and InN.
Solid State Communications, 116 (8),
421–425.
228 Karch, K. and Bechstedt, F. (1997) Ab
initio lattice dynamics of BN and AlN:
covalent versus ionic forces. Physical
Review B: Condensed Matter, 56 (12),
7404–7415.
229 Foutz, B.E., Oleary, S.K., Shur, M.S. and
Eastman, L.F. (1999) Journal of Applied
Physics, 85, 7727.
230 Matsuoka, T., Okamoto, H. and Nakao, M.
(2003) Growth of wurtzite InN using
MOVPE and its optical characteristics.
Physica Status Solidi c, 0 (7), 2806– 2809.
231 Matsuoka, T. (2004) MOVPE growth
and characteristics of nitride
semiconductors from GaN to InN, in
Advanced Materials in Electronics
(ed. Q. Guo), pp. 46–83.
232 Osamura, K., Naka, S. and Murakami, Y.
(1975) Preparation and optical properties
of Ga1 xInxN thin films. Journal of
Applied Physics, 46 (8), 3432–3437.
233 Puychevrier, N. and Menoret, M. (1976)
Thin Solid Films, 36, 141.
234 Tyagai, V.A., Evstigneev, A.M., Krasiko,
A.N., Andreeva, A.F. and Malakhov, V.Ya.
(1977) Soviet Physics: Semiconductors,
11, 1257.
235 Tansley, T.L. and Foley, C.P. (1986) Journal
of Applied Physics, 59, 3241.
236 Westra, K.L. and Brett, M.J. (1990) Thin
Solid Films, 192, 227.
237 Bhuiyan, A.G., Hashimoto, A. and
Yamamoto, A. (2003) Indium nitride
(InN): a review on growth,
References
238
239
240
241
242
243
244
characterization, and properties. Journal
of Applied Physiology, 94 (5), 2779–2808.
See Butcher, K.S. (ed.) (2006) Proceedings
of the 2nd ONR International Indium
Nitride Workshop Kailua-Kona, January
9–13, 2005, Hawaii, Journal of Crystal
Growth, 288 (2), 217–304.
Davydov, V.Yu., Klochikhin, A.A., Emtsev,
V.V., Ivanov, S.V., Vekshin, V.V.,
Bechstedt, F., Furthm€
uller, J., Harima,
H., Mudryi, A.V., Hashimoto, A.,
Yamamoto, A., Aderhold, J., Graul, J. and
Haller, E.E. (2002) Band gap of InN and
In-rich InxGa1xN alloys (0.36 < x < 1).
Physica Status Solidi b: Basic Research, 230
(2), R4–R6.
Davydov, V.Yu., Klochikhin, A.A., Seisyan,
R.P., Emtsev, V.V., Ivanov, S.V., Bechstedt,
F., Furthmuller, J., Harima, H., Mudryi,
A.V., Aderhold, J., Semchinova, O. and
Graul, J. (2002) Physica Status Solidi b:
Basic Research, 229, R1.
Davydov, V.Yu., Klochikhin, A.A., Emtsev,
V.V., Kurdyukov, D.A., Ivanov, S.V.,
Vekshin, V.A., Bechstedt, F., Furthm€
uller,
J., Aderhold, J., Graul, J., Mudryi, A.V.,
Harima, H., Hashimoto, A., Yamamoto,
A. and Haller, E.E. (2002) Band gap of
hexagonal InN and InGaN alloys. Physica
Status Solidi b: Basic Research, 234 (3),
787–795.
Davydov, V.Yu., Klochikhin, A.A., Seisyan,
R.P., Emtsev, V.V., Ivanov, S.V., Bechstedt,
F., Furthm€
uller, J., Harima, H., Mudryi,
A.V., Aderhold, J., Semchinova, O. and
Graul, J. (2002) Absorption and emission
of hexagonal InN. Evidence of narrow
fundamental band gap. Physica Status
Solidi b: Basic Research, 229 (3), R1–R3.
Wu, J., Walukiewicz, W.W., Yu, K.M.,
Ager, J.W., III, Haller, E.E., Lu, H., Schaff,
W.J., Saito, Y. and Nanishi, Y. (2002)
Unusual properties of the fundamental
band gap of InN. Applied Physics Letters, 80
(21), 3967–3969.
Matsuoka, T., Okamoto, H., Nakao, M.,
Harima, H. and Kurimoto, E. (2002)
Optical bandgap energy of wurtzite InN.
Applied Physics Letters, 81 (7), 1246–1248.
245 Gwo, S., Wu, C.-L., Shen, C.-H., Chang,
W.-H., Hsu, T.M., Wang, J.-S. and Hsu,
J.-T. (2004) Heteroepitaxial growth of
wurtzite InN films on Si (1 1 1) exhibiting
strong near-infrared photoluminescence
at room temperature. Applied Physics
Letters, 84 (19), 3765–3767.
246 Nag, B.R. (2002) Comment on band gap of
InN and In-rich InxGa1xN alloys
(0.36 < x < 1). Physica Status Solidi b:
Basic Research, 233 (3), R8–R9.
247 Davydov, V.Yu., Klochikhin, A.A., Emtsev,
V.V., Bechstedt, F., Mudryi, A.V. and
Haller, E.E. (2002) Reply to Comment
on band gap of InN and In-rich
InxGa1xN alloys (0.36 < x < 1). Physica
Status Solidi b: Basic Research, 233 (3),
R10–R11.
248 Inushima, T., Mamutin, V.V., Vekshin,
V.A., Ivanov, S.V., Sakon, T., Motokawa,
M. and Ohoya, S. (2001) Physical
properties of InN with the band gap
energy of 1.1 eV. Journal of Crystal Growth,
227–228, 481–485.
249 Arnaudov, B., Paskova, T., Paskov, P.P.,
Magnusson, B., Valcheva, E., Monemar,
B., Lu, H., Schaff, W.J., Amano, H. and
Akasaki, I. (2004) Energy position of nearband-edge emission spectra of InN
epitaxial layers with different doping
levels. Physical Review B: Condensed
Matter, 69, 115216.
250 Araki, T., Saito, Y., Yamaguchi, T.,
Kurouchi, M., Nanishi, Y. and Naoi, H.
(2004) Radio frequency-molecular beam
epitaxial growth of InN epitaxial films on
(0 0 0 1) sapphire and their properties.
Journal of Vacuum Science & Technology B:
Microelectronics and Nanometer Structures,
22 (4), 2139–2143.
251 Xu, K., Terashima, W., Hata, T.,
Hashimoto, N., Yoshitani, M., Cao, B.,
Ishitani, Y. and Yoshikawa, A. (2003)
Comparative study of InN growth on Gaand N-polarity GaN templates by
molecular-beam epitaxy. Physica Status
Solidi c, 0 (7), 2814–2817.
252 Xu, K. and Yoshikawa, A. (2003) Effects of
film polarities on InN growth by
j311
j 2 Electronic Band Structure and Polarization Effects
312
253
254
255
256
257
258
259
260
261
molecular-beam epitaxy. Applied Physics
Letters, 83 (2), 251–253.
Yoshikowa, A., Xu, K., Hashimoto, N.,
Terashima, W., Yoshitani, M., Choe, S.
and Ishitani, Y. (2003) Effects of film
polarities on InN growth by RF-molecular
beam epitaxy, MRS 8th Wide-Bandgap IIINitride Workshop, September
29–October 1, Richmond, VA.
Xu, K. and Yoshikawa, A. (2003) Effects of
film polarities on InN growth by
molecular-beam epitaxy. Applied Physics
Letters, 83 (2), 251–253.
Ishitani, Y., Xu, K., Terashima, W.,
Masuyama, H., Yoshitani, M.,
Hashimoto, N., Che, S.B. and Yoshikawa,
A. (2004) Temperature dependence of the
optical properties for InN films grown by
RF-MBE. GaN and Related Alloys – 2003
Symposium, Warrendale, PA, USA,
Material Research Society Symposium
Proceedings, 798, 207–212.
Ishitani, Y., Masuyama, H., Terashima,
W., Yoshitani, M., Hashimoto, N., Che,
S.B. and Yoshikawa, A. (2005) Bandgap
energy of InN and its temperature
dependence. Physica Status Solidi c, 2 (7),
2276–2276.
Lan, Z.H., Wang, W.M., Sun, C.L., Shi,
S.C., Hsu, C.W., Chen, T.T., Chen, K.H.,
Chen, C.C., Chen, Y.F. and Chen, L.C.
(2004) Growth mechanism, structure and
IR photoluminescence studies of indium
nitride nanorods. Journal of Crystal
Growth, 269, 87–94.
Chen, L.C. Center for Condensed Matter
Sciences, Academia Sinica, private
communication.
ODonnell, K.P., Pereira1 S., Martin,
R.W., Edwards, P.R., Tobin, M.J. and
Mosselmans, J.F.W. (2003) Physica Status
Solidi a: Applied Research, 195, 532.
Anderson, P.A., Lee, T.E., Kendrick, C.E.,
Diehl, W., Kinsey, R.J., Kennedy, V.J.,
Markwitz, A., Reeves, R.J. and Durbin,
S.M. (2004) Proceedings of the SPIE,
5277, 90.
Butcher, K.S.A., Wintrebert-Fouquet, M.,
Chen, P.P., Timmers, H. and Sherestha,
262
263
264
265
266
267
268
269
270
S.K. (2003) Detailed analysis of the
absorption data for InN. Presented at the
International Symposium on Point
Defects and Nonstoichiometry, March
2003, Sendai, Japan, Material Science in
Semiconductor Processing, 6, 351.
Shubina, T.V., Ivanov, S.V., Jmerik, V.N.,
Solnyshkov, D.D., Vekshin, V.A., Kopev,
P.S., Vasson, A., Leymarie, J., Kavokin, A.,
Amano, H., Shimono, K., Kasic, A. and
Monemar, B. (2004) Mie resonances,
infrared emission, and the band gap of
InN. Physical Review Letters, 92 (11),
117407–117407.
Juza, R. and Hahn, H. (1938) Zeitschrift f€
ur
Anorganishe und Allegemeine Chemie, 239,
282.
Hahn, H. and Juza, R. (1940) Zeitschrift f€
ur
Anorganishe und Allegemeine Chemie, 244,
III.
MacChesney, J.B., Bridenbaugh, P.M.,
OConnor, P.B. (1970) Materials Research
Bulletin, 5, 783.
Tomizawa, J., Takaim, O., Tsujikawa, S.
and Goto, S. (1983) Reactive evaporation
technique assisted by low temperature
plasma for preparation of ultrafine nitride
particles. Proceedings of the International
Ion Engineering Congress, Institute of
Electrical Engineering, Kyoto, Japan,
p. 1411.
Butcher, K.S.A. (2004) InN, a historic
review – from obscurity to controversy, in
Advanced Materials in Electronics,
Research Signpost (ed. Q. Guo), p. 1
Butcher, K.S.A. and Tansley, T.L. (2005)
InN, latest development and a review of
the band-gap controversy. Superlattices
and Microstructures, 38 (1), 1–37.
Hamberg, I. and Granqvist, C.G. (1986)
Journal of Applied Physics, 60, R123.
Davydov, V.Yu., Klochikhin, A.A.,
Emtsev, V.V., Kurdyukov, D.A., Ivanov,
S.V., Vekshin, V.A., Bechstedt, F.,
Furthm€
uller, J., Aderhold, J., Graul, J.,
Mudryi, A.V., Harima, H., Hashimoto, A.,
Yamamoto, A. and Haller, E.E.
(2002) Physica Status Solidi b: Basic
Research, 234, 787.
References
271 Kumar, S., Motlan, L.M. Tansley, T.L.
(1996) Japanese Journal of Applied Physics,
35, 2261–2267.
272 Butcher, K.S.A., Wintrebert-Fouquet, M.,
Chen, P.P.-T., Tansley, T.L., Dou, H.,
Shrestha, S.K., Timmers, H., Kuball, M.,
Prince, K.E. and Bradby, J.E. (2004)
Nitrogen rich indium nitride. Journal of
Applied Physics, 95, 6124.
273 Yoshimoto, M., Vamamoto, V. and Saraie,
J. (2003) Fabrication of InN/Si
heterojunctions with rectifying
characteristics. Physica Status Solidi c, 0,
2794–2797.
274 Westra, K.L., Lawson, R.P.W. and Brett,
M.J. (1988) Journal of Vacuum Science &
Technology A: Vacuum Surfaces and Films,
6, 1730.
275 Foley, C.P. and Lyngdal, J. (1987) Journal
of Vacuum Science & Technology A: Vacuum
Surfaces and Films, 5, 1708.
276 Butcher, K.S.A., Dou, H., Goldys, E.M.,
Tansley, T.L. and Srikeaw, S. (2002)
Ultraviolet Raman and optical
transmission studies of rf sputtered
indium nitride. Physica Status Solidi c, 0,
373–376.
277 Trainor, J.W. and Rose, K. (1974) Journal of
Electronic Materials, 3, 821.
278 Ivanov, S.V., Jmerik, V.N., Shubina, T.V.,
Vekshin, V.A., Kopev, P.S. and Monemar,
B. (2003) Plasma assisted MBE growth
and characterisation of InN on sapphire.
Presentation at the 1st International InN
Workshop, November 16–20, Fremantle,
Australia.
279 Paszkowicz, W., Cerny, R. and Krukowski,
S. (2003) Powder Diffraction, 18, 114.
280 Lagerstedt, O. and Monemar, B. (1979)
Physical Review B: Condensed Matter, 19,
3064.
281 Yamaguchi, T., Saito, Y., Kano, K., Araki,
T., Teraguchi, N., Suzuki, A. and Nanishi,
Y. (2002) Study of epitaxial relationship in
InN growth on sapphire (0 0 0 1) by RFMBE. Physica Status Solidi b: Basic
Research, 228, 17–20.
282 Walukiewicz, J.Wu, Yu, K.M., Shan, W.,
Ager, J.W., III, Hailer, E.E., Lu, H., Schaff,
283
284
285
286
287
288
289
290
291
292
W.J., Metzger, W.K. and Kurtz, S. (2003)
Journal of Applied Physics, 94, 6477.
Lang, D.V. (1977) Review of RadiationInduced Detects in III–V Compounds,
Institute of Physics Conference Series 31,
Chapter 1, p. 70.
Shrestha, S.K., Timmers, H., Butcher,
K.S.A. and Wintrebert-Fouquet, M. (2004)
Accurate stoichiometric analysis of
polycrystalline indium nitride films with
elastic recoil detection. Current Applied
Physics, 4, 237–240.
Butcher, K.S.A., Wintrebert-Fouquet, M.,
Chen P.P.-T., Prince, K.E., Timmers, H.,
Shrestha, S.K., Shubina, T.V., Ivanov, S.V.,
Wuhrer, R., Phillips, M.R. and Monemar,
B. (2005) Non-stoichiometry and nonhomogeneity in InN. 2004 International
Workshop on Nitride Semiconductors,
Pittsburgh, Physica Status Solidi c, 2 (7),
2263–2266.
Shrestha, S.K., Butcher, K.S.A.,
Wintrebert-Fouquet, M. and Timmers, H.
(2004) Reliable ERD analysis of group-III
nitrides despite severe nitrogen depletion.
Nuclear Instruments and Methods in Physics
Research B, 219–220, 686–692.
Kosiba, R., Ecke, G., Cimalla, V., Spieb, L.,
Krischok, S., Schaefer, J.A., Ambacher, O.
and Schaff, W.J. (2004) Sputter depth
profiling of InN. Nuclear Instruments and
Methods in Physics Research B, 215, 486.
Shubina, T.V., Ivanov, S.V., Jmerik, V.N.,
Solnyshkov, D.D., Vekshin, V.A., Kopev,
P.S., Vasson, A., Leymarie, J., Kavokin, A.,
Amano, H., Shimono, K., Kasic, A. and
Monemar, B. (2004) Mie resonances
infrared emission, and the band gap of
InN. Physical Review Letters, 92 (11),
117407–117407.
Xu, K. and Yoshikawa, A. (2003) Applied
Physics Letters, 83 251.
Kreibig, U. and Vollmer, M. (1995) Optical
Properties of Metal Clusters, Springer,
Berlin.
Mie, G. (1908) Annals of Physics (Leipzig),
25, 377.
Sobolev, V.V. and Zlobina, M.A. (1999)
Semiconductors, 33, 395.
j313
j 2 Electronic Band Structure and Polarization Effects
314
293 Nolte, D.D. (1994) Journal of Applied
Physics, 76, 3740.
294 Maxwell-Garnett, J.C. (1904) Philosophical
Transactions of the Royal Society of London,
203, 385.
295 Wei, S.-H., Nie, X., Batyrev, I.G. and
Zhang, S.B. (2003) Breakdown of the
band-gap-common-cation rule: the origin
of the small band gap of InN. Physical
Review B: Condensed Matter, 67, 165209.
296 Tsen, K.T., Liang, W., Ferry, D.K., Lu, H.,
Schaff, W.J., Özg€
ur, Ü., Fu, Y., Moon, Y.T.,
Yun, F., Morkoç, H., and Everitt, H.O.
(2005) Optical studies of carrier dynamics
and non-equilibrium optical phonons in
nitride-based wide bandgap
semiconductors. Superlattices and
Microstructures, 38 (2), 77–77.
297 Kasic, A., Schubert, M., Saito, Y., Nanishi,
Y. and Wagner, G. (2002) Effective
electron mass and phonon modes in ntype hexagonal InN. Physical Review B:
Condensed Matter, 65, 115206.
298 Xu, Y.-N. and Ching, W.Y. (1993) Physical
Review B: Condensed Matter, 48, 4335.
299 Tsen, K.T. (2001) Ultrafast Physical Processes
in Semiconductors, Semiconductors and
Semimetals, vol. 67 (ed. K.T. Tsen),
Academic Press, New York.
300 Wu, J., Walukiewicz, W., Shan, W., Yu,
K.M., Ager, J.W., III, Li, S.X., Haller, E.E.,
Lu, H. and Schaff, W.J. (2003) Journal of
Applied Physics, 94, 4457.
301 Arnaudov, G., Vilkotskii, V.A.,
Domanevskii, D.S., Evtimova, S.K. and
Tkachev, V.D. (1977) Fizika i Tekhnika
Poluprovodnikov, 11, 1799; Soviet Physics:
Semiconductors, 1977, 11, 1054.
302 De-Sheng, J., Makita, Y., Ploog, K. and
Queisser, H.J. (1982) Journal of Applied
Physics, 53, 999.
303 Bugajski, M. and Lewandovski, W. (1985)
Journal of Applied Physics, 57, 521.
304 Averkiev, N.S., Kalinin, B.N., Losev, A.V.,
Rogachev, A.A. and Filipenko, A.S. (1990)
Physica Status Solidi a: Applied Research,
121, K129.
305 Arnaudov, B., Paskova, T., Goldis, E.M.,
Evtimova, S. and Monemar, B. (2001)
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
Physical Review B: Condensed Matter, 64,
045213.
See, for example, Shklovskii, B.I. and
Efros, A.L. (1984) Electronic Properties of
Doped Semiconductors, Springer, Berlin,
Chapter 11.
Nag, B.R. (2003) On the band gap of
indium nitride. Physica Status Solidi b:
Basic Research, 237 (2), R1–R2.
Inushima, T., Yaguchi, T., Nagase, A., Iso,
A., Shiraishi, T. and Ooya, S. (1995)
Proceedings of 7th International
Conference on InP and Related Materials,
p. 187.
Inushima, T., Shiraishi, T. and Davydov,
V.Y. (1999) Solid State Communications,
110, 491.
Wu, J., Walukiewicz, W., Shan, W., Yu,
K.M., Ager, J.W., III, Haller, E.E., Lu, H.
and Schaff, W.J. (2002) Effects of the
narrow band gap on the properties of InN.
Physical Review B: Condensed Matter, 66,
201403.
Strite, S. and Morkoç, H. (1992) GaN, AlN
and InN: a review. Journal of Vacuum
Science & Technology B: Microelectronics
and Nanometer Structures, 10 (4),
1237–1266.
Tisch, U., Finkman, E. and Salzman, J.
(2002) Applied Physics Letters, 81, 463.
Tomic, S. and OReilly, E.P. (2003)
IEEE Photonics Technology Letters, 15, 6.
Groves, W.O., Herzog, A.H. and Craford,
M.G. (1971) Applied Physics Letters, 19, 184.
Wu, J., Shan, W. and Walukiewicz, W.
(2002) Semiconductor Science and
Technology, 17, 860.
Lindsay, A., Tomic, S. and OReilly, E.P.
(2003) Solid-State Electronics, 47, 443.
Suemune, I., Uesugi, K. and Walukiewicz,
W. (2000) Applied Physics Letters, 77, 3021.
OReilly, E.P. and Lindsay, A. (1999)
Physica Status Solidi b: Basic Research, 216,
131.
Lindsay, A. and OReilly, E.P. (1999) Solid
State Communications, 112, 443.
Choulis, S.A., Tomic, S., OReilly, E.P. and
Hosea, T.J.C. (2003) Solid State
Communications, 125, 155.
References
321 Choulis, S.A., Hosea, T.J.C., Tomic, S.,
Kamal-Saadi, M., Adams, A.R., OReilly,
E.P., Weinstein, B.A. and Klar, P.J. (2002)
Physical Review B: Condensed Matter, 66,
165321.
322 Gil, B. (2000) Solid State Communications,
114, 623.
323 Shtinkov, N., Desjardins, P. and Masut,
R.A. (2003) Physical Review B: Condensed
Matter, 67, 081202.
324 Kent, P.R.C., Bellaiche, L. and Zunger, A.
(2002) Semiconductor Science and
Technology, 17, 851.
325 Kent, P.R.C. and Zunger, A. (2001)
Physical Review Letters, 86, 2613.
326 Kent, P.R.C. and Zunger, A. (2001)
Physical Review B: Condensed Matter, 64,
115208.
327 Szwacki, G. and Boguslawski, P. (2001)
Physical Review B: Condensed Matter, 64,
161201.
328 Weyers, M., Sato, M. and Ando, H. (1992)
Japanese Journal of Applied Physics, Part 2:
Letters, 31, L853.
329 Weyers, M. and Sato, M. (1993) Applied
Physics Letters, 62, 1396.
330 Kuroiwa, R., Asahi, H., Asami, K., Kim,
S.-J., Iwata, K. and Gonda, S. (1998)
Applied Physics Letters, 73, 2630.
331 Kondow, M., Uomi, K., Hosomi, K. and
Mozume, T. (1994) Japanese Journal of
Applied Physics, Part 2: Letters, 33, L1056.
332 Pozina, G., Ivanov, I., Monemar, B.,
Thordson, J.V. and Andersson, T.G. (1998)
Journal of Applied Physics, 84, 3830.
333 Bellaiche, L. and Zunger, A. (1998)
Physical Review B: Condensed Matter, 57,
4425.
334 Sakai, S., Ueta, Y. and Terauchi, Y. (1993)
Japanese Journal of Applied Physics, Part 1:
Regular Papers, Short Notes & Review
Papers, 32, 4413.
335 Wei, S.-H. and Zunger, A. (1996) Physical
Review Letters, 76, 664.
336 Neugebauer, J. and Van de Walle, C.G.
(1995) Physical Review B: Condensed
Matter, 51, 10568.
337 Rubio, A. and Cohen, M.L. (1995) Physical
Review B: Condensed Matter, 51, 4343.
338 Bellaiche, L., Wei, S.-H. and Zunger, A.
(1996) Physical Review B: Condensed
Matter, 54, 17568.
339 Bellaiche, L., Wei, S.-H. and Zunger, A.
(1997) Applied Physics Letters, 70, 3558.
340 Mattila, T., Wei, S.-H. and Zunger, A.
(1999) Physical Review B: Condensed
Matter, 60, R11245.
341 Bellaiche, L., Wei, S.-H. and Zunger, A.
(1997) Physical Review B: Condensed
Matter, 56, 10233.
342 Bi, W.G. and Tu, C.W. (1997) Applied
Physics Letters, 70, 1608.
343 Salzman, J. and Temkin, H. (1997)
Materials Science & Engineering B:
Solid State Materials for Advanced
Technology, 50, 148.
344 Ougazzaden, A., Le Bellego, Y., Rao,
E.V.K., Juhel, M., Leprince, L. and
Patriarche, G. (1997) Applied Physics
Letters, 70, 2861.
345 Makimoto, T., Saito, H., Nishida, T. and
Kobayashi, N. (1997) Applied Physics
Letters, 70, 2984.
346 Malikova, L., Pollak, F.H. and Bhat, R.
(1998) Journal of Electronic Materials,
27, 484.
347 Uesugi, K., Morooka, N. and Suemune, I.
(1999) Applied Physics Letters, 74, 1254.
348 Sik, J., Schubert, M., Leibiger, G.,
Gottschalch, V., Kirpal, G. and
Humlicek, J. (2000) Applied Physics
Letters, 76, 2859.
349 Kozhevnikov, M., Narayanamurti, V.,
Reddy, C.V., Xin, H.P., Tu, C.W.,
Mascarenhas, A. and Zhang, Y. (2000)
Physical Review B: Condensed Matter, 61,
R7861.
350 Uesugi, K., Suemune, I., Hasegawa, K.,
Akutagawa, T. and Nakamura, T. (2000)
Applied Physics Letters, 76, 1285.
351 Perlin, P., Subramanya, S.G., Mars, D.E.,
Kruger, J., Shapiro, N.A., Siegle, H. and
Weber, E.R. (1998) Applied Physics Letters,
73, 3703.
352 Shan, W., Walukiewicz, W., Ager, J.W., III,
Haller, E.E., Geisz, J.F., Friedman, D.J.,
Olson, J.M. and Kurtz, S.R. (1999) Physical
Review Letters, 82, 1221.
j315
j 2 Electronic Band Structure and Polarization Effects
316
353 Jones, E.D., Modine, N.A., Allerman,
A.A., Kurtz, S.R., Wright, A.F., Tozer, S.T.
and Wei, X. (1999) Physical Review B:
Condensed Matter, 60, 4430.
354 Kurtz, S.R., Modine, N.A., Jones, E.D.,
Allerman, A.A. and Klem, J.F. (2002)
Semiconductor Science and Technology, 17,
843.
355 Skierbiszewski, C., Perlin, P.,
Wisniewski, P., Knap, W., Suski, T.,
Walukiewicz, W., Shan, W., Yu, K.M.,
Ager, J.W., Haller, E.E., Geisz, J.F. and
Olson, J.M. (2000) Large, nitrogeninduced increase of the electron effective
mass in InyGa1yNxAs1x. Applied Physics
Letters, 76 (17), 2409.
356 Skierbiszewski, C., Perlin, P.,
Wisniewski, P., Suski, T., Walukiewicz,
W., Shan, W., Ager, J.W., Haller, E.E.,
Geisz, J.F., Friedman, D.J., Olson, J.M.
and Kurtz, S.R. (1999) Effect of nitrogeninduced modification of the conduction
band structure on electron transport in
GaAsN alloys. Physica Status Solidi b: Basic
Research, 216, 135.
357 Yu, K.M., Walukiewicz, W., Shan, W.,
Ager, J.W., III, Wu, J., Haller, E.E., Geisz,
J.F., Friedman, D.J. and Olson, J.M.
(2000) Physical Review B: Condensed
Matter, 61, R13337.
358 Hai, P.N., Chen, W.M., Buyanova, I.A.,
Xin, H.P. and Tu, C.W. (2000) Applied
Physics Letters, 77, 1843.
359 Pan, Z., Li, L.H., Lin, Y.W., Sun, B.Q.,
Jiang, D.S. and Ge, W.K. (2001) Applied
Physics Letters, 78, 2217.
360 Buyanova, I.A., Chen, W.M. and Tu, C.W.
(2002) Semiconductor Science and
Technology, 17, 815.
361 Young, D.L., Geisz, J.F. and Coutts, T.J.
(2003) Applied Physics Letters, 82, 1236.
362 Young, L., Geisz, J.F. and Coutts, T.J.
(2003) Applied Physics Letters, 82, 1236.
363 Chtourou, R., Bousbih, F., Ben Bouzid, S.,
Charfi, F.F., Harmand, J.C., Ungaro, G.
and Largeau, L. (2002) Applied Physics
Letters, 80, 2075.
364 Perkins, J.D., Mascarenhas, A., Zhang, Y.,
Geisz, J.F., Friedman, D.J., Olson, J.M.
365
366
367
368
369
370
371
372
373
374
375
376
and Kurtz, S.R. (1999) Physical Review
Letters, 82, 3312.
Shan, W., Yu, K.M., Walukiewicz, W.,
Ager, J.W., III, Haller, E.E. and Ridgway,
M.C. (1999) Applied Physics Letters, 75,
1410.
Zhang, Y., Mascarenhas, A., Xin, H.P. and
Tu, C.W. (2000) Physical Review B:
Condensed Matter, 61, 7479.
Zhang, Y., Mascarenhas, A., Geisz, J.F.,
Xin, H.P. and Tu, C.W. (2001)
Physical Review B: Condensed Matter,
63, 085205.
Zhang, Y., Mascarenhas, A., Xin, H.P. and
Tu, C.W. (2001) Physical Review B:
Condensed Matter, 63, 161303.
Klar, P.J., Gruning, H., Heimbrodt, W.,
Koch, J., Hohnsdorf, F., Stolz, W.,
Vicente, P.M.A. and Camassel, J. (2000)
Applied Physics Letters, 76, 3439.
Zhang, Y., Mascarenhas, A., Xin, H.P. and
Tu, C.W. (2000) Physical Review B:
Condensed Matter, 61, 4433.
Ya, M.H., Chen, Y.F. and Huang, Y.S.
(2002) Journal of Applied Physics, 92, 1446.
Egorov, A.Yu., Odnoblyudov, V.A.,
Mamutin, V.V., Zhukov, A.E.,
Tsatsulnikov, A.F., Kryzhanovskaya, N.V.,
Ustinov, V.M., Hong, Y.G. and Tu, C.W.
(2003) Valence band structure of GaAsN
compounds and band-edge lineup in
GaAs/GaAsN/InGaAs heterostructures.
Journal of Crystal Growth, 251 (1–4),
417–421.
Kitatani, T., Kondow, M., Kikawa, T.,
Yazawa, Y., Okai, M. and Uomi, K. (1999)
Japanese Journal of Applied Physics, Part 1:
Regular Papers, Short Notes & Review
Papers, 38, 5003.
Sun, B.Q., Jiang, D.S., Luo, X.D., Xu, Z.Y.,
Pan, Z., Li, L.H. and Wu, R.H. (2000)
Applied Physics Letters, 76, 2862.
Buyanova, I.A., Pozina, G., Hai, P.N.,
Chen, W.M., Xin, H.P. and Tu, C.W.
(2000) Physical Review B: Condensed
Matter, 63, 033303.
Krispin, P., Spruytte, S.G., Harris, J.S. and
Ploog, K.H. (2001) Journal of Applied
Physics, 90, 2405.
References
377 Egorov, A.Yu., Odnoblyudov, V.A., Krizhanovskaya, N.V., Mamutin, V.V. and
Ustinov, V.M. (2002) Semiconductors, 36,
1355.
378 Klar, P.J., Gr€
uning, H., Heimbrodt, W.,
Weiser, G., Koch, J., Volz, K., Stolz, W.,
Koch, S.W., Tomic, S., Choulis, S.A.,
Hosea, T.J.C., OReilly, E.P., Hofmann
M., Hader, J. and Moloney, J.V. (2002)
Interband transitions of quantum wells
and device structures containing Ga(N,
As) and (Ga, In)(N, As). Semiconductor
Science and Technology, 17, 830–830.
379 Egorov, A.Yu., Odnoblyudov, V.A.,
Krizhanovskaya, N.V., Mamutin, V.V. and
Ustinov, V.M. (2002) Band-edge line-up in
GaAs/GaAsN/InGaAs heterostructures.
Fizika i Tekhnika Poluprovodnikov, 36 (12),
1440–1444.
380 Yang, T., Nakajima, S. and Sakai, S. (1997)
Japanese Journal of Applied Physics, Part 2:
Letters, 36, L320.
381 Tit, N. and Dharma-wardana, M.W.C.
(2000) Applied Physics Letters, 76, 3576.
382 Naoi, H., Naoi, Y. and Sakai, S. (1997)
Solid-State Electronics, 41, 319.
383 Beresford, R., Stevens, K.S. and Schwatzman, A.F. (1998) Journal of Vacuum Science
& Technology B: Microelectronics and
Nanometer Structures, 16, 1293.
384 Fan, J.C., Hung, W.K., Chen, Y.F., Wang,
J.S. and Lin, H.H. (2000) Physical Review
B: Condensed Matter, 62, 10990.
385 Hung, W.K., Cho, K.S., Chern, M.Y.,
Chen, Y.F., Shih, D.K., Lin, H.H., Lu, C.C.
and Yang, T.R. (2002) Applied Physics
Letters, 80, 796.
386 Hang, D.R., Huang, C.F., Hung, W.K.,
Chang, Y.H., Chen, J.C., Yang, H.C.,
Chen, Y.F., Shih, D.K., Chu, T.Y. and Lin,
H.H. (2002) Shubnikov–de Haas
oscillations of two-dimensional electron
gas in an InAsN/InGaAs single quantum
well. Semiconductor Science and
Technology, 17, 999–999.
387 Bi, W.G. and Tu, C.W. (1996) Journal of
Applied Physics, 80, 1934.
388 Yu, K.M., Walukiewicz, W., Wu, J.,
Beeman, J.W., Ager, J.W., III, Haller, E.E.,
389
390
391
392
393
394
395
396
397
398
399
400
Shan, W., Xin, H.P. and Tu, C.W.
(2001) Applied Physics Letters,
78, 1077.
Murdin, B.N., Kamal-Saadi, M., Lindsay,
A., OReilly, E.P., Adams, A.R., Nott, G.J.,
Crowder, J.G., Pidgeon, C.R., Bradley, I.V.,
Wells, J.-P.R., Burke, T., Johnson, A.D.
and Ashley, T. (2001) Auger
recombination in long-wavelength
infrared InNxSb1x alloys. Applied Physics
Letters, 78 (11), 1568–1568.
Murdin, B.N., Adams, A.R., Murzyn, P.,
Pidgeon, R., Bradley, I.V., Wells, J.-P.R.,
Matsuda, Y.H., Miura, N., Burke, T. and
Johnson, A.D. (2002) Band anticrossing
in dilute InNxSb1x. Applied Physics
Letters, 81 (2), 256–256.
Ashley, T., Burke, T.M., Price, G.J.,
Adams, A.R., Andreev, A., Murdin, B.N.,
OReilly, E.P. and Pidgeon, C.R. (2003)
Solid-State Electronics, 47, 387.
Baillargeon, J.N., Cheng, K.Y., Hofler,
G.E., Pearah, P.J. and Hsieh, K.C. (1992)
Applied Physics Letters, 60, 2540.
Liu, X., Bishop, S.G., Baillargeon, J.N. and
Cheng, K.Y. (1993) Applied Physics Letters,
63, 208.
Miyoshi, S., Yaguchi, H., Onabe, K., Ito, R.
and Shiraki, Y. (1994) Journal of Crystal
Growth, 145, 87.
Bi, W.G. and Tu, C.W. (1996) Applied
Physics Letters, 69, 3710.
Shan, W., Walukiewicz, W., Yu, K.M., Wu,
J., Ager, J.W., III, Haller, E.E., Xin, H.P.
and Tu, C.W. (2000) Applied Physics Letters,
76, 3251.
Wu, J., Walukiewicz, W., Yu, K.M.,
Ager, J.W., III, Haller, E.E., Hong, Y.G.,
Xin, H.P. and Tu, C.W. (2002)
Physical Review B: Condensed Matter,
65, 241303.
Rudko, G.Yu., Buyanova, I.A., Chen,
W.M., Xin, H.P. and Tu, C.W. (2002)
Applied Physics Letters, 81, 3984.
Rudko, G.Yu., Buyanova, I.A., Chen,
W.M., Xin, H.P. and Tu, C.W. (2003) SolidState Electronics, 47, 493.
Xin, H.P. and Tu, C.W. (2000) Applied
Physics Letters, 77, 2180.
j317
j 2 Electronic Band Structure and Polarization Effects
318
401 Xin, H.P., Tu, C.W., Zhang, Y. and
Mascarenhas, A. (2000) Applied Physics
Letters, 76, 1267.
402 Xin, H.P., Welty, R.J. and Tu, C.W. (2000)
Applied Physics Letters, 77, 1946.
403 Zhang, Y., Fluegel, B., Mascarenhas,
A., Xin, H.P. and Tu, C.W. (2000)
Physical Review B: Condensed Matter,
62, 4493.
404 Leibiger, G., Gottschalch, V., Schubert,
M., Benndorf, G. and Schwabe, R. (2002)
Physical Review B: Condensed Matter, 65,
245207.
405 Reddy, C.V., Martinez, R.E., II,
Narayanamurti, V., Xin, H.P. and Tu, C.W.
(2002) Physical Review B: Condensed
Matter, 66, 235313.
406 Buyanova, I.A., Pozina, G., Bergman, J.P.,
Chen, W.M., Xin, H.P. and Tu, C.W.
(2002) Applied Physics Letters, 81, 52.
407 Sun, B.Q., Jiang, D.S., Pan, Z., Li, L.H.
and Wu, R.H. (2000) Applied Physics
Letters, 77, 4148.
408 Polimeni, A., Capizzi, M., Geddo, M.,
Fischer, M., Reinhardt, M. and Forchel, A.
(2001) Physical Review B: Condensed
Matter, 63, 195320.
409 Gokhale, M.R., Wei, J., Wang, H. and
Forrest, S.R. (1999) Applied Physics Letters,
74, 1287.
410 Zhukov, A.E., Kovsh, A.R., Semenova,
E.S., Ustinov, V.M., Wei, L., Wang, J.-S. and
Chi, J.Y. (2002) Semiconductors, 36, 899.
411 Choulis, S.A., Hosea, T.J.C., Tomic, S.,
Kamal-Saadi, M., Weinstein, B.A.,
OReilly, E.P., Adams, A.R. and Klar, P.J.
(2003) Physica Status Solidi b: Basic
Research, 235, 384.
412 Sun, H.D., Dawson, M.D., Othman, M.,
Yong, J.C.L., Rorison, J.M., Gilet, P.,
Grenouillet, L. and Million, A. (2003)
Applied Physics Letters, 82, 376.
413 Bellaiche, L. (1999) Applied Physics Letters,
75, 2578.
414 Al-Yacoub, A. and Bellaiche, L. (2000)
Physical Review B: Condensed Matter, 62,
10847.
415 Pinault, M.-A. and Tournie, E. (2001)
Applied Physics Letters, 78, 1562.
416 Klar, P.J., Gr€
uning, H., Koch, J., Sch€afer,
S., Volz, K., Stolz, W. and Heimbrodt, W.
(2001) Physical Review B: Condensed
Matter, 64, 121203.
417 Hong, Y.G., Andre, R. and Tu, C.W. (2001)
Journal of Vacuum Science & Technology B:
Microelectronics and Nanometer Structures,
19, 1413.
418 Ungaro, G., Le Roux, G., Teissier, R. and
Harmand, J.C. (1999) Electronics Letters,
35, 1246.
419 Harmand, J.C., Ungaro, G., Largeau, L.
and Le Roux, G. (2000) Applied Physics
Letters, 77, 2482.
420 Harmand, J.-C., Caliman, A., Rao, E.V.K.,
Largeau, L., Ramos, J., Teissier, R.,
Travers, L., Ungaro, G., Theys, B. and
Dias, I.F.L. (2003) Semiconductor Science
and Technology, 17, 778.
421 Bastard, G. (1982) Physical Review B:
Condensed Matter, 25, 7584.
422 Marzin, J.Y. (1986) Strained superlattices,
in Heterojunction and Semiconductor
Superlattices (eds A. Allan, G. Bastard, N.
Boccara, M. Lannoo and M. Voos),
Springer, Berlin, pp. 161–176.
423 Pollak, F.H. and Cardona, M. (1968)
Physical Review B: Condensed Matter, 172,
816.
424 Gil, B., Hamdani, F. and Morkoç, H.
(1996) Oscillator strengths for band to
band optical processes in GaN epilayers.
Physical Review B: Condensed Matter, 54
(11), R7678.
425 Ahn, D. (1996) Optical gain of InGaP and
cubic GaN quantum-well lasers with very
strong spin–orbit coupling. Journal of
Applied Physics, 79 (10), 7731.
426 Uenoyama, T. and Suzuki, M. (1995)
Applied Physics Letters, 67, 2527.
427 Weisbusch, C. and Vinter, B. (1991)
Quantum Semiconductor Structures:
Fundamentals and Applications, Academic
Press, San Diego, CA.
428 For a review of electronic states in
semiconductor quantum wells, see
Bastard, G. and Brum, J.A. (1986) IEEE
Journal of Selected Topics in Quantum
Electronics, 22, 1625.
References
429 Jeon, J.B., Lee, B.C., Sirenko, Yu.M., Kim,
K.W. and Littlejohn, M.A. (1997) Journal of
Applied Physics, 82, 386.
430 Ivchenko, E.L. and Pikus, G.E. (1997)
Superlattices and other Heterostructures,
Springer Series in Solid-State Sciences,
2nd edn, vol. 110, Springer, Berlin.
431 Bigenwald, P., Christol, P., Konczewicz,
L., Testud, P. and Gil, B. (1997) Presented
at EMRS, Strassburg, France.
432 Bigenwald, P. and Gil, B. (1994) Solid State
Communications, 91, 33.
433 Bykhovski, A., Gelmont, B. and Shur, M.
(1993) Applied Physics Letters, 63, 2243.
434 Bykhovski, A., Gelmont, B. and Shur, M.
(1993) Journal of Applied Physics, 74, 6734.
435 Bykhovski, D., Gelmont, B.L. and Shur,
M.S. (1997) Journal of Applied Physics, 81,
6332.
436 Gualtieri, J.G., Kosinski, J.A. and Ballato,
A. (1994) IEEE Transactions on Ultrasonics
Ferroelectrics and Frequency Control, 41, 53.
437 OClock, G.D. and Duffy, M.T. (1973)
Applied Physics Letters, 23, 55.
438 Burns, G. (1985) Solid State Physics,
Academic Press, New York, pp. 88–92.
439 King-Smith, R.D. and Vanderbilt, D.
(1993) Theory of polarization of
crystalline solids. Physical Review B:
Condensed Matter, 47 (3), 1651–1651.
440 Resta, R. (1994) Reviews of Modern Physics,
66, 899.
441 Bernardini, F., Fiorentini, V. and
Vanderbilt, D. (2001) Accurate calculation
of polarization-related quantities in
semiconductors. Physical Review B:
Condensed Matter, 63, 193201.
442 Bernardini, F., Fiorentini, V. and
Vanderbilt, D. (2001) Accurate calculation
of polarization-related quantities in
semiconductors. Physical Review B:
Condensed Matter, 63, 193201.
443 Littlejohn, M.A., Hauser, J.R. and
Glisson, T.H. (1975) Applied Physics
Letters, 26, 625.
444 Bykhovski, A., Gelmont, B. and Shur, M.
(1995) Journal of Applied Physics, 77, 1616.
445 Shur, M., Gelmont, B. and Khan, A. (1996)
Journal of Electronic Materials, 25, 777.
446 For a review, see Resta, R. (1994)
Macroscopic polarization in crystalline
dielectrics: the geometric phase approach.
Reviews of Modern Physics, 66, 899, and
references therein.
447 See, for example, Kittel, C. (1996)
Introduction to Solid State Physics, John
Wiley & Sons, Inc; Beam, W. (1965)
Electronics of Solids, 7th edn, McGrawHill.
448 Fiorentini, V., Della Sala, F., Di Carlo, A.
and Lugli, P. (1999) Effects of
macroscopic polarization in III–V nitride
multiple quantum wells. Physical Review
B: Condensed Matter, 60, 8849–8858.
449 Rode, D.L. (1970) Physical Review B:
Condensed Matter, 2, 4036.
450 Bykhovski, A.D., Kaminski, V.V., Shur,
M.S., Chen, Q.C. and Khan, M.A. (1996)
Applied Physics Letters, 69, 3254.
451 Yamaguchi, S., Kariya, M., Nitta, S.,
Takeuchi, T., Wetzel, C., Amano, H. and
Akasaki, I. (1998) Observation of
photoluminescence from Al1 xInxN
heteroepitaxial films grown by
metalorganic vapor phase epitaxy. Applied
Physics Letters, 73, 830.
452 Ambacher, O., Smart, J., Shealy, J.R.,
Weimann, N.G., Chu, K., Murphy, M.,
Schaff, W.J., Eastman, L.F., Dimitrov, R.,
Wittmer, L., Stutzmann, M., Rieger, W.
and Hilsenbeck, J. (1999) Journal of
Applied Physics, 85, 3222.
453 Bernardini, F. and Fiorentini, V. (2002)
Nonlinear behavior of spontaneous and
piezoelectric polarization. International
Workshop on Physics of Light-Matter
Coupling in Nitrides (PLMCN-1),
September 26–29, 2001, Rome, Italy,
Physica Status Solidi a: Applied Research,
190 (1), 65–73.
454 Al-Yacoub, A., Bellaiche, L. and Wei, S.-H.
(2002) Piezoelectric coefficients of
complex semiconductor alloys: the case of
Ga1 xInxN. Physical Review Letters, 89 (5),
057601.
455 Al-Yacoub, A. and Bellaiche, L. (2001)
Piezoelectricity of ordered (Ga0.5In0.5)N
alloys. Applied Physics Letters, 79 (14), 2166.
j319
j 2 Electronic Band Structure and Polarization Effects
320
456 Shimada, K., Sota, T., Suzuki, K.
and Okumura, H. (1998) Japanese
Journal of Applied Physics, Part 2: Letters,
37, L1421.
457 Smith, D.L. and Mailhiot, C. (1988)
Journal of Applied Physics, 63, 2717.
458 Wang, J., Jeon, J.B., Sirenko, Yu.M. and
Kim, K.W. (1997) IEEE Photonics
Technology Letters, 9, 728.
459 Bernardini, F. and Fiorentini, V. (1998)
Macroscopic polarization and band
offsets at nitride heterojunctions. Physical
Review B: Condensed Matter, 57, R9427.
460 Di Carlo, A., Della Sala, F., Lugli, P.,
Fiorentini, V. and Bernardini, F. (2000)
Doping screening of polarization fields in
nitride heterostructures. Applied Physics
Letters, 76 (26), 3950.
461 Della Sala, F., Di Carlo, A., Lugli, P.,
Bernardini, F., Fiorentini, V., Scholz, R.
and Jancu, J.-M. (1999) Free-carrier
screening of polarization fields in
wurtzite GaN/InGaN laser structures.
Applied Physics Letters, 74 (14), 2002.
462 Di Carlo, A. (1998) Tightbinding Approach
to Computational Materials Science (eds
P.E.A. Turchi, A. Gonis and L Colombo),
Materials Research Society, Pittsburgh,
Materials Research Society Symposium
Proceedings, 491, 389.
463 Monroy, E., Gogneau, N., Enjalbert, F.,
Fossard, F., Jalabert, D., Bellet-Amalric,
E., Dang, L.S. and Daudin, B. (2003)
Molecular-beam epitaxial growth and
characterization of quaternary III-nitride
compounds. Journal of Applied Physiology,
94 (5), 3121.
464 Aumer, M.E., LeBoeuf, S.F., McIntosh,
F.G. and Bedair, S.M. (1999) Applied
Physics Letters, 75, 3315.
465 Dimakis, E., Georgakilas, A.,
Androulidaki, M., Tsagaraki, K., Kittler,
G., Kalaitzakis, F., Cengher, D., BelletAmalric, E., Jalabert, D. and Pelekanos,
N.T. (2003) Plasma-assisted MBE growth
of quaternary InAlGaN quantum well
heterostructures with room temperature
luminescence. Journal of Crystal Growth,
251 (1–4), 476.
466 Wright, A.F., Leung, K. and van
Schilfgaarde, M. (2001) Effects of biaxial
strain and chemical ordering on the band
gap of InGaN. Applied Physics Letters, 78
(2), 189.
467 Pereira, S., Correia, M.R., Monteiro, T.,
Pereira, E., Alves, E., Sequeira, A.D. and
Franco, N. (2001) Compositional
dependence of the strain-free optical band
gap in InxGa1xN layers. Applied Physics
Letters, 78 (15), 2137.
468 McCluskey, M.D., Van der Walle, C.G.,
Romano, L.T., Krusor, B.S. and Johnson,
N.M. (2003) Effect of composition on
the band gap of strained InxGa1xN
alloys. Journal of Applied Physics, 93,
4340–4342.
469 Wright, A.F. and Nelson, J.S. (1995) Firstprinciples calculations for zinc-blende
AlInN alloys. Applied Physics Letters, 66
(25), 3465.
470 Onuma, T., Chichibu, S.F., Uchinuma, Y.,
Sota, T., Yamaguchi, S., Kamiyama, S.,
Amano, H. and Akasaki, I. (2003)
Recombination dynamics of localized
excitons in Al1 xInxN epitaxial films on
GaN templates grown by metalorganic
vapor phase epitaxy. Journal of Applied
Physics, 94 (4), 2449.
471 Lukitsch, M.J., Danylyuk, Y.V., Naik, V.M.,
Huang, C., Auner, G.W., Rimai, L. and
Naik, R. (2001) Optical and electrical
properties of Al1 xInxN films grown by
plasma source molecular-beam epitaxy.
Applied Physics Letters, 79 (5), 632.
472 Bernardini, F., Fiorentini, V. and
Vanderbilt, D. (1997) Polarization-based
calculation of the dielectric tensor of polar
crystals. Physical Review Letters, 79 (20),
3958–3958.
473 Yu, L.S., Qiao, D.J., Xing, Q.J., Lau, S.S.,
Boutros, K.S. and Redwing, J.M. (1998) Ni
and Ti Schottky barriers on n-AlGaN
grown on SiC substrates. Applied Physics
Letters, 73 (2), 238.
474 Morkoç, H., Ünl€
u, H. and Ji, G. (1991)
Principles and Technology of MODFETS,
vol. II, John Wiley & Sons, Ltd, Chichester,
UK, p. 317.
References
475 Chu, R.M., Zhou, Y.G., Zheng, Y.D., Han,
P., Shen, B. and Gu, S.L. (2001) Influence
of doping on the two-dimensional
electron gas distribution in AlGaN/GaN
heterostructure transistors. Applied
Physics Letters, 79 (14), 2270.
476 Ambacher, O., Foutz, B., Smart, J., Shealy,
J.R., Weimann, N.G., Chu, K., Murphy,
M., Sierakowski, A.J., Schaff, W.J.,
Eastman, L.F., Dimitrov, R., Mitchell, A.
and Stutzmann, M. (2000) Journal of
Applied Physics, 87, 334.
477 Ridley, B.K., Ambacher, O. and Eastman,
L.F. (2000) Semiconductor Science and
Technology, 15, 270.
478 Reale, A., Massari, G., Di Carlo, A., Lugli,
P., Vinattieri, A., Alderighi, D., Colocci,
M., Semond, F., Grandjean, N. and
Massies, J. (2003) Comprehensive
description of the dynamical screening of
the internal electric fields of AlGaN/GaN
quantum wells in time-resolved
photoluminescence experiments. Journal
of Applied Physics, 93 (1), 400.
479 Fiorentini, V., Bernardini, F. and
Ambacher, O. (2002) Evidence for
nonlinear macroscopic polarization in
III–V nitride alloy heterostructures.
Applied Physics Letters, 80 (7), 1204.
480 G€
orgens, L., Ambacher, O., Stutzmann,
M., Miskys, C., Scholz, F. and Off, J.
(2000) Applied Physics Letters, 76, 577.
481 Grandjean, N., Damilano, B., Dalmasso,
S., Leroux, M., La€
ugt, M. and Massies, J.
(1999) Journal of Applied Physics, 86, 3714.
482 Langer, R., Simon, J., Ortiz, V., Pelekanos,
N.T., Barski, A., Andre, R. and Godlewski,
M. (1999) Applied Physics Letters, 74, 3827.
483 Kim, H.S., Lin, J.Y., Jiang, X.H., Chow,
W.W., Botchkarev, A. and Morkoç, H.
(1998) Applied Physics Letters, 73, 3426.
484 Vaschenko, G., Patel, D., Menoni, C.S.,
Ng, H.M. and Cho, A.Y. (2002) Nonlinear
485
486
487
488
489
490
491
492
493
494
macroscopic polarization in GaN/
AlxGa1xN quantum wells. Applied
Physics Letters, 80 (22), 4211.
Simon, J., Langer, R., Barski, A., Zervos,
M. and Pelekanos, N.T. (2001) Physica
Status Solidi a: Applied Research, 188, 867.
Perlin, P., Suski, T., Lepkowski, S.,
Teisseyre, H., Grandjean, N. and Massies,
J. (2001) Physica Status Solidi a: Applied
Research, 188, 839.
Wetzel, C., Takeuchi, T., Amano, H. and
Akasaki, I. (1999) Japanese Journal of
Applied Physics, 38, L163.
Shi, C., Asbeck, P.M. and Yu, E.T. (1999)
Piezoelectric polarization associated with
dislocations in wurtzite GaN. Applied
Physics Letters, 74, 573.
Cartney, M.R., Ponce, F.A., Cai, J. and
Bour, D.P. (2000) Mapping electrostatic
potential across an AlGaN/InGaN/AlGaN
diode by electron holography. Applied
Physics Letters, 76, 3055.
Cherns, D., Jiao, C.G., Mokhtari, H., Cai,
J. and Ponce, F.A. (2002) Physica Status
Solidi b: Basic Research, 234, 924.
Cai, J. and Ponce, F.A. (2002)
Determination by electron holography of
the electronic charge distribution at the
threading dislocation in epitaxial GaN.
Physica Status Solidi a: Applied Research,
192, 407.
Barghout, K. and Chaudhuri, J. (2004)
Calculation of residual thermal stress in
GaN epitaxial layers grown on
technologically important substrates.
Journal of Materials Science, 39 (18),
5817–5823.
Morkoç, H., Ünl€
u, H. and Ji, G. (1991)
Fundamentals and Technology of
MODFETs, vols I and II, John Wiley &
Sons, Ltd, Chichester, UK.
Olsen, G.H. and Ettenberg, M. (1977)
Journal of Applied Physics, 48, 2543.
j321
j323
3
Growth and Growth Methods for Nitride Semiconductors
Introduction
Although the synthesis of GaN goes to back more than a half century, there are several
pivotal developments, which, in the opinion of the author, are responsible for laying
the technological framework and paving the way for the tremendous commercial and
scientific interest in nitrides. They are as follows: synthesis of AlN by Tiede et al. [1],
synthesis of GaN through the reaction of Ga, and ammonia by Johnson et al. [2]
synthesis of InN by Juza and Hahn [3], epitaxial deposition of GaN using the hydride
VPE technique by Maruska and Tietjen [4] employment of nucleation buffer layers by
Amano et al. [5] and Yoshida et al. [6] and achievement of p-type GaN by Akasaki
et al. [7]. A more recent development that paved the way for all the commercial activity
is the preparation of high-quality InGaN by Nakamura et al. [8], which followed the
synthesis of InGaN by Osamura et al. [9]. Nearly every crystal-growth technique,
substrate-type, and orientation has been tried in an effort to grow high-quality group
III–V nitride thin films. Increasingly, researchers have successfully taken advantage
of the hydride vapor phase epitaxy (HVPE), organometallic vapor phase epitaxy
(OMVPE), and molecular beam epitaxy (MBE) techniques, which have yielded greatly
improved film quality. All of these epitaxial methods must contend with two main
problems: the lack of native GaN substrates and difficulty with nitrogen incorporation and concomitant high ammonia flow rates needed particularly for In-containing nitride semiconductors.
A major drawback of GaN is that native substrates are not yet available in large
quantities. This is, in part, owing to the low solubility of nitrogen in bulk Ga and the
high vapor pressure of nitrogen over GaN at the growth temperature of bulk crystals.
The best alternatives now lie in development of sapphire, SiC, or AlN substrates.
Interest in AlN substrates has increased recently owing to the closer lattice match
over sapphire, matched stacking order, and high thermal conductivity. These factors
make AlN one of the best choices for growth of detectors requiring high AlN content
AlGaN and backside illumination. Early work on producing bulk AlN looked
promising [10].
The problem of nitrogen is endemic in epitaxial deposition techniques as well.
Regardless of the growth method employed, the major difficulty in growing group III
Handbook of Nitride Semiconductors and Devices. Vol. 1. Hadis Morkoc
Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 978-3-527-40837-5
j 3 Growth and Growth Methods for Nitride Semiconductors
324
nitrides arises from the need to incorporate stoichiometric quantities of nitrogen into
the film. This is accomplished in vapor phase processes at high substrate temperatures by decomposing a nitrogen-containing molecule, such as ammonia, on the
substrate surface. It should be noted that as the N vapor pressure increases, going
from AlN to GaN and then to InN, the ammonia flow rate must be increased reaching
tens of liters per minute for InGaN and V/III ratios of 200 000 for InN. It can also be
accomplished at lower temperatures in MBE growth by increasing the reactivity of
nitrogen through remote plasma excitation or ionization or using ammonia as the
reactive nitrogen source.
A great deal of effort has been spent in trying to overcome the problems arising
from lack of GaN substrates. Some of the best device results have been achieved
through use of buffer layers such as thick HVPE, including freestanding, variety, or
lateral epitaxial overgrowth variety.
3.1
Substrates for Nitride Epitaxy
As there is lack of a commercial native substrate, a plethora of substrates have been
employed in the growth of GaN films [11–14]. Recognizing the thermodynamic
bottleneck with regard to native substrates, unconventional methods employing
various implementations of compliant substrates and lateral epitaxy have been
explored. The most promising results on more conventional substrates so far have
been obtained on sapphire and SiC, with SiC making substantial inroads. Also
coming on the scene are thick freestanding GaN templates grown by HVPE and then
separated from the sapphire substrates. GaN, AlN, and InN have been grown
primarily on (0 0 0 1) sapphire but also on the ð2 1 3 1Þ, ð1 1 0 1Þ, ð1 1 0 2Þ, and
ð1 1 2 0Þ surfaces. In addition, III–V nitrides have been grown on Si, NaCl, GaP, InP,
SiC, W, ZnO, MgAl2O4, TiO2, and MgO. Other substrates as well have been used for
nitride growth, including Hf and LiAlO2, and LiGaO2. Table 3.1 is a compilation of the
lattice parameters and thermal characteristics of a number of prospective substrate
materials for nitride growth.
Lattice-mismatched substrates lead to a substantial density of misfit and threading
dislocations (in the range of 108 and 1010 cm2), though selective epitaxy followed by
coalescence, which goes by many names such as epitaxial lateral overgrowth (ELO),
lateral epitaxial overgrowth (LEO), and epitaxial lateral overgrowth (ELOG), is a
promising method for reducing dislocations down to 106 cm2. In comparison, the
extended defect densities are essentially zero for silicon homoepitaxy and
102–104 cm2 for gallium arsenide homoepitaxy [13]. Additional crystalline defects
besetting the layers include inversion domain boundaries (IDBs) and stacking faults.
Such defects are either directly or indirectly responsible for the creation of nonradiative recombination centers, which manifest themselves as energy states in the
forbidden energy bandgap reducing the quantum efficiency as well as producing
scattering centers. Other adverse effects of structural and point defects are that
impurities diffuse more readily along threading dislocations, and carrier transport is
3.104
3.189
4.758
AlN (hexagonal)
GaN (hexagonal)
Al2O3 (sapphire)
(rhombohedral)
4H-SiC (hexagonal)
6H SiC (hexagonal)
ZnO (hexagonal)
ScAlMgO4 (hexagonal)
c-LiAlO2 (tetragonal)
LiGaO2 (orthorhombic)
MgAl2O4 (cubic/spinel)
Si (cubic)
GaAs (cubic)
b-SiC (cubic)
MgO (cubic/rock salt)
6.372
—
Conventional
b (Å)
10.053
15.1123
5.2065
25.195
6.2679
5.407
4.966
5.175
12.991
c (Å)
3.1340
6.372
3.0817
3.2496
3.104
3.19
2.747
Matched a
(Å)
1.5
0.5
4.9
4.9
0.3–0.4
3.2
2.3
0.3–0.5
Thermal conductivity,
j (W cm1 K1)
10.5
4.2, 4.68
4.75, 2.9
6.2, 12.2
7.1, 15
a ¼ 6, b ¼ 9, 7
7.45
3.59
6
4.2, 5.3
5.59, 3.17
7.5, 8.5
Da/a, Dc/c (·106 K1)
In part from Landolt-B€
ornstein, vol. 17, Springer, New York, 1982. ZnO data are from W. Harsch of Eagle Picher.
3.073
3.0817
3.2496
3.246
5.1687
5.402
8.083
5.4301
5.6533
4.36
4.216
a (Å)
Crystal
Table 3.1 Lattice parameters and thermal characteristics of a number of the prospective
substrate materials for nitride growth and their lattice mismatch with GaN.
P63mc
P63mc
R3m
P41212
Pna21
Fd3m
Fd3m
F 43m
F 43m
Fm3m
P63mc
P63mc
R3c
Space group
3.63%
3.36%
þ1.9%
þ1.8%
1.7%
0.18%
2.7%
0%
49% (13%)
Mismatch
3.1 Substrates for Nitride Epitaxy
j325
j 3 Growth and Growth Methods for Nitride Semiconductors
326
either impeded, as in lateral transport, or aided, as in vertical transport. The high
density of defects also leads to boundary-limited transport making the important
basic parameters such as diffusion constant and mobility nearly impossible to
measure. The extended defects in nitrides also lead to inhomogeneities in electric
potential because of the high piezoelectric constants of GaN. Electrically active
defects induced either directly or indirectly by extended defects cause excess leakage
that is detrimental to both optical devices (in the form of dark current in detectors and
reduced quantum efficiency in emitters) and electrical devices (in the form of
increased gate current leakage and output conductance in field-effect transistors).
Details regarding the piezoelectric properties can be found in Chapter 2.
Lattice-mismatched substrates are commonly used at present in spite of efforts to
produce GaN [15,16] and AlN [10] bulk materials. Despite the lack of matched
substrates, remarkable progress in the growth of high-quality epitaxial III nitride
films has been achieved by a variety of methods such as hydride vapor phase epitaxy
(inorganic VPE or hydride VPE also goes with the acronym HVPE) [11,17],
OMVPE [18], and reactive molecular beam epitaxy (RMBE) [19,20]. Moreover, thick
freestanding GaN templates for further epitaxy have been prepared by HVPE [21]. By
far the most frequently used methods are the VPE methods with heterojunction
capability. The most versatile among the VPE methods is the metalorganic chemical
vapor deposition (OMVPE). OMVPE is the primary method employed in the
investigation and production of optoelectronic devices, such as LEDs and lasers,
albeit the quality of MBE films comes close to that grown by OMVPE. Electronic
devices with higher quality interfaces are achieved principally with OMVPE and
MBE. Inorganic VPE was the first method used to grow epitaxial III-N semiconductors but was nearly abandoned [22]. The technique, however, got revived recently by
growing very high-quality and thick GaN buffer layers and templates [23] for the
growth of device structures using MBE or OMVPE [11]. Efforts are underway to
expand the method to the growth of AlGaN. Below, a discussion of the class of
substrates that have been explored is followed by a discussion of the properties of and
processing steps for the conventional substrates before growth for each of these
substrates.
3.1.1
Conventional Substrates
GaN, as the most studied member of the semiconducting group III nitrides, has been
grown on many substrates. Many of the major problems that have hindered the
progress in GaN and related semiconductors can be traced back to the lack of a
suitable substrate material that is lattice and thermally matched to GaN. Lattice
mismatch is responsible for stacking faults and dislocations. Thermal mismatch
causes the epilayer to crack on cooling. Specifically, the semiconductors GaN, AlN,
and InN have been grown primarily on sapphire, most commonly in the c (0 0 0 1)
orientation but also on the a- ð1 1 2 0Þ and R- ð1 1 0 2Þ planes [14]. Growth on a-plane
produced c-plane GaN, but growth on R-plane sapphire produces a nonpolar, a-plane
3.1 Substrates for Nitride Epitaxy
GaN. In addition, the group III–V nitrides have been grown on SiC, ZnO, MgAl2O4,
Si, GaAs, MgO, NaCl, W, and TiO2. As high-resistivity SiC substrates became
available, it became the proffered substrate for transistor work, in part owing to its
high thermal conductivity. In addition, freestanding GaN prepared by HVPE is at or
nearing production capacity primarily for low threshold lasers that need low-defect
material. Some of the suitable substrate materials have become commercially
available only recently. Almost all the group III–V nitride semiconductors have been
deposited on sapphire despite its poor structural and thermal match to the nitrides.
The preference for sapphire substrates can be ascribed to its wide availability,
hexagonal symmetry, and ease of handling and pregrowth cleaning. Sapphire is
also stable at high temperatures (1000 C) required for epitaxial growth using the
various CVD techniques commonly employed for GaN growth. Owing to thermal and
lattice mismatches between sapphire and the group III–V nitrides, it is necessary to
grow a thick epilayer to obtain good-quality material.
3.1.2
Compliant Substrates
When large mismatch exists between an epilayer and its substrate, the misfit is
typically accommodated by the introduction of misfit dislocations at the interface,
which are accompanied by threading dislocation segments in the epilayer. To
overcome this problem, a compliant substrate is used in high-misfit systems. The
role of the compliant substrate is to accommodate the large mismatch either by
plastic deformation of the compliant substrate in a manner that avoids the formation
of dislocations in the heteroepitaxial film or by homogeneous elastic strain of the
threading dislocation, which also avoids formation of the threading dislocations.
Both mechanisms are facilitated by a compliant substrate whose stiffness constants
are well below those of the epilayer and the supporting bulk substrate. As for the
former mechanism, the soft and thin nature of the compliant substrate energetically
favors the capture of dislocations resulting from the mismatch by the substrate rather
than by the stiffer epilayer, thus paving the way for predilection toward misfit
accommodation (MA) by homogeneous elastic strain rather than misfit dislocations.
An attractive approach is to insert a pillarlike interfacial layer that is capable of
accommodating thermal strain, the effectiveness of which depends on the height of
the pillars and the size of the wafer. An effort has been made to find a universal
substrate onto which any epitaxial layer can be grown with a very low density of
structural defects. Some experimental success has been achieved, but only in specific
cases, since 1991 when Lo and colleagues [24,25] introduced the basic idea of a
compliant substrate. Different kinds of epitaxial layers have been grown on compliant
substrates of GaAs twist bonded to bulk substrates of GaAs. Among them are InGaP,
In0.22Ga0.78As, GaSb, and InSb where the respective misfits with GaAs are 1, 1.5,
8, and 15%, respectively [26,27], with positive figures denoting tensile stresses in
the layer on GaAs. The use of a thin compliant substrate is not limited to the epitaxial
growth of III–V compounds but may also be found in SiGe grown on a thin compliant
j327
j 3 Growth and Growth Methods for Nitride Semiconductors
328
substrate of Si on a viscous SiO2 layer [28]. Other approaches pursue compliancy by
twist bonding, that is, deposition of a low melting temperature interlayer. Twist
bonding Si to SOI provides a high density of interlayer dislocations that can elastically
accommodate misfit between the compliant substrate and the heteroepitaxial film.
Otherwise, if the bonding between the compliant layer and substrate is strong, an
array of screw dislocations form, transforming to edge/screw dislocations upon
deposition of a lattice-mismatched film by elastic deformation. Various explanations
have been offered to understand the mechanisms of misfit accommodation [29].
The term compliant is used liberally to describe an approach or a set of approaches
to grow lattice mismatch materials where the substrate or some interface layer
accommodates the mismatch by expansion/contraction or generating defects within
itself as opposed to epitaxial layers. Specifically, the concept behind the latter case is to
force the defects caused by mismatch to propagate into the substrate as opposed to the
epilayer, requiring generation of misfit dislocations in the thin, weakly bound
template layer rather than the growing epitaxial layer.
In case of nitride, the initial approach proposed was that Si on an insulator be utilized
for GaN growth. Here, a thin Si layer on silicon dioxide, which, in turn, is on Si would be
employed. The downside is that the quality of GaN on compliant Si has been poor at
best. The modified approach to overcome this barrier is to carbonize the Si to convert it
to SiC. If one utilizes the (1 1 1) orientation, one would then get the wurtzitic phase of
GaN. To be specific, Yang et al. [30] suggested that before the beginning of the
carbonization process, SiO2, Si, and C need to be deposited successively on a Si
substrate. By exposing the new composite substrate to a flux of acetylene or carbon
particles at 900 C, a thin layer (less than 50 nm) of Si (on SiO2) will be partially or
completelyconverted into SiC. GaNis then grownonthis SiC. Again, the problemhere,
setting aside the problems associated with the growth on SiC, is that SiC so formed is
not contiguous and is extremely defective both in terms of bulk and surface structural
properties. In addition, air gaps form beneath the layer surface. Consequently, this
technique has not yet lived up to the original proposal and expectations. Efforts still
continue to exploit this approach despite the lack of progress so far.
Compliance based on expansion and/or contraction is a very neat idea and may be
workable for small-sized wafers. However, it is impractical for larger wafers. For
example, if the lattice mismatch between the compliant substrate is 4% (epitaxial
layer having the larger lattice constant) and the wafer is 50 mm, an expansion in the
substrate required for producing defect-free epitaxial layer is 2 mm, which is
substantial and unlikely.
3.1.3
van der Waals Substrates
To get around the lattice-mismatch problem, a new growth method called van der
Waals epitaxy has been proposed [31], which delivers strain-free films. In this
approach, the substrate and epitaxial film are separated by an intermediate epitaxial
two-dimensional (2D) buffer material such as MoS2, WS2, or other materials such as
II–VI (ZnTe) or III–VI compounds (GaSe, InSe, etc.) having weak van der Waals
3.2 A Primer on Conventional Substrates and their Preparation for Growth
bonding to the substrate and the film. Strain from lattice mismatch between the
epitaxial film and the substrate is completely relieved in the region between the layer
and the buffer. As in the case of the compliant substrate scheme, this approach has
not been very successfully applied to nitrides.
3.2
A Primer on Conventional Substrates and their Preparation for Growth
A substrate is like the foundation of a building. As such, substrate preparation deserves
the mostattention.Though thedetailsof the proceduresemployed vary fromone growth
method to the next, a chemical preparation before loading into the growth reactor is
common. In the OMVPE technique, this is followed by either a simple heat treatment or
a combination of heat treatment with gas-phase etching, where temperatures for heat
treatment in the vicinity of 1200 C are possible. In the case of vacuum-deposition
techniques where it is not always possible to achieve sufficiently high temperatures, dry
processing techniques are employed. One of the dry processing techniques is utilizing
ECR remote plasma etching with a mixture of hydrogen and helium, as discussed below.
The purpose of the He gas is to take advantage of its energetic metastable states with long
mean free paths. In addition to a clean surface, the goal is to get as flat a surface as
possible, because the nitride stacking order, AaBbAaBb, is different from the stacking
order found in most of the substrates under investigation. The exception is ZnO whose
stacking order matches that of the nitrides. Because the atomic steps on the (0 0 0 1)
surface are of the bilayer type, the surface terraces would have the same surface polarity
so that stacking mismatch boundaries (SMB) can be avoided.
The surfaces of the substrates used have to be prepared for epitaxial growth, a
process that includes degreasing followed by chemical etching when possible.
Surfaces of as-received sapphire and SiC substrates contain mechanical polishing
damage that must be removed. Chemical etches are not yet available for this purpose.
Consequently, a high-temperature treatment under a controlled environment is
employed, as will be discussed below.
The degreasing procedure, which is the first step for growth, for Si, sapphire, SiC,
ZnO, LiGaO2 and LiAlO2, and GaN and AlN, whose specifics will be discussed below,
is the same. The substrate is first dipped in a solution of trichloroethane (TCE) kept at
300 C, for 5 min. It is then rinsed for 3 min each in acetone and methanol. This is
followed by a 3-min rinse in deionized (DI) water. The above process is repeated three
times to complete the degreasing process. The substrates are then etched, which is a
substrate dependent procedure. Following degreasing, a variety of substrate-specific
methods are employed to get various substrates growth ready as discussed below.
3.2.1
GaAs
GaAs, as a substrate for GaN epitaxy, is justified on the premise of obtaining pure
(wurtzite-free) zinc blende GaN on GaAs(1 0 0), attaining thick wurtzite GaN films on
j329
j 3 Growth and Growth Methods for Nitride Semiconductors
330
GaAs(1 1 1) substrates, and dilute GaAsN films for infrared applications. The bulk of
the research on GaAs based nitrides is on (1 0 0) surface for cubic GaN. GaAs(1 0 0) is
one of the few semiconductor substrates on which metastable zinc blende GaN
epitaxial films readily form, and many researchers have investigated the best ways of
avoiding any inclusion of the wurtzite polytype into these films. GaAs is much more
readily wet etched than any of the other substrates used for nitride epitaxy, which also
makes GaN films easier to separate from GaAs than sapphire. Thus, GaAs(1 1 1)
substrates are considered a template for creating freestanding thick GaN films for
subsequent epitaxy. Because the decomposition rate of GaAs in NH3 or an ultrahigh
vacuum (UHV) rapidly increases at temperatures above 700 C, a deposition process
including multiple temperatures is required. Unless the substrate temperature is
high, the maximum growth rate (GR) attainable is limited. Moreover, even a small
amount of GaAs decomposition could be detrimental, as surface roughening or
faceting enhances the onset of mixed polarity growth. Because MBE is capable of
depositing epitaxial GaN films at a lower temperature as compared to vapor phase
methods, it has been more commonly employed in this respect. The maximum
allowed temperature could be increased once the GaAs substrate is completely
encased with GaN deposited at a low temperature (LT), thereby making OMVPE and
HVPE more viable.
3.2.1.1 A Primer on GaAs
GaAs has the zinc blende crystal structure with the symmetry group of F 43m.
Figure 3.1 displays the perspective view of the GaAs crystal along [1 0 0], [1 1 0], and
[1 1 1] directions. Table 3.2 lists the physical, chemical, thermal, mechanical, and
optical properties of GaAs important for the GaN epitaxy. For additional details, see
Liu and Edgar [13].
GaAs substrates are grown with either liquid encapsulated Czochralski (LEC) or
vertical gradient freeze (VGF) methods. GaAs has seen a spectacular improvement
over a period of two decades in that crystal defects, impurities, and micro-inhomogeneities have been reduced. In fact, GaAs wafers with diameters greater than
Figure 3.1 The perspective view of the GaAs crystal (a) along
[1 0 0] (1 · 1 · 1 unit), (b) [1 1 0] (2 · 2 · 2 units), and (c) [1 1 1]
(2 · 2 · 2 units) directions [13]. (Please find a color version of this
figure on the color tables.)
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Table 3.2 Properties of GaAs at room temperature (partially after Ref. [13]).
Parameter
Value
Lattice constant (Å)
Density (g cm3)
Melting point ( C)
Heat capacity (J g1 K1)
Thermal conductivity (W cm1 K1)
Thermal diffusivity (cm2 s1)
Thermal expansion (linear) (·106 K1)
Percent change in lattice (300–1200 K)
Bulk modulus (GPa)
Youngs modulus (GPa)
Poissons ratio
Refractive index
Relative dielectric constant
Electrical resistivity (undoped)
5.6536
5.32
1240
0.327
0.45
0.26
6.03
Da/a0 ¼ 0.5876
75.0
85.5
0.31
3.66 near band edge
e0 ¼ 13.1
1.0 · 104 O cm, nonstoichiometric
defect compensated
150 mm and with various doping types and concentrations are commercially
available. Silicon and tellurium are common n-type dopants with resultant electron
concentrations in the range of 1016–1018 cm3. On the contrary, zinc is the standard
p-type dopant with resultant hole concentrations in the range of 1018–1019 cm3.
Commercial LEC GaAs has a typical etch pit density (EPD) of less than 104 cm2 and
an electron mobility greater than 4000 cm2 V1 s1. But, VGF grown GaAs offers a
lower defect density, with EPD less than 103 cm2. Both (1 0 0) and (1 1 1) types of
GaAs with different vicinal degrees are available.
3.2.1.2 Surface Preparation of GaAs for Epitaxy
The substrate preparation, following degreasing, includes etching in acid such as
H2SO4 : H2O2 : H2O. After rinsing, the surface can be treated in dilute HF for H
passivation of the surface. The H passivation layer can be desorbed in the deposition
chamber. However, owing to the well-advanced nature of GaAs technology, epi-ready
substrates with protective oxides are commercially available. Once in the deposition
reactor, the oxide layer can be removed by thermal desorption.
Direct nucleation of GaN on GaAs is difficult owing to great chemical and
mechanical mismatch with GaN (the large lattice match of about 20% caused by
5.65 Å vs. 4.51 Å for zinc blende GaN), but it is a task mitigated somewhat with the
deposition of a GaAs prelayer. Zinc blende GaN on (2 · 4) GaAs can be grown by first
establishing an atomically smooth GaAs surface with minimized step density. This is
accomplished by depositing a GaAs prelayer, on the order of 100 nm to as high as
1 mm, and choosing appropriate nucleation conditions. Without a GaAs prelayer, the
more stable wurtzite polytype of GaN grows preferentially once (1 1 1) facets of the
GaN film are generated on the rough substrates. The content of hexagonal GaN phase
j331
j 3 Growth and Growth Methods for Nitride Semiconductors
332
is drastically reduced with an epitaxial GaAs prelayer in MBE growth. The growth
temperature for a GaAs prelayer is typically about 600 C.
It is imperative that the GaAs surface is nitridated prior to GaN growth for chemical
and mechanical transitioning. Nitridation of the GaAs substrate results in a thin
nitride surface layer, which provides a cubic template for growth, improving the
quality of the GaN layer and suppressing GaAs decomposition at higher substrate
temperatures needed for GaN epitaxy. GaAs substrates are nitridated either by
exposure to nitrogen plasma (MBE) or by annealing in ammonia (OMVPE and
RMBE).
At temperatures below 200 C, the nitridation is hindered by kinetic limitations. At
temperatures above 600 C, simultaneous etching of the surface may occur along
with the nitridation process. Nitridation of GaAs(0 0 1) does not take place homogeneously but proceeds along {1 1 1} facets into the underlying GaAs layer. Complete
nitridation can lead to a highly facetted interface between the GaN layer and GaAs
substrate, which, in turn, leads to the nucleation of the wurtzite phase and could lead
to polycrystalline GaN. Using the highest growth rate possible to quickly bury the
interface or maintaining an As flux during growth of the first few monolayers of GaN
helps to prevent the deterioration of the GaAs surface. A buffer layer of GaN at low
temperature following the initial nitridation generally improves the eventual quality.
AlN is not used as buffer because its zinc blende polytype is extremely difficult to
nucleate.
3.2.2
Si
Si is the most perfected and least expensive substrate that is available in sizes up to
300 mm. Unlike GaAs, ZnO, and a few others, silicon has good thermal stability
under conditions used for GaN epitaxy. However, Wz GaN and AlN grown on Si(1 1 1)
are highly defective. The incentives for using Si substrates remain high, however, and
good progress in reducing the defect density by using epitaxial lateral overgrowth or
pendeo-epitaxy has been reported.
3.2.2.1 A Primer on Si
Si has a diamond-lattice structure with the space group of Fd
3m (No. 227) and can be
thought of as two interpenetrating fcc sublattices with one sublattice displaced from
the other by one quarter
of the distance along a body diagonal of the cube (i.e., the
pffiffiffi
displacement of a 3=4, where a ¼ 5.43102 Å is the lattice constant). Each atom in the
lattice is surrounded by four equidistant nearest neighbors that lie at the corners of a
tetrahedron. Figure 3.2 illustrates the perspective view along the [0 0 1], [0 1 1], and
[1 1 1] directions of a Si cell. Table 3.3 lists physical, chemical, thermal, mechanical,
and optical properties of Si at room temperature.
Single crystalline ingots are produced by the Czochralski (CZ) method (over 85%
of silicon crystals are grown by this method) or the Float Zone (FZ) method, used
mostly for purification. These ingots eventually become thin Si wafers through the
processes of shaping, slicing, lapping, etching, polishing, and cleaning. Impurities
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Figure 3.2 The perspective view along (a) the [0 0 1], (b) [0 1 1],
and (c) [1 1 1] directions of a Si cell. (Please find a color version of
this figure on the color tables.)
can be added directly to the melt to create p-type and n-type silicon. The only
discernible half-drawback of the CZ method is that oxygen (typical at a level of
1018 cm3) and carbon (typical at a level of 1016 cm3) can be incorporated because of
the reduction of the quartz crucible and contamination by graphite fixtures. It should
be noted that these impurities are not without benefits in that oxygen increases the
yield strength or acts as internal getter to tie up metallic contaminants. The FZ
method does not use any crucible, and thus the impurity level is markedly reduced,
making it easier to grow high-resistivity material. For more details, refer to Ref. [13].
3.2.2.2 Surface Preparation of Si for Epitaxy
For wurtzitic GaN growth, (1 1 1) plane Si is used. The (0 0 1) surface is also used for
cubic GaN growth, albeit in only a few cases. As-received Si surface is already
Table 3.3 Properties of Si at room temperature (partially after Ref. [13]).
Parameter
Value
Lattice constant (Å)
Density (g cm3)
Melting point ( C)
Heat capacity (J g1 K1)
Thermal conductivity (W cm1 K1)
Thermal diffusivity (cm2 s1)
Thermal expansion (linear)
Percent change in lattice (298–1311 K)
Shear modulus (GPa)
Bulk modulus (GPa)
Youngs modulus (GPa)
Poissons ratio
Refractive index
Relative dielectric constant
Electrical resistivity (undoped)
5.43102
2.3290
1410
0.70
1.56
0.86
2.616 · 106 K1
Da/a0 ¼ 0.3995
680
97.74
165.6
0.218
3.42
e0 ¼ 11.8
Up to 50 kO cm
j333
j 3 Growth and Growth Methods for Nitride Semiconductors
334
excellent and removal of only a very thin surface layer, using the RCA etch followed by
hydrogenation of surface dangling bonds, is sufficient. This is accomplished by
immersing Si for 10 min in a 1 : 1 : 5 solution of HCl : H2O2 : H2O kept at 60 C, which
grows a porous oxide, followed by a rinse in deionized water. The resulting oxide layer
is then removed by dipping the substrate in a 10 : 1 solution of H2O : HF for 20 s. The
hydrogenation process takes place through a short exposure of the wafer to an HF
solution.
3.2.3
SiC
The cohesive bond strength of SiC is so large that it was once considered an element
under the name of carborendum. Owing to its large thermal conductivity and dearth
of defect causing in-plane rotation of GaN with respect to SiC lattice, and availability
of high-resistivity substrates, SiC is continually gaining recognition as a very viable
substrate for epitaxy for both optical and electronic devices. Much of the early
drawbacks having to do with pre-epitaxy surface preparation, micropipes, size, and, to
an extent, cost issues have been mitigated to the point that some commercial LEDs
and almost all of high power field-effect transistors utilize nitride heterostructures on
SiC.
3.2.3.1 A Primer on SiC
A basic unit of crystalline SiC is a covalently bonded tetrahedron of C atoms with a Si
atom at its center or vice versa, that is, either SiC4 or CSi4, as illustrated in Figure 3.3.
Variation in the stacking order of SiC along the c-direction leads to more than 250
Figure 3.3 Tetragonal bonding of a carbon atom with its four
nearest silicon neighbors. The bond lengths depicted with a and
C–Si (the nearest neighbor distance) are approximately 3.08 and
1.89 Å, respectively. The right side is the three-dimensional
structure of 2H-SiC structure. (Please find a color version of this
figure on the color tables.)
3.2 A Primer on Conventional Substrates and their Preparation for Growth
A
B
C
Carbon
Si
Base
B
A
C
A
B
C
A
C
A
A
C
B
B
B
A
A
B
A
A
3C
A
2H
4H
6H
Figure 3.4 Stacking sequence of cubic and three polytypes of wurtzitic SiC.
polytypes, of which a few prominent ones are shown in Figure 3.4. (A basic
discussion of stacking is given in Section 1.1.) By observing the SiC crystal from
the side, the stacking sequence can be projected as in Figure 3.5. The distance a
between neighboring silicon or carbon atoms is approximately 3.08 Å for all polytypes. The height of the unit cell c varies with the different polytypes, as tabulated in
Table 3.4. Consequently, the c/a ratio varies from polytype to polytype but is always
close to the ideal for a close packed structure. This ratio is approximately 1.641, 3.271,
and 4.908 for the 2H-, 4H-, and 6H-SiC polytypes, respectively, whereas
theffiffiffiffiffiffiffi
equivapffiffiffiffiffiffiffiffi p
ffi
lent
ideal
ratios
for
these
polytypes
are
1.633,
3.266,
and
4.899
(
8=3
,
2
8=3
, and
pffiffiffiffiffiffiffiffi
3 8=3), respectively [13].
Each polytype has a unique set of electronic and optical properties. The bandgaps at
liquid helium temperature of the different polytypes range between 2.39 eV for 3CSiC and 3.33 eV for the 2H-SiC polytype. The two most important polytypes as
substrates for GaN epitaxy, 6H-SiC and 4H-SiC, have bandgaps at liquid helium
temperature of 3.02 and 3.27 eV, respectively.
The hexagonal polytypes of SiC, such as 4H- and 6H-SiC, belong to the same space
group, P63mc (No. 186), as wurtzite GaN. The most studied substrates for GaN
epitaxy are the 3C-SiC/Si(1 0 0) and 6H-SiC, as these polytypes have been the most
readily prepared or commercially available for the longest time. With 4H-SiC now
commercially available, its use will become more common. Table 3.4 shows the
physical, chemical, thermal, mechanical, and optical properties of SiC at room
temperature. The thermal expansion coefficient of SiC in c- and a-planes as a
function of temperature is shown in Figure 3.6.
Bulk SiC crystals are produced by sublimation in the modified Lely process,
developed by Tairov and Tsvetkov [33], which employs a SiC seed crystal for the
control of polytype and orientation. Growth is achieved by the vapor transport of Si,
Si2C, and SiC2 driven by a temperature difference in an argon atmosphere in a
graphite, tantalum, or tantalum carbide crucible at 20–500 Torr and at about 2200 C.
The 4H-SiC and 6H-SiC(0 0 0 1) varieties both on- and off-axis (typically 3.5 for 6HSiC and 8 for 4H-SiC), silicon and carbon face, are available in sizes up to 100 mm in
j335
j 3 Growth and Growth Methods for Nitride Semiconductors
336
Figure 3.5 Views of the ½1 1 2 0 planes for the 3C-, 2H-, 4H-, and 6H-SiC polytypes.
diameter. Screw dislocations occur in high densities and depending on the magnitude of their Burgers vector, the core of a screw dislocation can be hollow (nanopipes
or micropipes) or closed and run through the entire wafer. Hollow core screw
dislocations take place when the Burgers vector (b) is two or more times the c-lattice
constant (c) for 6H-SiC or three times the lattice constant for 4H-SiC. Because the best
micropipe density has been reduced to about 1 cm2, research has shifted on to
closed-core screw dislocations, which occur in densities of approximately
103–104 cm2. Both 4H-SiC and 6H-SiC wafers are available in low resistivity nand p-type forms with concentrations in the range of 1015–1019 cm3. The resistivities for n- and p-type material for the aforementioned doping range lie in the range
0.01–0.10 and 1–10 O cm, respectively. Interest in semi-insulating SiC is driven by
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Table 3.4 Properties of SiC at room temperature (after Ref. [13]).
Parameter
Polytype
Value
Lattice constant (Å)
3C
2H
4H
6H
3C
2H
6H
3C
6H
3C
4H
6H
3C
6H
6H
3C
3C
Ceramic
3C
2H
4H
6H
3C
6H
a ¼ 4.3596
a ¼ 3.0753, c ¼ 5.0480
a ¼ 3.0730, c ¼ 10.053
a ¼ 3.0806, c ¼ 15.1173
3.166
3.214
3.211
2793
0.71
3.2
3.7
3.8
3.9
4.46 for a-axis, 4.16 for c-axis
Da/a0 ¼ 0.4781, Dc/c0 ¼ 0.4976
Da/a0 ¼ 0.5140
440
0.183–0.192
2.6916 at l ¼ 498 nm
2.6686 at l ¼ 500 nm
2.6980 at l ¼ 498 nm
2.6894 at l ¼ 498 nm
e(0) ¼ 9.75, e(1) ¼ 6.52
e(0) ¼ 9.66, e(1) ¼ 6.52 ? c-axis
e(0) ¼ 10.3, e(1) ¼ 6.70 || c-axis
102–103, higher in V doped
Density (g cm3)
Melting point ( C)
Heat capacity (J g1 K1)
Thermal conductivity (W cm1 K1)
Linear thermal expansion coefficient (·106 K1)
Percent change in lattice (300–1400 K)
Youngs modulus (GPa)
Poissons ratio
Refractive index (ordinary ray)
Dielectric constant
Electrical resistivity (undoped)
4H
Thermal expansion coefficient (%)
1.8
α−SiC
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
–0.2
0
200 400
600
800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800
Temperature (K)
Figure 3.6 Thermal expansion coefficient of SiC in c and a planes
as a function of temperature [32].
j337
j 3 Growth and Growth Methods for Nitride Semiconductors
338
FETs. The resistivities close to 1010 O cm in both V-doped and intrinsically compensated SiC are possible.
3.2.3.2 Surface Preparation of SiC for Epitaxy
The surface preparation of SiC prior to deposition takes on a different meaning in MBE
as opposed to OMVPE. Lacking high temperature capability and H, MBE growth relies
on ex situ cleaning procedures. In contrast, SiC surfaces can be epi readied in situ
during growth by vapor phase. However, one of the best, if not the best, approach
involves gas phase preparation of SiC surface in which the surface is exposed to in H
and/or HCl gases at high temperatures, the details of which are discussed below.
Unless SiC substrates are H polished a priori or in the growth reactor, as can be
done in HVPE or OMVPE methods, it is recommended that approximately 3 mm
from the surface be removed in a hot KOH solution (300–350 C). If the substrate
quality is not high, the etch rate in defective regions is high and smooth surfaces do
not follow. Assuming that the previous step is successful, it is followed by a DI rinse
for 3 min and the wafer is blow dried by N2. The SiC substrate is then subjected to a
series of oxidation and passivation procedure. The substrate is immersed for 5 min in
a 5 : 3 : 3 solution of HCl : H2O2 : H2O at 60 C, followed by a 30 s DI rinse. The
resulting oxide layer is then removed by dipping the substrate, for 20 s, in a 10 : 1
solution of H2O : HF. This procedure is repeated several times (three to five times)
after which the substrate should not be exposed to the atmosphere for longer than
30 min, otherwise another oxidation–passivation procedure would be required.
Skipping the chemical etching step leaves the surface with residual damage from
mechanical polishing.
As mentioned above, techniques have been developed to remove the surface
damage by plasma or vapor etching. One such technique is the mechanical chemical
polish, which has made substantial progress [34] (commercial service is available
from Novasic), and the other is etching in H and/or Cl environments at very high
temperatures. The surface morphology of the as-received SiC substrate that underwent a standard mechanical chemical polish (MCP) contains much surface and
subsurface damage as characterized by many grit scratches shown in Figure 3.7a.
Special MCP procedures have been developed to obtain smoother surfaces (see
Ref. [34]), two example of which obtained by Eagle Picher and Novasic are shown in
Figure 3.7b and c. Schottky barrier diodes on as-received and Novasic MCP-treated
substrates indicated as much as four orders of magnitude reduction in the reverse
bias current, albeit with a considerable nonuniformity.
Another method that leads to atomically smooth SiC surfaces and much improved
GaN overlayers, in terms of reduced extended defect concentration as shown by plan
Figure 3.7 (a) AFM image of as-received SiC
surface following a standard mechanical
chemical polish. Image size is 10 mm · 10 mm
and vertical scale is 50 nm. Note the presence of
scratches; (b) AFM image of SiC surface after a
mechanical chemical polish performed at Eagle
"
Picher. Note that scratches are no longer present.
Image 10 mm · 10 mm, vertical 5 nm; (c) a
2 mm · 2 mm AFM image of a Cree 6H-SiC wafer
MCP polished by Novasic showing a root mean
square roughness of 0.134 nm.
3.2 A Primer on Conventional Substrates and their Preparation for Growth
j339
j 3 Growth and Growth Methods for Nitride Semiconductors
340
view transmission electron microscopy (TEM) images, is the high temperature H
annealing [35]. Those investigating various issues dealing with SiC have recently
developed and exploited the in situ hydrogen etching [36]. H2, H2 þ HCl, or H2 þ C3H8
etching at temperatures between 1300 and 1550 C removes the scratches caused by
mechanical polishing [37]. This is more effective than wet HF etching of SiO2 after
oxidation. Other wet etching techniques in bases such as molten salts (Na2O2, NaOH,
KOH, etc. at temperatures approaching or at 500 C) reveal the defect features of SiC
surface and thus are not suitable for surface preparation for epitaxy. Even though
nitridation has been shown to improve the smoothness of SiC substrate owing to a
combination of nitrogen chemisorption and etching at 1050 C in NH3 flow for 30 min,
the utility of long nitridation processes is questioned because of SixNy formation.
An atomic force microscopy (AFM) image of a SiC substrate polished by a hightemperature H treatment (1500 C), similar to that reported in Ref. [38], and used for
GaN growth in Ref. [35], is shown in Figure 3.8, which clearly shows well-ordered and
unbroken atomic terraces indicative of superb surface quality. Similar results can be
obtained by a H treatment in a typical SiC growth reactor at about 1500 C. A light
follow-up etch in molten KOH solution ensures atomically smooth terraced surfaces
if the H polishing steps are not ideal.
As previously mentioned, vacuum deposition equipment, such as the one used in
MBE, is not compatible with the high-temperature H or HCl treatment, but remote
plasma etching techniques can be employed to at least remove the damaged surface
layer if H-etched samples are not available. To circumvent the need for high
temperatures and exotic treatments incompatible with conventional MBE setups,
a preparation procedure adapted from conventional Si technology, and augmented by
H plasma cleaning, has been shown to work for SiC. In the first step of this procedure
reported by Lin et al. [39], the surface is hydrogen passivated using an HF dip before
Figure 3.8 AFMimageofSiCsurfaceaftera5 min1600 C hydrogen
polishing step. Note that scratches are no longer present. The step
height seen in a full c-direction lattice parameter for 6H-SiC.
3.2 A Primer on Conventional Substrates and their Preparation for Growth
being introduced into vacuum. In the second step, the substrate is treated with
hydrogen plasma, which reduces the CO level (oxygen–carbon bonding) to a value
below the X-ray photoemission detection limit.
Detailed investigation of SiC surface after H anneal have been undertaken [40]
using such techniques as low-energy electron diffraction (LEED) experiments and
Auger electron spectroscopy (AES) in addition to reflection high-energy electron
diffraction (RHEED). The emphasis is to determine the surface chemical and
structural properties taking it beyond what may be needed simply for epitaxy but
looking at it with the precision needed for MOS-like structures with AlN/SiC
composite. When (0 0 0 1) SiC samples subjected to a H polish at 1500 C for 5 min
flow
3000 scum are introduced into the UHV system, they exhibited a
in
2 ffiffi
pffiffiaffi Hp
ffi
( 3 3) R30 LEED pattern with bright and sharp superstructure spots. For
comparison on the C-polarity surfaces, ð0 0 0 1Þ, no background was observed, which
indicates a high degree of order. On the (0 0 0 1) surfaces, however, always a faint
background was visible. The ratio between the average intensities of fractional and
integer order beams was above 0.5 for both surfaces, indicating strong surface
reconstruction in both cases. The typical Auger spectra displayed a strong OKLL peak
in addition to the typical SiLVV and CKLL peaks [40]. The SiLVV signal on the ð0 0 0 1Þ
surface showed both bulk-related peak at 90 eV and a feature at around 65 eV
attributed to oxygen-bonded silicon. The SiLVV signal of the (0 0 0 1) surface is more
complex as expected because of differently coordinated Si for both surfaces. The
LEED pattern on the ð0 0 0 1Þ surface changed to (3 3) structure following a 30-min
annealing at 1050
pffiffiffi C.pIn
ffiffiffi addition, both the oxygen and the Si–O AES signals
vanished. The ( 3 3) R30 structure remained for the (0 0 0 1) surface after a
30-min annealing at 1000 C. However, OKLL peak disappeared and SiLVV signal
returned the bulklike shape. The simultaneous structural evolution and oxygen
removal from both surfaces are indicative
oxide on the surface, which was
pffiffiffi of
pffiffisilicon
ffi
confirmed by LEED analysis of the ( 3 3) R30 phase of the presence of Si2O3
overlayer, referred to as the honeycomb silicate adlayer, on the SiC surface.
The honeycomb silicate adlayer is formed by two Si atoms per unit cell and each
oxygen atom connects two of the Si atoms completing a ring-type structure, as shown in
Figure
3.9
pffiffiffi p
ffiffiffi depicting the top view or the projection on the c-plane. The silicate-related
( 3 3) R30 structure on the (0 0 0 1) surface, shown as a side view, in Figure 3.9 is
identically arranged with Si atoms also oriented toward the substrate. Interestingly, the
silicate layer and substrate are linearly bridged by oxygen, Si–O–Si, not connected via
Si–Si bonds. This simply implies that the surface as it stands is not optimum for nitride
growth. It is possible that in OMVPE environment, the silicate adlayer may be
removed, a statement that cannot be unequivocally made for MBE growth. In fact,
the growth of AlN on such SiC surface leads to three-dimensional (3D) growth and, as
discussed earlier, exposureto Ga athigh temperature removes thesilicate layer [41–44].
Treating the surface of ex situ HCl-treated (1300 C) SiC with in situ Ga spray,
Onojima et al. [41] obtained SiC surfaces free of the silicate adlayer, as shown
schematically in Figure 3.10, and were able to obtain a 2D AlN growth. Owing
pffiffiffi topGa
ffiffiffi
deposition on the SiC surface and subsequent flash-off, an oxygen-free ( 3 3)
R30 surface structure was achieved and initial 2D growth with an evident RHEED
j341
j 3 Growth and Growth Methods for Nitride Semiconductors
342
[1 1 0 0]
(a)
[1 1 2 0]
[0 0 0 1]
Top Si
Second Si
(b)
Silicate adlayer
C
Top O
SiC surface
Second O
Figure 3.9 (a) Top view of the oxide structure on
SiC ð0 0 0 1Þ. The Si2O3 silicate adlayer
consisting of a honeycomb structure with
SiOSi bonds. At the center of the hexagons,
one carbon atom of the topmost substrate
bilayer is visible [the dark shaded area indicates
the (1 1; 1) unit cell and light shaded the
pffiffiffi pffiffiffi
ð 3 3ÞR30 -unit cell]; (b) side view of the
oxide structure on the SiC (0 0 0 1) in ð0 1 1 0Þ SiC
projection. Linear SiOSi bonds connect the
silicate layer and the underlying SiC substrate.
Courtesy of N. Onojima (patterned after
Ref. [40]). (Please find a color version of this
figure on the color tables.)
intensity oscillation was demonstrated. The initial growth mode of AlN closely
correlated with the crystalline quality of AlN layer.
Figure 3.11 shows the RHEED images of HCl-treated surface with a silicate
adlayer, HF treated surface with has residual O, and finally in situ Ga spray treated
SiC,
of O on the surface for the ½1 1 2 0 and ½1 1 0 0 azimuths. Note the
pffiffiffi which
pffiffiffi is void
for
the
HCl-treated
surface, 1 · 1
( 3 3) R30 RHEED surface reconstruction
pffiffiffi pffiffiffi
RHEED reconstruction, and again ( 3 3) R30 RHEED surface reconstruction
for the in situ Ga spray treated surface.
3.2.4
Sapphire
Owing to its relatively low cost, availability in large area, and continual improvement
in its quality, both in terms of bulk and surface properties, sapphire has become the
[1 1 0 0]
[1 1 2 0]
[0 0 0 1]
(a)
Si
(b)
1/3 ML Si ad atom
ad-Si
C
SiC surface
Figure 3.10 (a) Top view (projection on the Si-plane of the basal
plane of SiC) and (b) side view of SiC after an in situ Ga exposure
indicating of the lack of silicate adlayer. Courtesy of N. Onojima.
(Please find a color version of this figure on the color tables.)
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Figure 3.11 RHEED images along the ½1120 and ½1100 azimuths
for HCl, HF,pand
in ffiffisitu
pffiffiSi-polarity
ffi pffiffiffi SiC surface
ffiffiffi p
ffi Ga spray treated
indicating ð 3 3ÞR30 , 1 · 1, and ð 3 3ÞR30 ,
respectively. Courtesy of N. Onojima.
dominant substrate material for epitaxy. Although there are other reasons, sapphire is
transparent for most of the bandgaps of nitride alloys; thus, it affords certain benefits
in detectors, for example, for back illumination and in LEDs, for lack of absorption.
3.2.4.1 A Primer on Sapphire
Sapphire has the space group of R3c (No. 167), as provided in the International
Tables for Crystallography, and is primarily of ionic bond nature. It can be represented by both rhombohedral unit cells, with volume 84.929 Å3, and hexagonal unit
cell, with volume 254.792 Å3, which is displayed in Figure 3.12 [13]. In the rhombohedral unit cell there are 10 ions in total, 4 Al3 þ ions and 6 O2 ions. The hexagonal
unit cell has 30 ions in all, 12 Al3 þ ions and 18 O2 ions. Oxygen is located at (x, y, z)
¼ (0.306, 0, 0.25). If this position is approximated to (x, y, z) (1/3, 0, 1/4), the anion
framework forms an hcp lattice with a ¼ 0.476 nm and c ¼ 1.299 nm.
The unit cell described by Miller–Bravais indices consists of six close-packed (0 0 0 1)
planes of O2 ions sandwiching 12 planes of Al3þ ions that occupy two thirds of the
available octahedral voids created by the O2 ions. An Al3þ ion is located at (x, y, z)
¼ (0, 0, 0.352) instead of (0, 0, 1/3), thus the cations are shifted by 0.025 nm along the
c-axis from the ideal octahedral sites. The oxygen ion is larger than the aluminum ion
by a factor of about 3 in terms of its radius; therefore, the steps on the substrate
are limited to those in the oxygen sublattice, leading to step heights in multiples of
c/6 (d(0006) 0.216 nm). The (0 0 0 1) Al2O3 surfaces are oxygen terminated and
present steps along f1 1 2 0g and f1 1 0 0g planes [45]. Two crystallographically
equivalent surfaces are related by a symmetry operation of the space group. Along
the [0 0 0 1] direction, A–A or B–B surfaces are separated by c/3, 2c/3, and c steps. Steps
separating two A surfaces are noted as A–A, and c/3 steps of height c/6, c/2, or 5c/6
j343
j 3 Growth and Growth Methods for Nitride Semiconductors
344
separate the two surfaces related by a glide symmetry operator. Such steps are dubbed
demi-steps and are noted as A–B, c/6 [46,47].
The unreconstructed basal c-plane perspective views for both unit cells are given in
Figure 3.13 [13], where the cell boxes are polyhedra. A schematic representation of
sapphire unit cell indicating the six O layers in the unit cell is shown in Figure 3.14. The
oxygen ions form a pseudohexagonal lattice. The small Al ions occupy the octahedral
sites. The labeling of planes and directions in the context of sapphire substrates are
shown in Figure 1.5. Properties of sapphire are provided in Table 3.5 [13]. All common
surfaces employed for GaN epitaxy including the (0 0 0 1) and ð1 1 0 0Þ are nonpolar.
Thus, the polarity control on sapphire depends on the particulars of growth conditions
employed with the ominous inversion domain formation always a possibility. Because
Figure 3.13 Perspective views in (2 · 2 · 1) unit cells: (a) along the
[0 0 0 1] direction in a rhombohedral unit cell; (b) along the
3.2 A Primer on Conventional Substrates and their Preparation for Growth
0.287 nm
0.252 nm
b
B
O 2–
Al3+
a
A
0.052 nm
b
0.0797 nm
C
0.1358 nm
a
B
b
0.1441 nm
0.1661 nm
[0 0 0 1]
A
a
[1 0 1 0]
Figure 3.14 A schematic diagram of the Al2O3 sapphire unit cell,
there are six oxygen layers in the unit cell, the distances between
the various atomic layers change as shown in the figure. The
oxygen ions form a pseudohexagonal lattice. The small Al ions
occupy the octahedral sites. Courtesy of P. Ruterana and Ref. [47].
Table 3.5 Properties of sapphire (in part after Ref. [13] and references therein).
Parameter
Value
Condition
Lattice constant (Å)
Melting point ( C)
Density (g cm3)
Thermal expansion coefficient (K1)
a ¼ 4.765, c ¼ 10.2982
2030
3.98
6.66 · 106 || c-axis
9.03 · 106 || c-axis
5.0 · 106 ? c-axis
a/a0 ¼ 0.83, c/c0 ¼ 0.892
20 C
0.23 || c-axis
0.25 || a-axis
77.9
452–460 in [0 0 0 1]
direction, 352–484 in
½1 1 2 0 direction
190
0.25–0.30
23.9 2.0
296 K
299 K
298 K
300 K
300 K
300 K
8.1–8.6
>1011
Experimental value
300 K
Percent change in lattice
constants with DT
Thermal conductivity (W cm1 K1)
Heat capacity (J K1 mol1)
Youngs modulus (GPa)
Tensile strength (MPa)
Poissons ratio
Hardness: Knoop
nanoindentation (GPa)
Energy band gap (eV)
Resistivity (O cm)
20 C
20–50 C
20–1000 C
20–1000 C
293–1300 K
j345
j 3 Growth and Growth Methods for Nitride Semiconductors
Thermal expansion coefficient (%)
346
2.4
2.2
2.0
1.8
Polycrystalline
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
–0.2
0
c-axis
Al2O 3
a-axis
Si
200
400
600
800
100
1200
1400 1600
1800 2000
Temperature (K)
Figure 3.15 Thermal expansion coefficient of sapphire on the cplane (along the a-axis) and along the c-axis, and that of Si as a
function of temperature [48].
of heteroepitaxy of GaN on sapphire, it is useful to display the thermal expansion
coefficient of sapphire as done in Figure 3.15.
3.2.4.2 Surface Preparation of Sapphire for Epitaxy
As-received sapphire substrates contain scratches caused by mechanical polishing
with root mean square (RMS) roughness values between 0.8 and 2.1 nm over l mm2
areas. Wet chemical etches such as phosphoric acid (H3PO4), sulfuric–phosphoric
acid combination (H2SO4–H3PO4), fluorinated and chlorofluorinated hydrocarbons,
tetrafluorosulfur (SF4), and sulfur hexafluoride (SF6) have been employed. None of
these techniques, however, produces a surface free of damage and scratches. For
MBE growth, which does not allow in situ cleaning of the surface in H at high
temperatures, a 3 : 1 solution of H2SO4 : H3PO4 is used as the etchant. The substrate
is dipped in this solution and kept at 300 C for 20 min. This is followed by a rinse in
DI water for 3 min. Although the hot etching removes some material, the resultant
surface still bears the scratches caused by mechanical polishing. However, the
surface becomes flatter after etching, with the RMS roughness being reduced from
0.323 nm in image to 0.211 nm in image in one case.
A high-temperature annealing technique after wet chemical etching of mechanically polished sapphire substrates has been shown to result in atomically smooth
surfaces [49]. Figure 3.16 shows the AFM images of two c-plane sapphire surfaces, (a)
before and (b) after the chemical etching. In an OMVPE or HVEP environment, the
typical process is to simply heat the sapphire under flowing hydrogen at temperatures
between 1000 and 1100 C. This process etches sapphire slightly leading to the
formation of hexagonal pits if there are residual amounts of gallium left in the reactor
from prior runs. The crystal quality of subsequently deposited GaN films was
insensitive to the presence of these pits [13].
To eliminate surface damage altogether, a high-temperature annealing step has
been employed, which gives rise to atomically smooth surfaces. A very high
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Figure 3.16 (a) AFM image of an as-received sapphire substrate.
Note the scratches caused by mechanical chemical polishing; (b)
AFM image of a sapphire substrate after a 180 C etch in sulfuric/
phosphoric acid. Some improvements are apparent, but the
scratches remain and are accentuated to some extent. Image size
2 mm · 2 mm.
temperature annealing investigation of sapphire substrates was recently undertaken.
Annealing experiments in air at 1000, 1100, 1200, 1300, and 1380 C (the ceiling of
the furnace employed) for 30- and 60-min periods were conducted to determine the
best conditions with the aid of AFM images of the finished surface. This was followed
by observation of RHEED patterns once in an MBE system.
A small, but progressive, improvement was observed in the reduction of scratches
up to 1300 C. However, annealing at 1380 C for 1 h led to scratch-free and smooth
surfaces to the point where the only noticeable feature in AFM images were the
atomic steps about 0.15 nm in height. AFM images indicated that annealing at
j347
j 3 Growth and Growth Methods for Nitride Semiconductors
348
Figure 3.17 An AFM image of sapphire following a 1380 C–1 h
annealing in atmosphere. Atomically flat surface is clearly visible.
Atomic step heights are about 0.15 nm, which represent the only
roughness in the image. The diagonal lines, from left to right, are
the artifacts of AFM.
1380 C for 1 h leads to atomically smooth surfaces as shown in Figure 3.17. An
atomically smooth surface is maintained after nitridation as well. RHEED images
typically show extended and bright rods associated with sapphire at temperatures as
low as 600 C during the ramp-up as shown in Figure 3.18.
Figure 3.18 A RHEED image at about 800 C of an annealed
sapphire at 1380 C for 1 h (½1 1 2 0 azimuth). Clear streaky
RHEED pattern observed at temperatures as low as 600 C
indicates that the high temperature annealing step produces clean
epiready surfaces. Without the annealing procedure, the RHEED
images are not as clear and elongated and not reproducible.
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Sapphire is nitridated by exposing it to nitrogen plasmas or thermally cracked
ammonia (a practice that has been abandoned) in MBE reactors or by to ammonia/
hydrogen gas mixtures in OMVPE reactors. Sapphire substrates that have not
undergone a heat treatment in O, as described above, exhibit the polishing damage
in the form of random scratches when they undergo MBE-like pregrowth in situ heat
treatment followed by an exposure to ammonia [50]. Considering the stacking-order
mismatch between sapphire and nitrides, these features are likely to have deleterious
effects on growth. A low density (108 cm2) of surface outgrowths was observed
after 30 min of nitridation. The presence of surface damage does not appear to have
influenced the formation of protrusions. There is no clear correlation between the
positions at which the protrusions have formed and the local surface topography.
Uchida et al. [51] observed similar protrusions after 5 min of nitridation at 1050 C in
an OMVPE system, but at a much higher density than that observed in the MBE
process. It is likely that a combination of a higher substrate temperature and the
background ammonia pressure promotes a more rapid nitridation reaction leading to
a higher density of protrusions in OMVPE-grown samples. Although what is
reported for these particular samples may hold, it should again be noted that
atomically smooth surfaces following annealing in air, as described above, do not
show discernible change after nitridation. Noting that AlOxN1x would be unstable at
the nitridation temperatures employed, the nitridation of sapphire should result in
the formation of AlN. In fact, in the MBE process, this can be observed with RHEED
in that the pattern associated with the ½1 1 2 0 azimuth of sapphire gives way to the
pattern associated with the ½1 1 2 0 azimuth of AlN but with a 30 rotation to
minimize misfit strain. Nitridation has direct consequences in the quality of the
low-temperature buffer layer and final layer(s). The ultimate test whether nitridation
was done properly requires going through the buffer and final layer growth. The
particulars of the final layer are then used to draw conclusions about the nitridation
process as was done by Wickenden et al. [52], the details of which are discussed in
Section 3.5.5.1.
In the study of Uchida et al. [51], nitridation was reported to occur very rapidly for
times less than 3 min and then slowed considerably. For short nitridation times, of
<3 min, the surface was reported to be relatively smooth, but stress-induced
protrusions developed for longer nitridation times. The density of the protrusions
increased with time, making the surface progressively rougher. Uchida et al. [51]
posed the argument that nitridation produced an amorphous AlNxO1x layer via the
exchange of oxygen atoms from sapphire and nitrogen atoms from ammonia.
However, this amorphous layer was not seen in subsequent TEM micrographs after
the deposition of a 4.0 mm thick GaN layer, which they attributed to diffusion of N and
O atoms into the crystalline layer. X-ray photoelectron spectroscopy (XPS) analyses
were carried out to confirm the incorporation of nitrogen atoms into the sapphire
substrate during the nitridation process. Sapphire substrates exposed to ammonia
even for as little as 1 min did indeed show the 1s nitrogen peak; the same is expected
from RF nitrogen exposure as well. This indicates that nitrogen atoms react with the
surface. The downshifting of the oxygen 1s line by about 0.25 eV, relative to the
spectra obtained from a bare sapphire sample, and the sample nitrided for 1 min
j349
j 3 Growth and Growth Methods for Nitride Semiconductors
350
suggests the generation of a significant bonding between oxygen and nitrogen atoms
in the nitrided layer [50]. The RHEED observations during nitridation indicate that
AlN forms on sapphire during nitridation and its orientation along the c-direction is
rotated by 30 to accommodate the strain caused by lattice mismatch.
The duration of nitridation is an important factor in terms of the quality of the
eventual GaN layer. Increase in nitridation times from 60 to 400 s has been
reported [53] to result in mobility reduction in GaN films, the overall features of
which are consistent with the study of Kim et al. [50,54]. The nitridation process, both
in OMVPE and MBE, has a great impact on the subsequent layers. In MBE, if
nitridation is done well, which can clearly be observed by RHEED image changing
from sapphire to AlN, the subsequent AlN layer is of high quality. In OMVPE, the
picture is more complex in that nitridation is followed by the low-temperature buffer
layer that is then annealed prior to the commencement of growth of subsequent
layers. The structure of the low-temperature buffer layer and the change following
annealing are such that the morphology of the subsequently deposited GaN buffer
layer changes from rough to highly faceted (possibly mixed with the zinc blende
structure) and to smooth films of the wurtzite structure. The details of the lowtemperature buffer and other buffer layers as well as other issues related to GaN
growth in general are discussed in Section 3.5.5.1.
The calculated lattice mismatch between the basal GaN before the in-plane rotation
and the basal plane of sapphire is about 49%. However, the actual lattice mismatch of
nitride layers with sapphire is reduced by the rotation of the nitride lattice with
respect to the substrate unit cell by 30 . Consequently, the lattice mismatch is reduced
to 13% to AlN, 16% to GaN, and 29% to InN. This large mismatch would cause
even the very thin layers to be fully relaxed at growth temperatures. When the
samples are cooled down after the growth, a residual thermal strain is created. In
general, films grown on the basal plane show either very little or none of the cubic
GaN phase.
Among the faces of sapphire that have been utilized for nitrides are the c-, a-, and
r-planes. The stacking configurations perpendicular to the c- and a-planes are
displayed in Figure 3.19a and b. The atomic arrangements on the c- and a-planes of
sapphire are also shown in Figure 3.19. Figure 3.20 depicts the stacking arrangement perpendicular to the r-plane (a) and the atomic arrangement on the r-plane of
sapphire (b).
3.2.5
ZnO
Unlike sapphire and polytypes of SiC that are available, ZnO and GaN have a
common stacking order and a small lattice mismatch as shown in Table 3.1. In
addition, high-quality substrates, nearly 2 in. in diameter, are available. ZnO is up
against the fact that most laboratories select sapphire substrates with SiC making
inroads. The reasons are that the layers grown on sapphire and SiC have in many
cases better quality, and sapphire is available up to 6 in. in diameter and is inexpensive. The temporary problems plaguing the SiC approach are high cost, fluctuating
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Basal or c-plane sapphire
[0 0 0 1]
O O O
Al
Al
O O O
Al
Al
O O O
Al
Oxygen
(a)
Al
a-plane (1 1 2 0) sapphire
[1 1 2 0]
O
O
Al Al
O
O
Oxygen
Al
(b)
Figure 3.19 (a) The atomic arrangement and stacking order (left)
and the top view (right) of the c-plane sapphire. (b) The atomic
arrangement and stacking order (left) and the top view (right) of
the a-plane sapphire.
quality, and poor surface finish. A more permanent, fundamental problem is the
stacking-order mismatch on SiC and sapphire. As mentioned earlier, ZnO has the
desired stacking order and a reasonably close lattice match to GaN. Moreover, as in
the case of LiGaO2, it can be selectively removed from GaN followed by GaN being
transferred to a template with good thermal conductivity.
3.2.5.1 A Primer on ZnO
The thermal expansion coefficients and the lattice constants of ZnO are Da/a ¼
4.8 · 106 K1, Dc/c ¼ 2.9 · 106 K1, a ¼ 3.2426 Å, and c ¼ 5.1948 Å, respectively.
The thermal expansion coefficient of ZnO in the c-plane as a function of temperature
j351
j 3 Growth and Growth Methods for Nitride Semiconductors
352
Side view of
sapphire r-plane
{1 0 1 2}
Oxygen
(a)
Al
Al2O3 [1 0 1 1]
3.2 A Primer on Conventional Substrates and their Preparation for Growth
1.2
Thermal expansion coefficient (%)
ZnO
1.0
a-axis
0.8
Polycrystalline
0.6
0.4
c-axis
0.2
0.0
–0.1
0
200
400
600
800
1000 1200 1400 1600 1800 2000
Temperature (K)
Figure 3.21 Thermal expansion coefficient of ZnO on the c-plane
(along the a-xais), along the c-axis as a function of temperature;
the thermal expansion coefficients of polycrystalline ZnO [48].
is depicted in Figure 3.21. Also shown are the thermal expansion coefficients of polycrystalline ZnO.
High-quality ZnO substrates have recently become available in laboratory quantities. While stable in air and O environments at temperatures as high as 900 C,
perhaps even higher, exposure to ammonia etches ZnO even at temperatures as low
as 600 C. It is believed that atomic hydrogen reacts with O forming a volatile water
vapor. The Zn metal is removed from the surface by evaporation.
3.2.5.2 Substrate Preparation for Epitaxy
The chemical preparation of ZnO involves a 5 min acetone bath followed by the same
procedure in methanol while using ultrasound agitation to remove particulates. The
wafers are then rinsed in deionized water, followed by blow drying with filtered
nitrogen prior to introduction into the growth chamber. The details of the growth
processes are given in Section 3.5.7.
ZnO is similar to the SiC and sapphire substrates and yet very different. It
shares very similar problems, chiefly the scratches caused by mechanical polishing.
3
Figure 3.20 (a) Sapphire r-plane stacking
sequence showing O atoms in larger clear circles
and Al atoms in smaller, filled circles. The salient
feature is that each Al layer has an O layer above
and below it. (b) The atomic arrangement on
three layers (the uppermost one is O,
immediately below is Al and third layer down is
another O layer) on the r-plane of sapphire. The
lines are there just guides to eye and do not
represent bonds. (Please find a color version of
this figure on the color tables.)
j353
j 3 Growth and Growth Methods for Nitride Semiconductors
354
Figure 3.22 An AFM image of a ZnO surface after a 3-h annealing
procedure in air at Ta ¼ 900 C. The RMS is about 0.1 nm. The
image is 500 nm · 500 nm.
Chemical etches have not yet been developed to deal with this problem. In fact, it may
not even be possible to accomplish this task with chemical etches. However, as in the
case of sapphire, annealing in oxygen appears to lead to improved surfaces over
mechanically polished substrate. Annealing at 900 C for 3 h in air leads to atomic
bilayer steps, which is about the best that can be achieved, as shown in Figure 3.22.
Annealing in air at 850 and 950 C also led to very smooth surfaces [55]. This implies
that the process is not very temperature sensitive and that reproducible results should
be obtained without extreme control over the temperature employed. In an MBE
system, the RHEED patterns also bear this out by showing sharp 1 · 1 diffraction rods
during the ramp-up of the temperature, as shown in Figure 3.23.
Figure 3.23 RHEED image of ZnO taken at 780 C (½1 1 2 0 azimuth.
3.2 A Primer on Conventional Substrates and their Preparation for Growth
3.2.6
LiGaO2 and LiAlO2
There are other nontraditional substrates that are beginning to appear; among them
are LiGaO2 (lithium gallium oxide (LGO), which is orthorhombic with space group
Pna21 No. 33), and LiAlO2 (lithium aluminum oxide, which is tetragonal with space
group P41212).
3.2.6.1 LiGaO2 Substrates
Lithium gallate (LiGaO2) is the most closely lattice-matched substrate currently being
considered for GaN heteroepitaxy, with an average lattice constant mismatch of only
0.9% in the basal plane. It is therefore expected that the quality of the very thin GaN
films on LiGaO2 would be better than those on other substrates discussed so far.
There is some experimental evidence to this effect. The GaN films grown on LiGaO2
are of Ga polarity. Although LiGaO2 is easily etched, which is useful for transferring
the GaN film to another substrate, it is not easily etched to a smooth surface for
epitaxy. As in the case of ZnO, etching for a few tens of seconds in H3PO4 removes the
surface damage caused by mechanical polishing and reduces the short-range roughness. The main disadvantages of LiGaO2 are its low thermal stability under OMVPE
growth conditions, low thermal conductivity, high thermal expansion coefficients, and
electrical insulation. However, when GaN layers of about 300 mm are grown by HVPE
followed by the removal of LiGaO2, the thermal and electrical conductivity of LiGaO2
are not relevant.
The structure of LiGaO2 is similar to the wurtzitic structure, but because Li and Ga
have different ionic radii, the crystal has an orthorhombic structure. The atomic
arrangement in the (0 0 1) face is hexagonal, which promotes the epitaxial growth of
(0 0 0 1) GaN, so that the epitaxial relationship (0 0 0 1) GaN/(0 0 1) LiGaO2 is expected.
The crystal structure deviates slightly from the hexagonal symmetry because of the
need to accommodate two different metallic atoms of Ga and Li. For the visualization
of LiGaO2, one can think of it as the I–III–V analogue of ZnO, with one half of the Zn
replaced by Li and the other half by Ga. The metal atoms are ordered, alternating in the
[0 1 0] direction between Li layers and Ga layers.
Unlike ZnO, LiGaO2 melts under atmospheric pressure, and hence large single
crystals can be pulled from the melt using the Czochralski method. The orthorhombic
lattice dimensions of LiGaO2 are a ¼ 5.402 Å, b ¼ 6.372 Å, and c ¼ 5.007 Å. The
distance between the nearest cations in LiGaO2 is in the range of 3.133–3.189 Å, while
the distance between nearest anions is in the range
pffiffiffiof 3.021–3.251 Å. For comparison,
the basal plane wurtzite lattice parameter h ¼ a= 3 ¼ b=2. The main virtue of LiGaO2
is that it has a very small lattice constant mismatch with GaN (averaging 0.9% at room
temperature: 1.9% in the a-direction and 0.19% in the b-direction). The orientation
relationship between GaN and LiGaO2 has been studied by several groups, yielding
½1 1 2 0 GaN||[0 1 0] LiGaO2 and [0 0 0 1] GaN||[0 0 1]LiGaO2. The atomic relationship
between GaN and LiGaO2 in the ideal case is shown in Figures 3.24 and 3.25.
Anion (oxygen) or cation (gallium and lithium) termination is available on the
(0 0 1) substrate surfaces. As is the case in polar surfaces in all compound substrates,
j355
j 3 Growth and Growth Methods for Nitride Semiconductors
356
a
2
m
C
b
m
LiGaO2
Lower O
Li
Upper O
Ga
Figure 3.24 Example of the exact fit of GaN atoms over the LiGaO2
lattice if there is no distortion. Courtesy H. Paul Maruska. (Please
find a color version of this figure on the color tables.)
the A-face (oxygen-terminated surface) can be etched easily, whereas the B-face
(metal-terminated surface) is difficult to etch in an aqueous solution of nitric acid
(H2O : HNO3 ¼ 1 : 1). This is attributed to surface oxygen atoms having a dangling
bond with two electrons, whereas surface metal atoms have no dangling bonds (hard
to etch). A maximum etch rate of 0.25 mm min1 for the cation face was obtained at
pH 9.2 and temperature 50 C. The etch is selective in relation to GaN, which means
that the entire LGO substrate could be chemically removed without affecting the GaN
layer.
As-received LGO surfaces contain scratches and pits as a result of polishing
damage and crystalline defects, as shown in Figure 3.26. However, following a
chemical cleaning in acetone, methanol, and DI water and etching in H3PO4 for 10 s
3.2 A Primer on Conventional Substrates and their Preparation for Growth
LGO: orthorhombic
a = 5.402 Å
b = 6.372 Å
c = 5.007 Å
c
(a)
b
a
LGO: orthorhombic
a = 5.402 Å
b = 6.372 Å
c = 5.007 Å
c
(b)
b
a
Projection on c-plane
Δb = –0.19 %
a
Ga
N
=3
.18
9Å
bLGO = 6.372 Å
Δa = +1.1%
Δa = +1.9%
Δb = –1.1%
(c)
aLGO = 5.402 Å
Figure 3.25 Structure of (a) orthorhombic LiGaO2 (LGO), (b)
GaN, and (c) a detailed view of the relative orientation of GaN with
respect to LGO. Courtesy of H. Paul Maruska. (Please find a color
version of this figure on the color tables.)
j357
j 3 Growth and Growth Methods for Nitride Semiconductors
358
Figure 3.26 AFM images of LiGaO2 (LGO): (a) as-received,
(b) after etching in H3PO4 for 10 s at 80 C, and (c) H3PO4 for 20 s
at 80 C.
at 80 C, the surface is improved, with more improvement resulting when the etching
time is increased to 20 s as shown in Figure 3.26b and c, with reduced pits and shortrange roughness. Compared to the as-received LGO, which exhibited a RMS roughness of 1.12 nm, a 20-s etching reduces this value to 0.78 nm. The RHEED analysis
also shows a marked improvement in the sharpness and intensity of the reconstruction rods.
The bandgap of LGO is 5.6 eV. The Youngs modulus of LGO is about 150 GPa and
its density is 4.175 g cm3. The hardness is 7.5–10 GPa, as measured by nanoindentation. Because LiGaO2 is asymmetric along the [1 0 0] and [0 1 0] directions, the
coefficients along these two directions are different. The thermal expansion coefficients are Da/a ¼ 6 · 106 K1, Db/b ¼ 9 · 106 K1, and Dc/c ¼ 7 · 106 K1.
Optically, it is biaxial with a transparency ranging from 0.3 to 6 mm and refractive
indices of na ¼ 1.7617, nb ¼ 1.7311, and nc ¼ 1.7589 at 620 nm. The crystal has no
natural cleavage planes. The structure of interest is the only stable form of the
compound in the range from room temperature to its melting point.
Because LiGaO2 melts congruently at 1585 C, the requirements for its crystal
growth are relatively modest – no high-temperature or high-pressure apparatus is
necessary. Therefore, good-quality wafers are available with relatively large diameters
at a price comparable to that of sapphire. Crystals up to 50 mm in diameter and
200 mm in length have been produced at pull rates of 2–5 mm h1 by the Czochralski
method from the mixture of Li2CO3 and Ga2O3 [56]. Crystals have been grown in
[1 0 0], [0 1 0], and [0 0 1] orientations.
3.2.6.2 LiAlO2 Substrates
The lattice constants of LiAlO2 are a ¼ 5.1687 Å and c ¼ 6.2679 Å with a density of
2.615 and hardness of 8 GPa, as determined by nanoindentation. The thermal
expansion coefficients are Da ¼ 7.1 · 106 K1, and will cause a compressive strain
in GaN g/cm3 and Dc ¼ 15 · 106 K1. Optically, it is uniaxial with transparency
ranging from 0.2 to 4 mm and refractive indices of ne ¼ 1.6014 and n0 ¼ 1.6197 at
3.2 A Primer on Conventional Substrates and their Preparation for Growth
633 nm. The crystal has no natural cleavage planes requiring, as in the case of LGO,
the facet development with a chemically assisted ion etching or chemical mechanical
polishing. Of course, the former is better in terms of performance and cost reduction.
The structure of interest is the high temperature form (or g form) of the compound. It
melts congruently around 1700 C and is stable at room temperature. Single crystals
can be grown by the conventional Czochralsky melt pulling method.
The epitaxy relationships between GaN and LiAlO2 are expected to be ð0 1 1 0Þ
GaN/(1 0 0) LiAlO2 with ½2 1 1 0 GaN/[0 0 1] LiAlO2. Unlike Al2O3 and 6H-SiC
substrates with very smooth surfaces, except the scratches caused by mechanical
chemical polishing, the LiAlO2 substrate exhibited a wavelike surface with equidistant grooves about 10 nm deep, which could have originated from the mechanical
surface polishing.
Because the preferred growth direction of the GaN epitaxial film is [0 0 0 1], the
match along the a-axis is more critical for the film deposition than the c-axis.
Considering the a-axis lattice constant, it is obvious that LiGaO2 is preferred for
GaN and LiAlO2 for Al1xGaxN. Another important factor is that because these two
crystals have exactly the same structure as GaN, the growth orientation may not have
to be limited just to the c-axis or the [0 0 0 1] direction. In fact, epitaxial growth can be
achieved at any orientation. The degree of lattice matching may vary slightly
depending on the structure of epitaxial nitrides in exact orientation.
3.2.7
AlN and GaN
It is well known that nitride semiconductors do not enjoy native substrates,
notwithstanding considerable efforts to produce them. The main impediment is
the large vapor pressure of N on AlN, GaN, and InN, in ascending order, coupled with
a low solubility of N in the molten metal at reasonable temperatures and pressures. It
is thus imperative to consider the phase diagrams of these binaries. Shown in
Figure 1.18 are the partial pressures of N2 over AlN and Al liquid as a function
of temperature, as determined by Slack and McNelly [57], on GaN by Karpinski
et al. [58], and on InN by Porowski and Grzegory [59]. The calculated values for the N2
vapor pressure on AlN by Slack and McNelly are 1, 10, and 100 atm at about 2550,
2800, and 3120 C, respectively. The GaN data, however, tell a different story in that
the partial pressure of N2 is very high, necessitating high-pressure experiments to
collect data. Karpinski et al. employed a tungsten carbide anvil cell and pressures of
up to 60 kbar to collect the data presented. The GaN data deviate from the calculations
of Thurmond and Logan [60] most noticeably at high temperatures. The equilibrium
N2 pressure data were calculated from the measurement of the equilibrium ammonia
pressure over GaN assuming N2 to be an ideal gas and thus would predict a linear
dependence in the log vs. 1/Tscale. The partial pressure data alone are indicative that
AlN would be easier to synthesize than GaN. In fact, Slack and McNelly [10] obtained
growth rates in the millimeters per hour range under very moderate pressures with a
duration of growth determined by the reaction rate of Al with a tungsten crucible. The
same partial pressure data also indicate the difficulties associated with an epitaxial
j359
j 3 Growth and Growth Methods for Nitride Semiconductors
360
deposition going from AlN to GaN and then to InN requiring an increasing amount
of nitrogen overpressure necessary to avoid decomposition.
The data of the phase diagrams of GaN, AlN, and InN are limited and contradictory
by reason of the high melting temperatures (TM) and the high nitrogen dissociation
pressures (P dis
N2 ), particularly for InN and to a lesser extent for GaN. Dissociation
pressure of MN, where M represents a metal species such as Al, Ga, or In, is defined
as the nitrogen pressure at the thermal equilibrium of the reaction [61]
MClðgÞ þ NH3 ðgÞ ! MNðsÞ þ HClðgÞ þ H2 ðgÞ;
ð3:1Þ
where s, l, and g indicate solid, liquid, and gas state, respectively. Reported values
for P dis
N2 of GaN are plotted in Figures 1.18 and 1.19. Examining the available data,
Sasaki and Matsuoka [61] concluded that the data of Madar et al. [62] and Karpinski
et al. [58] are most reliable. The data of Karpinski et al. are therefore shown. As can be
deduced from Figure 1.18, the nitrogen dissociation pressure equals 1 atm at
approximately 850 C and 10 atm at 930 C. At 1250 C, GaN is unstable and
decomposes even under a pressure of 10 000 bar of N2. The case of InN is even
more problematic at the decomposition temperature of about 700 C [59]. It should
therefore come as no surprise that the incorporation of nitrogen at high temperatures
is a nontrivial problem at best. For pressures below equilibrium at fixed temperatures, the thermal dissociation occurs at a slow and apparently constant rate,
suggesting a diffusive process of dissociation. Despite the fact that equilibrium
growth of InN appears nearly impossible, InGaN grows at temperatures in the range
of 700–900 C with very high amounts of reactive nitrogen on the surface. The
difficulties of growing bulk GaN and especially InN are alleviated somewhat with AlN
synthesis.
Judging from the N pressure at equilibrium, AlN is the closest to successful
production of all the group III–V nitride semiconductors under discussion for bulk
growth in the conventional sense. The most satisfactory method of growing highpurity AlN is the one in which AlN itself is the starting material. Slack and
McNelly [57,63] employed a technique in which high-purity AlN is produced through
an intermediate AlN powder formed by utilizing AlF3. Sublimation converts the AlN
powder into single crystals in a closed tungsten crucible or in an open tube with a gas
flow. The main problem with this growth technique is perhaps the surface oxidation
of the powder owing to the strong reactivity of oxygen and aluminum. If this oxidation
is minimized, then AlN could be produced with only 100 ppm of oxygen and with
lower amounts of other impurities. The purest AlN prepared by Slack and McNelly
employing a tungsten crucible had 350 ppm oxygen. However, when W or Re was
used for the crucible, very little contamination of the AIN with metal impurities was
found. Furthermore, the crystals grown in W or Re crucibles generally showed
uniform amber color indicating that, indeed, both oxygen and carbon contaminations were scrupulously minimized. GaN is more difficult to grow than AlN, but not
as difficult as InN. A much lower equilibrium pressure of N on AlN, on the contrary,
lends itself to the growth of this alloy utilizing the sublimation technique. Samples
with dimensions of some 4 mm diameter and 12 mm length have already been
prepared [64] and used to determine much of the AIN thermal data discussed in this
3.2 A Primer on Conventional Substrates and their Preparation for Growth
chapter. The deposition temperature is around 2250 C. Deterioration of the W boat
determines the size of the crystal.
The thermodynamic properties of GaN [65], in particular its melting conditions, are
so extreme that the application of the common growth methods utilizing stoichiometric solutions is technically impossible. To increase the solubility of N in Ga melt
and also to reduce N desorption, nitride crystal growth can be attempted under high N2
pressures as has been done by Leszczynski et al. [66,67], Porowski et al. [68,69], and
Grzegory et al. [70,71].
The GaN templates are crystallized in gas pressure vessels with volumes up to
1500 cm3 and with a workable crucible volume of 50–100 cm3. The high pressure–
high temperature reactor consists of a pressure chamber and a multizone furnace. It
also has features for in situ annealing in vacuum and electronics for stabilizing and
programming of pressure and temperature. The walls of the vessel are water cooled
considering the high temperatures involved. The pressure in the chamber is
stabilized to a precision better than 10 bar. The temperature is measured by an
array of thermocouples in the furnace. This allows a stabilization of temperature to
0.2 C and programmable variations of the temperature distribution in the crucible.
GaN crystals presented are grown from nitrogen solutions in pure liquid gallium
or in Ga alloyed with 0.2–0.5 at.% of Mg or Be at pressures in the range of 10–20 kbar
and temperatures of 1400–1600 C. Magnesium and beryllium, being the most
efficient acceptors in GaN, are added to the growth solutions to obtain p-type crystals.
Supersaturation in the growth solution is obtained by a deliberate application of
temperature gradient of 2–20 C cm1 along the long axis of the crucible. This
approach assures a continuous flow of nitrogen from the hotter part of the solution to
the cooler parts. Crystallization experiments performed without intentional seeding
resulted in crystals nucleating spontaneously on the internal surfaces of polycrystalline GaN crusts in the cooler zone of the solution. Typical duration of the growth
process was 120–150 h.
Leszczynski et al. [15], Porowski et al. [16], and Grzegory et al. [70] performed nitride
crystal growth from a solution under high N2 pressure. The growth experiments under
high pressure were carried out in a gas pressure chamber of 30 mm internal diameter
with a furnace dimension of 14 mm (1500 C) or 10 mm (1800 C) and with a boron
nitride (BN) crucible containing Al, Ga, or In. The temperature was stabilized to a
precision better than 10 C. Measures were made to optimize the pressure range for
growth. With this optimization, the crystals grew only at a pressure for which the
nitride was stable over the entire temperature range. In the case of GaN, single crystals
were grown from a solution in liquid Ga under a N2 pressure of 8–17 kbar at
temperatures ranging between 1300 and 1600 C. The quasi-linear temperature
gradient in the process, which spanned over 5–24 h, was 30–100 C cm1. The
nucleation and growth of single-crystal GaN took place through the process of
dissociation and transport of thin polycrystalline GaN film deposited on a Ga surface
into the cooler part of the crucible.
The technique has also been applied to AlN and InN as well, with GaN being the
most successful. At high N2 pressure, the synthesis rate of AlN is high and that of InN
is extremely low. The rate of AlN growth is so high that, at a pressure lower than
j361
j 3 Growth and Growth Methods for Nitride Semiconductors
362
6.5 kbar, thermal explosion takes place during heating of a bulk Al sample. Owing to a
low stability, the crystallization rate of AlN at 1600–1800 C is marginally low
(0.5 mm h1 in the literature). On the contrary, owing to kinetic (low-temperature)
and thermodynamical (low-stability) barriers, crystal-growth experiments of InN
result in very small crystallites (5–50 mm), particularly when grown by slow cooling of
the system from the temperatures exceeding the stability of InN. A wide variety of
GaN, AlN, and InN crystals result from changes in temperature and pressure used
during growth.
3.2.7.1 Seedless Growth of GaN
3.2.7.1.1 Seedless Growth of GaN by High Nitrogen Pressure Solution Growth
(HNPSG) for Substrates The GaN crystals grown by the high nitrogen pressure
solution method without seeding are wurtzitic and mainly have the form of hexagonal
platelets. They grow at a rate of just below 0.1 mm h1, along the h1 0 1 0i like
directions and have nearly perfect morphology indicating a stable layer-by-layer growth
mode. As one can deduce from the form of the crystals, the growth is strongly
anisotropic in that it is much faster (about 100 times) in the c-plane, as can be deduced
from the size of crystals produced. This holds true for supersaturations corresponding
to an average growth rate in h1 0 1 0i directions of 0.05–0.1 mm h 1. The templates
are transparent with flat mirrorlike faces. The behavior of the crystals remains the
same for solutions containing Mg or Be. The average size of crystals, grown without
any intentional seeding, scales with the diameter of the high-pressure reactor.
A photograph of a slice of one such sample prepared at UNIPRES in Warsaw [72]
is displayed in Figure 3.27.
Growth rate anisotropy in favor of enhanced rate along the c-plane is desirable for
acceptable size templates. However, this at the expense of a much reduced growth rate
in the c-direction leads to only template growth as opposed to a boule growth, which
would allow many substrates to be sliced. The vertical growth rate in the c-direction can
Figure 3.27 GaN crystals grown in high pressure chambers of
different sizes. The grid size corresponds to 1 mm · 1 mm. The
schematic cross section of the hexagonal platelet is shown [71].
3.2 A Primer on Conventional Substrates and their Preparation for Growth
be increased by increasing the supersaturation of the Ga melt, but this has the
undesirable effect of unstable needlelike forms. The supersaturation of the solution is
determined primarily by the temperature of growth and its gradient, mass transport
mechanisms in Ga, and by the local competing processes such as neighboring crystals
because we are dealing with seedless, or spontaneous, growth. For large GaN
templates to be obtained without accelerated growth near the edges and corners, it
is imperative that the supersaturation be attained. It should be noted that for extremely
high supersaturation, edge nucleation on the hexagonal faces of GaN platelets often
occurs, which seeds the unstable growth on that particular face. The tendency toward
unstable growth is stronger for the Ga-face for undoped templates and N-face for
doped templates (with sufficient Mg to render the crystal p-type), resulting in rough
surfaces leaving the unaffected face mirrorlike. However, if Mg concentration is not
high, the aforementioned instability has no effect in that the N-face remains mirrorlike [73]. The instability is characterized as macrosteps, periodic inclusions of solvent
or cellular growth structures. The polarity of the crystal surfaces can be identified by
etching in hot alkali solutions, because the Ga-polar surface is inert to etching, whereas
the N-polar one etches very rapidly. The validity of the etching method has been
verified by performing convergent beam electron diffraction (CBED) [74] and XPS [75]
measurements in the same samples. The topic is further discussed in Section 4.2, in
conjunction with extended defects.
The low doping level, with no bearing on stability, may be related to the position of
the Fermi level that may influence the microscopic nature of growing surfaces, which
is consistent with ab initio calculations and underscores the importance of both native
and impurity-related point defect formation in GaN [76,77]. This would presuppose
that the instabilities mentioned above may have their genesis in point defect
formation for which there is no direct evidence as yet.
Summarizing, the seedless growth method leads to rates of 0.05–0.1 mm h1
along the h1 0 1 0i directions and the crystal is morphologically stable. However, the
crystal is morphologically stable along the h1 0 2 0i directions even for higher growth
rates. This explicates that the size of the stable platelets would depend on the volume
of the solution and growth time. One can argue that increasing the volume of the
crucible and growth time might lead to scaling up the size of the crystal.
3.2.7.1.2 Seeded Growth of GaN by HNPSG Method for Substrates To circumvent at
least some of the scalability-related issues, seeded growth can be attempted using the
templates prepared by the seedless method. However, seeding growth along the c-axis
seems to be much more challenging because the observed growth rates are small and
the growth tends toward instability [71]. Parameters such as seed preparation and the
configuration of the experiment in the context of N flow could be optimized to
enhance the in-plane or out-of-plane growth rates. For example, the macrosteps at the
edges result from exposure to higher nitrogen fluxes, because the supply is from the
hotter part of the solution. This can be alleviated by configuring the seed to melt
relation, which can also lead to much more uniform supersaturation across the
growth front. Other complications result from constitutional supercooling of the
solution, caused by a very low temperature gradient and high thermal and low
j363
j 3 Growth and Growth Methods for Nitride Semiconductors
364
solute diffusivity [78]. This leads to the creation of a depleted zone at the growth front,
which cannot be remedied because the concentration cannot be recovered by
diffusion owing to low solute diffusivity.
N-type GaN platelets have been used for seeded crystallization to suppress the
cellular growth on the Ga-polar (0 0 0 1) surface. The N-polar ð0 0 0 1Þ surfaces have
also been used for comparison. Large positive temperature gradients of the order of
100 C cm 1 have been applied at a nominal average crystallization temperature of
about 1500 C. After 20–50 h growth, the substrates with the new crystals were
removed from the solution and investigated [71]. Essentially, the new growth, which
was a hillock 6 mm in diameter, was transparent, colorless, and void of periodic and
cellular structures. The dominant feature was the propagation of the macrosteps
from the hillock center. Growth on the N-polar surface was mainly by the propagation
of the macrosteps [71]. Several growth centers were also noted for similar experimental conditions. It is still not clear if this is related to differences in surface
nucleation mechanisms for the different polarities or due to inadequate surface
preparation.
The average growth rates in these experiments were between 4 and 8 mm h1 for
both polarities, which depends on supersaturation at the growth front, which, in turn,
is a function of temperature gradient and the amount of liquid Ga layer on the
substrate. Microscopic observation of cross sections of the samples indicates a stable
growth in terms of continuity of the newly grown material, that is, inclusions of the
solvent and/or voids were not observed. The interface between template (80–100 mm
thick) and the newly grown crystals was not noticeable, indicating that the surface
preparation and the wetting procedures were adequate. The X-ray diffraction (XRD)
analysis indicated similar quality for the template and overgrowth. Future efforts
should include crystallization experiments for finding the optimum configuration for
a stable growth along the h0 0 0 1i directions. Of the particular issues remaining to be
addressed adequately are step bunching and its reduction, and uniform supersaturation across growing surface. The uniform supersaturation can be achieved by
reducing the radial temperature gradient and/or decreasing the width of the Ga layer
over the substrate.
Summarizing, an examination of the crystal morphology indicates that the crystal
shape and size depend on the pressure, the temperature range, and the supersaturation during growth. For pressures and temperatures lying deeply in the GaN
stability field (e.g., higher pressure and lower temperature), the crystals are hexagonal
prisms elongated in the c-direction. Under conditions close to the equilibrium curve,
the dominating shape of the crystals is a hexagonal platelet. The crystals grown slowly
(slower than 0.1 mm h1) at smaller temperature gradients, exhibited higher crystalline quality. Typical full width at half maximum (FWHM) of the X-ray rocking
curves for (0 0 4) Cu Ka. reflections are 23–32 arcsec.
Probably owing to the nonuniform distribution of nitrogen in the solution across
the growing crystal face, the quality of GaN crystals deteriorates with increasing
growth rate (high supersaturation) and with increasing dimensions of the crystals.
This is apparent especially when the size of the face becomes comparable to the size
of the crucible. The deterioration of quality of 5–10 mm crystals grown at a rate of
3.2 A Primer on Conventional Substrates and their Preparation for Growth
0.5–1 mm h1 was evidenced by the broadening of the X-ray rocking curve. GaN
substrates are not available in large quantities. Extremely high N2 equilibrium
pressure on GaN will most likely preclude even pseudoconventional growth methods
to fall short of producing GaN substrates. Bulk GaN substrates have been prepared
under high pressures (12–20 kbar) and temperatures (1200–1600 C). Despite complications and challenges, the high-pressure method is capable of growing thicker
crystals, which can be sliced into platelets, as shown in Figure 3.28.
3.2.7.2 Pertinent Surfaces of GaN
Improve surface morphology of epitaxial layers, metal–semiconductor contacts,
processing and effect of surface features on transport, and optical properties of
GaN, requires an understanding of surface structure and growth processes on an
atomic scale. For the technologically relevant surfaces, that is, polar (0 0 0 1), ð0 0 0 1Þ
and nonpolar ð1 1 0 1Þ, ð1 1 0 0Þ, ð1 1 2 0Þ, experimental and theoretical investigations have led to considerable understanding of their atomic geometry, the driving
forces leading to these structures, and their effect on adatom kinetics, the latter is
discussed in Section 3.4.2.
The polar hexagonal {0 0 0 1} basal plane is the most common surface on which to
grow. Even though other surfaces have been explored for growth, the structures
grown on the basal plane have exhibited the best performance. Among the basal
planes, the Ga-polarity surface (0 0 0 1) is more stable and can be produced with
smoother surfaces than the N-polarity ð0 0 0 1Þ surface. Figure 3.29 shows the
schematic representations of top view of the ideal truncated GaN(0 0 0 1) surface
as presented by Neugebauer [79]. The dashed lines depict the boundary of a (2 2)
unit cell. High symmetry adsorbate sites are also marked as H3 (hollow) and T4 (atop a
second layer atom). The side views along the ð1 1 0 0Þm-plane and ð1 1 2 0Þa-plane are
shown in Figures 3.31 and 3.32.
Figure 3.28 GaN substrate crystal, obtained by slicing of the new
material, grown by directional crystallization along c-axis;
(a) before slicing where the bottom ledge corresponds to the
original template as shown in the cross-sectional artistic rendition
and (b) one sliced template. The grid is 1 mm · 1 mm [71].
j365
j 3 Growth and Growth Methods for Nitride Semiconductors
366
T4
H3
Ga
N
Figure 3.29 Schematic representations of top view of the ideal
truncated GaN (0 0 0 1) surface. The dashed lines depict the
boundary of a (2 · 2) unit cell. High-symmetry adsorbate sites are
also marked as H3 (hollow) and T4 (atop a second layer atom).
The darker circles depict Ga atoms, and the lighter circles show N
atoms, respectively. The side views along the ð1 1 0 0Þm-plane and
ð1 1 2 0Þa-plane are shown in Figures 3.31 and 3.32.
Experimentally, the most commonly observed reconstruction on (0 0 0 1) GaN is
(2 · 2), which corresponds to a stable growth front, yielding high-quality thin films, as
discussed in Section 3.5.6. Because this reconstruction has not been observed on
ð0 0 0 1Þ GaN, its presence is considered the domain of (0 0 01) GaN leading to
inversion domains, which are discussed in Section 4.1.3. Other reconstructions
observed by RHEED and STM on this surface are (1 2), (4 4), (5 5), and (6 4),
details of which are discussed in Section 3.5.6. The ideal truncated GaN(0 0 0 1)
surface with Ga in the top surface layer is shown in Figure 3.29. Tetrahedral bonds are
such that each Ga surface atom has one dangling bond and is bonded to three N
atoms in the second layer below. Adding one Ga or N layer on this surface would be
called a Ga or N adlayer structure and adding/removing a Ga atom would be called Ga
adatom/vacancy. Theoretically, this surface has been studied by several researchers
employing plane wave pseudopotential methods and ab initio tight-binding methods [79]. All studies are centered on (1 1) and (2 2) structures, specifically
adatoms on H3 and T4 sites, trimers, and Ga adlayers on various sites.
The calculated relative formation energies are such that the (2 · 2) H3 N adatom
model (with N at the He hollow site) is energetically the most stable under N-rich
conditions. The (2 · 2) Ga-vacancy model gives only slightly higher energy. In contrast,
under Ga-rich conditions, the (2 · 2) T4 Ga adatom structure (Ga adatom atop the atom
in the second layer below) is favored. Under N-rich conditions, the Ga-vacancy and N
adatom (on an H3 site) are the stable surfaces with energies close to one another. In the
case of Ga-rich conditions, the Ga adatom (on a T4 site) is the lower energy structure
and is thus preferred.
As for the ð0 0 0 1Þ N-face, four dominant reconstructions, (1 1), (3 3), (6 6),
and c(6 · 12) listed in order of increasing surface coverage, have been identified,
discussed in detail in Section 3.5.6. For N-rich conditions on this surface, a (2 · 2) H3
3.2 A Primer on Conventional Substrates and their Preparation for Growth
Ga adatom model is found to be most stable. In the case of Ga-rich conditions, a (1 · 1)
adlayer structure is energetically favored. In the stable (1 · 1) model, a full monolayer
(ML) of Ga atoms is situated directly atop the N atoms, with the Ga–N bond length
equal to 1.97 Å (compared to 1.94 Å in bulk GaN). The Ga–Ga separation in the
adlayer (3.19 Å) is considerably larger than a typical Ga–Ga separation of 2.7 Å in bulk
Ga. It should be pointed out that this is a completely novel structure with no known
analogue among other semiconductor surfaces. In fact, this structure violates most of
the empirical rules used to describe semiconductor surfaces in that it clearly disobeys
electron counting and maximizes the number of dangling bonds. One immediate
conclusion that can be drawn is that the surface must be metallic, which has indeed
been observed by scanning tunneling spectroscopy (STS) [79].
Regarding the (3 · 3) surface, structural models with one, two, or three additional Ga
adatoms on (or in) the Ga adlayer have been considered. One additional Ga adatom is
the best model for the observed (3 · 3) reconstruction where the extra Ga atom resides
only 0.9 Å above the adlayer plane. If no lateral relaxation is allowed, the Ga adatom
must be positioned 1.8 Å above the adlayer to preserve a reasonable Ga–Ga distance.
However, the extremely large inward relaxation of the adatom is enabled by a 0.5 Å
lateral relaxation of the nearest-neighbor Ga-adlayer atoms. This then allows the
adatom to be situated much closer to the adlayer plane, which stabilizes the structure,
adopting the nomenclature that it is an in-plane adatom model.
The GaN ð1 1 0 1Þ surface has been observed as a sidewall facet in lateral epitaxial
overgrowth (ELO), which is used for threading defect reduction and thus deserves
discussion. The ELO method is discussed in Section 3.5.5.2. These surfaces have also
been found as sidewalls in the inverted pyramid defects, which form at the
termination of some threading dislocations at the GaN surface during growth of
In-containing alloys [79]. Northrup et al. [80] have theoretically investigated this
surface by employing first-principles calculations. The schematic representation of a
stable surface structure under Ga-rich conditions is given in Figure 3.30. It consists of
Ga atoms in two distinct types of sites in the surface layer. These sites have been
labeled as B2 and T1 sites. The atoms in B2 sites are bonded to two N atoms in the layer
below. However, the atoms in T1 sites are bonded to one N atom each in the layer
below, as in the (1 · 1) Ga-adlayer structure existing on the ð1 1 0 1Þ surface [79]. The
structure stable under N-rich conditions contains one Ga atom per cell bonded in an
H3 site. This adatom is bonded to three N atoms in the layer below.
In terms of nonpolar surfaces, the ð1 1 0 0Þ and ð1 1 2 0Þ are important as they do
not exhibit the polarization endemic in layers grown on polar surfaces, see Section 2.12
for a discussion of polarization. Growth on these nonpolar surfaces has progressed
well, some details of which are discussed in Section 3.5.11. These surfaces also
represent the sidewalls of processed (0 0 0 1) surfaces as well as being central planes in
ELO. A schematic model of the GaN ð1 1 0 0Þ surface is shown in Figure 3.31. On the
ð1 1 0 0Þ surface, an equal number of threefold-coordinated Ga and N atoms exist in
the top surface layer. This allows charge neutrality without changes in stoichiometry or
reconstruction, hence the term nonpolar. The Ga and N atoms form an array of Ga–N
dimers. The ð1 1 0 0Þ surface has been studied by Northrup et al. [81] and Filippetti
et al. [82] and a review by Neugebauer [79] is available that employs density–functional
j367
j 3 Growth and Growth Methods for Nitride Semiconductors
368
(0 0 0 1) c-plane
B 2 site
T1 site
–
(1 0 1 1) facet
B 2 site
Ga
N
T1 site
Figure 3.30 Schematic representation of the GaN ð1101Þ Gaadlayer surface. Surface sites T1 and B2 are sites in which a Ga
atom makes one or two bonds with N atoms. The darker circles
depict Ga atoms and the lighter circles N atoms. Patterned after
Ref. [79].
theory calculations. The two main relaxation mechanisms in effect are a contraction
of the GaN bonds by 6% accompanied by a slight buckling rehybridization with N
atoms. The N atoms tend to adopt a p3 configuration, whereas Ga atoms adopt a sp2
configuration. The bond rotation angle is 7% and the surface energy is 118 meV Å2.
The structure of the ð1 1 2 0Þ surface can be construed as a chain of threefoldcoordinated Ga and N atoms, as shown in Figure 3.32. In each of the unit cells, there
are four surface atoms, namely two Ga and two N atoms. This surface also has been
Ga
N
Figure 3.31 Schematic top view of the Wz GaN ð1 1 0 0Þ surface.
Larger circles depict the atoms in the first layer and the smaller
ones portray those in the second layer. The dashed lines outline
the boundary of a unit cell (5.179 Å 3.171 Å). Patterned after
Ref. [79].
3.2 A Primer on Conventional Substrates and their Preparation for Growth
[0 0 0 1]
Ga
N
Figure 3.32 Schematic top view of the Wz GaN ð1 1 2 0Þ surface.
The dashed lines outline the boundary of a unit cell
(5.493 Å 5.179 Å). The smaller circles denote atoms in the
second layer. Patterned after Ref. [79].
investigated with density–functional theory calculations [81]. The calculated Ga–N
bond lengths in the surface chain are 1.85 Å (cis) and 1.87 Å (trans), representing a
contraction of approximately 4–5% compared to bulk. Similar to the ð1 1 0 0Þ surface,
the Ga atoms relax toward an sp2 configuration. The bond rotation angle is 7 and the
surface energy is slightly larger than that for the ð1 1 0 0Þ surface by 123 meV Å 2.
Contrary to the ð1 1 0 0Þ surface, the structure formed by replacing surface N atoms
with Ga atoms is not energetically favored even under Ga-rich conditions.
3.2.7.3 GaN Surface Preparation for Epitaxy
A very crucial step in the homoepitaxial growth of semiconductors is the attainment
of an atomically clean and smooth substrate surface, without which even severe
extended defects would form at the interface between the template and
epitaxial overlayer. Available methods for cleaning the surface prior to epitaxy are
characterized as external and internal ones. Ex-situ processes attempt to remove any
metallic and organic contaminants from the surface. However, these methods
always leave a thin oxide layer on the surface (of thickness similar to the native
oxide), which must be removed before growth. This is typically done in situ in the
epitaxial reactor. In the vapor phase techniques, the thin oxide is removed during
ramp to growth temperature under a reducing environment such as H. However,
removal within the MBE UHV system (in situ) usually includes a preliminary heating
for outgassing of adsorbed gases in a preparation chamber and a final heating for
oxide removal and preparation of an atomically clean surface within the growth
chamber [83].
j369
j 3 Growth and Growth Methods for Nitride Semiconductors
370
Overviews of the ex-situ and in-situ preparation of a GaN surface are found in
Refs [84,85]. Because high-quality GaN epitaxy by MBE, and perhaps by other
techniques as well, requires growth on GaN templates prepared by vapor phase
techniques, it is necessary to discuss this issue of GaN surface cleaning in the context
of MBE for the moment. Commonly, a GaN surface that has been exposed to the air is
contaminated with oxygen and carbon [84–86], which must be removed before
epitaxy.
XPS can be used to quantify the surface contaminants. Such studies on GaN
surface oxide led to the observation of two components of the O 1s peak centered at
531.3–531.6 and 532.4–532.7 eV, respectively [85–87]. The O 1s component centered
at 531.3 eV, which can be removed in HCl [86], NH4OH [87], HF, and UV/O3 (ozone
treatment) is attributed to stoichiometric Ga2O3, although the oxide may also contain
some component of mixed oxynitride of Ga, which is also soluble in alkali solutions.
The origin of the high-energy O 1s peak at 532.7 eV, which of course can be removed
with ion etching, is not yet clear. Prabhakaran et al. [87] reported only that it could be
removed by argon ion sputtering while the 531.3 eV O 1s peak remained. Ar ion
etching was also reported to remove the C 1s peak that, concurrently with the highenergy 532.7 eV O 1s peak, paved the way for speculation that C and O are the
components of C–O bonding [86]. The high-energy O peak has been attributed to
OH species (hydroxides) by King et al. [85]. This oxide component was dominant on
GaN surfaces after solvent cleaning and prior to UV/O3 exposure.
The carbon contamination on the GaN surface, which manifests itself with a main C
1s XPS peak at 285.7 eV, is indicative of a mixture of C–O and C–H bonding [85].
Shifting of the C 1s peak to the lower binding energy of 283.7 eV after sputter cleaning
led Prabhakaran et al. [87] to suggest that this is atomic carbon and could get
incorporated into the epitaxial layer. Shalish et al. [86] have observed a C 1s component
at 289.9 eV, which was attributed to the carboxylic group (COOH), and a peak at
287.7 eV, which was assigned to C–Cl bonds, appearing after etching in HCl. These
observations [86,87] point to the necessity of surface preparation with HCl or HF, or
possibly NH4OH to remove any native oxide from the surface prior to introducing the
sample into the growth system. As for C contamination, the UV/O3 oxidation
treatment was found most useful [85].
After chemical preparation, the sample is still exposed to air before loading which
brings us to in situ treatment. The C and O contamination could not be removed at
temperatures (900–1000 C) even higher than the decomposition temperature (800
C) [84,85]. This means that annealing of GaN must be conducted in N or Ga flux to
maintain the stoichiometry of the surface [85]. The difficulty associated with oxide
desorption has been attributed to the strength of the Ga–N bonds [85] in that oxides
probably desorb as either Ga–O or N–O species instead of O2 and this requires
breaking the strong Ga–N bonds. Most of the C–O-bonded carbon was reported to
desorb at temperatures between 500 and 600 C leaving behind only C–H-bonded
carbon, which apparently desorbs at much higher temperatures [85]. Efficient removal
of C components from the surface of GaN has been successfully conducted with NH3
and Ga fluxes at 800–900 C in the authors laboratories and elsewhere [84,85]. King
et al. [85] concluded that atomically clean surfaces obtained by annealing in NH3
3.2 A Primer on Conventional Substrates and their Preparation for Growth
at 800 C exhibited 2 · 2 reconstruction in LEED. However, Bermudez et al. [84] found
only partial removal of oxygen after annealing in NH3 up to 900 C. They attributed this
to the fact that the reaction
Ga2 O3 þ 2NH3 ! 2GaN þ 3H2 O
ð3:2Þ
is endothermic by 0.79 eV/O atom; hence, reduction of Ga2O3 by NH3 may require
temperatures above 900 C, while the reaction
3C þ 4NH3 ! 3CH4 þ 2N2
ð3:3Þ
is exothermic by about 6.7 eV/C atom.
In-situ RHEED observations of GaN surfaces that have been exposed to air show
the absence of any amorphous native oxide layer on the surface, in contrast to that for
the conventional arsenide-based semiconductors [88]. Although the GaN surface
adsorbs oxygen and other impurities, owing to the large number of unsaturated
dangling bonds [89], the oxidation is self limiting due to the chemical inertness of
GaN. Thus, it is possible to grow GaN overlayers by MBE after an ex situ and in situ
preparation consistent with the above discussion, a recipe for which is summarized
below, to the point that the interface between the epitaxial overlayer and underlying
template could not even be detected with TEM. Depending on the history of the
template, the procedure used at VCU consists of degreasing followed by HCl or
HF : H2O treatment, which, in turn, is followed by a few minutes treatment in boiling
aqua regia. After rinsing and drying, the sample is loaded into the introduction
chamber of MBE.
3.2.8
Other Substrates
Other substrates such as Hf, MgO, NaCl, W, and TiO2, and so on are not discussed
here as they do not represent any sustained effort. However, spinel at one point got a
good deal of attention and has been discussed here. Lasers employing films grown on
the c-plane of sapphire utilize etched cavities because sapphire does not cleave well.
The cleaving process is further complicated in that the GaN epilayers are rotated
(about 30 ) with respect to the underlying sapphire substrate making it impossible to
align the cleavage plane of GaN and sapphire. For this reason, Nakamura et al. [90,91]
explored lasers grown on the a-plane of sapphire where the c-plane of GaN aligns
normal to the substrate surface. The facets in GaN and underlying sapphire are
oriented on the a-plane and c-plane, respectively. Although the quality does not
compare to that on the c-plane, improved cavity formation along the m-plane (1 0 1 0)
or a-plane (1 1 2 0) outweighs its reduced material quality. Moreover, laser structures
on (1 1 1) MgAl2O4 (spinel substrates), which lead to wurtzite GaN along the c-plane
have also been explored for injection-laser experiments [91]. Spinel cleaves along the
(1 0 0) plane, inclined to the surface with cleavage following the m-plane of GaN about
where the epilayer is reached. Even though the facet quality in this scheme is the best
among the aforementioned approaches, material quality degradation is too severe to
pull it ahead of the other approaches. It is, however, very clear that a substrate with
j371
j 3 Growth and Growth Methods for Nitride Semiconductors
372
good cleavage characteristics and on which GaN can be grown without rotation is
desperately needed. Considerable progress in etched facets have been made to
circumvent this problem, as discussed in Volume 3, Chapter 2.
3.3
GaN Epitaxial Relationship to Substrates
Owing to the lack of native substrates for GaN epitaxy, GaN is grown on foreign
substrates, often with large lattice mismatch. Consequently, internal strain minimization, among other considerations, leads to an atomic arrangement different from
that of the substrate material. The degree of this difference is substrate dependent, as
discussed below.
3.3.1
Epitaxial Relationship of GaN and AlN with Sapphire
Sapphire is an ubiquitous substrate on which to grow any semiconductor, and GaN is
no exception. Sapphire remains the most frequently used substrate for group III
nitride epitaxial growth owing to its low cost, the availability of 3-in. diameter crystals
of good quality, its transparent nature, its stability at high temperatures, and a fairly
mature technology for nitride growth.
Before delving into the orientational relationship between the GaN epilayer and
underlying substrate, taken sapphire as default, a discussion of two notations and
translation between the two is warranted. Here it should be mentioned that Miller–
Bravais indices containing four indices are the most common and straightforward.
However, any plane and direction can be described with three indices for the
hexagonal system. These two systems are referred to as Miller indices (h k l) and
Miller (h j k l) indices. The latter can be obtained from the former by adding the first
two indices of the former, changing the sign and assigning it as the third Miller index
in the (h j k l) notation. For example, (1 1 0) in (h k l) notation would translate to
ð1 1 2 0Þ in the (h j k l) notation. Similarly, (1 0 0) would translate to ð1 0 1 0Þ and (1 0 0)
would translate to ð1 0 1 0Þ. Of interest are the commonly used X-ray diffraction
peaks of (0 0 2) and (1 0 2), which in the (h j k l) notation would be (0 0 0 2) and
ð1 0 1 2Þ. The index in the c-direction does not change, leading to a translation from
(1 0 2) in the (h k l) notation to ð1 0 1 2Þ in the (h j k l) notation. In the (h j k l) notation, a
dot is often placed after the second Miller index to indicate that the crystal symmetry
under question is hexagonal.
The orientation order of the GaN films grown on the main sapphire planes {basal,
c-plane (0 0 0 1), a-plane ð1 1 2 0Þ, and R-plane ð1 1 0 2Þ} by ECR-MBE has been
studied in great detail [92–94]. The epitaxial relationship between GaN and sapphire
is insensitive to the method of growth in that both MBE and OMVPE layers exhibit the
same relationship. A few examples of the film/substrate epitaxial relationships are
1:0 Al2O3 and ½1 1:0 GaN || ½1 2:0
(0 0 0 1) GaN || (0 0 0 1) Al2O3 with ½2 1:0 GaN || ½1 1:0 GaN ||
Al2O3, ð2 1:0Þ GaN || (0 1.2) Al2O3 with [0 0.1] GaN || ½0 1:1 Al2O3 and ½0 3.3 GaN Epitaxial Relationship to Substrates
Table 3.6 Crystallographic relationship between GaN films and
sapphire substrates ([13] and references therein).
Miller indices
Crystal plane (h j k l)
(h k l)
c
a
m
r
(0 0 0 1)
ð1 1 2 0Þ
ð1 0 1 0Þ
ð1 0 1 2Þ
(0 0 1)
(1 1 0)
(1 0 0)
(1 0 2)
GaN plane || sapphire GaN direction ||
surface plane (h j k l)
sapphire direction (h j k l)
(0 0 0 1) always
ð0 0 0 1Þ or ð1 0 1 0Þ
ð1 0 1 3Þ or ð1 2 1 2Þ
ð1 1 2 0Þ or ð1 2 1 0Þ
½1 2 1 0 jj½1 1 0 0, ½1 2 1 0 jj½1 1 0 0
½1 1 2 0 jj ½1 0 0 0, ½1 1 2 0 jj ½0 0 0 3
½1 2 1 0 jj½0 0 0 1, ½1 0 1 0 jj ½1 2 1 0
½0 0 0 1 jj½1 1 0 1, ½0 0 0 1 jj½1 0 1 1,
½1 1 0 0 jj ½1 1 2 0; or ½1 1 2 0 jj ½1 1 0 2
The data on AlN on sapphire are somewhat limited, but the available data indicate AlN exhibits
similar behavior to that of GaN.
½2 1:0 Al2O3 (Refs [10–12] in [95]). The aforementioned orientational relationships
1
1 0 GaN ||
in the (h j k l) notation become (0 0 0 1) GaN || (0 0 0 1) Al2O3 with ½2 ½1 1 0 0 Al2O3 and ½1 1 0 0 GaN || [1 2 1 0] Al2O3, ð2 1
1 0Þ GaN || ð0 1 1 2Þ Al2O3 with
[0 0 0 1] GaN || ½0 1 1 1 Al2O3 and ½0 1 1 0 GaN || ½2 1
1 0 Al2O3. The second
set deals with a-plane GaN on r-plane sapphire, which can also be expressed as
the epitaxial relationship of ð1 1 2 0Þa-plane GaN on ð1 1 0 2Þr-plane sapphire
with ½1 1 2 0GaNjj½1 1 0 2sapphire, ½0 0 0 1GaNjj½
1 1 0 1sapphire, and ½
1 1 0 0
GaNjj½1 1 2 0sapphire owing to hexagonal symmetry. A tabular representation of
epitaxial relationships of GaN grown on various planes of sapphire is given in
Table 3.6.
The calculated lattice mismatch between the basal GaN and the basal sapphire
plane is larger than 30%. However, the actual mismatch is smaller (16%), because the
small cell of Al atoms on the basal sapphire plane is oriented 30 away from the larger
sapphire unit cell. This smaller lattice mismatch can be calculated by adopting the
model explained in Figure 3.33.
pffiffiffi
3awGaN asapphire
¼ 0:16:
ð3:4Þ
asapphire
It is on this plane that the best films have been grown with relatively small in-plane
and out-of-plane misorientations. In general, films on this plane show either none or
nearly none of the cubic GaN phase.
GaN grown on the ð1 1 2 0Þa-plane turns out to be (0 0 0 1) oriented and anisotropically compressed. The effect of this uniaxial stress on the laser performance is
discussed in Volume 3, Chapter 2. The in-plane relationship of GaN and sapphire is
depicted in Figure 3.34 where the ½1 1 2 0 direction of GaN is aligned with the [0 0 0 1]
direction of sapphire. In this orientation, the bulk positions of both the substrate and
the GaN cations lie along the sapphire [0 0 0 1] direction. The mismatch between the
substrate and the film is given by
c sapphire 4awGaN
¼ 0:02;
c sapphire
ð3:5Þ
j373
j 3 Growth and Growth Methods for Nitride Semiconductors
374
GaN[1 2 1 0]
Sapphire[1 1 0 0]
GaN[1 0 1 0]
Sapphire[1 1 2 0]
Al
N
GaN cell
Sapphire cell
Figure 3.33 Projection of bulk basal plane sapphire and GaN
cation positions for the observed epitaxial growth orientation. The
circles mark Al-atom positions and the dashed lines show the
sapphire basal plane unit cells. The open circles mark the N-atom
positions and solid lines show the GaN basal plane unit cell. The Al
atoms on the sapphire plane sit at positions approximately 0. 5 Å
above and below the plane position [92].
and for the GaN ½1 1 0 0 direction parallel to the sapphire ½1 1 0 0 direction by
c sapphire 1:5awGaN
¼ 0:005:
c sapphire
ð3:6Þ
In one investigation [96], the range of the growth conditions leading to good films
on the a-plane was found to be wider than that on the c-plane. Another impetus for
exploring growth on the a-plane is the relative ease with which the sapphire could be
cleaved along the weak single bond in the c-plane. The GaN film on the top would
cleave along the ð1 1 2 0Þ a-plane or the ð1 1 0 0Þ m-plane depending on the in-plane
rotation of GaN with respect to sapphire. The aforementioned discussion is somewhat academic in that GaN grown on a-plane still has [0 0 0 1] direction normal to the
surface. However, growth on r-plane with OMVPE leads to a-plane GaN, the details of
which are discussed in Section 3.5.11.
GaN films have also been grown on the r-face f1 1 0 2g of sapphire purportedly
to achieve a lattice mismatch smaller than on the c-plane sapphire. Films grown on
the r-face has been reported to assume an orientation similar to f2 1
1 0g. The
arrangement in the case of the ð1102Þ face of sapphire and ð2 1 1 0Þ of GaN is
depicted in Figure 3.35a. Although of no immediate impact on the topic under
discussion, Figure 3.35b provides an image for us to gain some familiarity with the
r-plane of GaN in relation to the basic hexagonal lattice structure. The lattice
3.3 GaN Epitaxial Relationship to Substrates
GaN[1 1 2 0]
Sapphire[0 0 0 1]
GaN[1 1 0 0]
Sapphire[1 1 0 0]
Al
N
GaN cell
Sapphire cell
Figure 3.34 Projection of bulk a-plane sapphire
and basal plane GaN cation positions for the
observed epitaxial growth orientation. The solid
circles mark the Al-atom positions and the
dashed lines show the sapphire a-plan unit cells.
The open circles mark the N-atom positions and
the solid lines show the GaN basal plane unit cell.
The mismatch along the GaN ½1 1 2 0 direction
parallel to sapphire [0 0 0 1] direction is 2%. The
mismatch along the GaN ½1100 direction
parallel to sapphire ½1 1 0 0 direction is – 5%.
The lines drawn through the N atoms are for
underscoring hexagonal symmetry, not the
bonds as the bonds are formed between the Ga
atoms on plane above the nitrogen plane and N
atoms [92].
mismatch between the ½1101 direction of sapphire and the [0 0 0 1] direction of GaN
parallel to the sapphire ½1101 direction is equal to
r
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
3c GaN 3a2sapphire þ c 2sapphire
rffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
¼ 0:01:
ð3:7Þ
3a2sapphire þ c 2sapphire
In the case when the ½1 1 0 0 direction of GaN is parallel to the sapphire ð0 1 2 0Þ
direction, the lattice mismatch is
pffiffiffi
awGaN ðasapphire = 3Þ
pffiffiffi
¼ 0:16:
ð3:8Þ
ðasapphire = 3Þ
j375
j 3 Growth and Growth Methods for Nitride Semiconductors
376
GaN[1 1 0 0]
4.76 Å
Sapphire[1 1 2 0]
GaN[0 0 0 1]
5.12 Å
N
Sapphire[1 1 0 1]
Ga
Al
(a)
Figure 3.35 (a) Projection of bulk r-plane
sapphire and a-plane GaN anion and cation
positions for the observed epitaxial growth
orientation. During growth it is assumed that the
top O layer desorbs leaving behind the Al layer
with its nearly square unit cells (dimensions are
given in terms of angstroms). The figure should
be treated with some caution as the exact
placement of N atoms belonging to the GaN cell
has been done somewhat arbitrarily. Calculations
that can shed some light on the preferred
position of Ga and N atoms with respect to Al are
lacking. Because the a-plane of GaN is nonpolar,
only the N and Ga atoms on the same plane are
shown. The Ga atoms in the first layer would be
lacking a lattice site to bond, making dangling
bonds very plausible in addition to sever
distortion of the lattice as even the N atoms in the
GaN unit cells not to align everywhere with the
underlying Al atoms. Even though some N atoms
appear to vertically align with Al below, that bond
should in reality not be what is loosely referred as
the long bond as that would imply the c-direction
to be out of the plane, which is not the case.
However, the image provides a reasonably good
picture of how the a-plane GaN lattice is stacked
on the r-plane sapphire. The lines connecting Al
atoms represent only the unit cell not the bonds.
For a 3D view of a plane GaN on r-plane sapphire,
see Figure 3.161. (b) Although it is not of an
immediate application, this figure illustrates the
r-plane of GaN with its associated directions. In
addition, the figure on the lower left indicates the
placement of the r-plane in a hexagonal cell.
3.3 GaN Epitaxial Relationship to Substrates
One unit cell
r-plane, GaN[1 1 0 2]
GaN[1 1 2 0]
[1 1 0 1]
N
c
Ga
View direction
Bars represent Ga–N bonds
r-plane
(1 1 0 2)
c/2
b
(b)
a
Figure 3.35 (Continued)
The mismatch along the [0 0 0 1] direction of GaN parallel to the ½
1 1 0 1 direction
of sapphire is 1%, which is much smaller than the 16% mismatch along ½1 1 0 0
direction of GaN parallel to the ½1 1 2 0 direction of sapphire. Growth on the r-face
exhibits ridgelike features that allow relaxation of the mismatch. It is assumed, as in
the case of c-plane of sapphire, the topmost O layer is desorbed and the Al layer of
sapphire is then exposed. Another mechanism accomplishing the same is also
implicitly assumed. A closer look, however, indicates that while the above argument
about lattice mismatch would hold for only the Al–N bonds at the corners of the unit
cell of sapphire, shown as rectangles in Figure 3.35a, it does not necessarily hold for
the Al–N bond at the center of the unit cell. In addition, the Ga atoms that must be on
the same plane as N in GaN (true for a nonpolar a-plane that is the topic of discussion
here) will not have one of its bonds satisfied and would be left dangling. We must
hasten to state that the representation in Figure 3.35a is a very simple one intended
only to give the reader a first-order glimpse as to how the a-plane GaN might be
organized on the r-plane sapphire with no consideration to energy minimization.
This awaits calculations for additional insight, which is missing. More details on the
r-plane sapphire can be found in Figure 3.20.
The lattice mismatch between GaN and all the other substrates, inclusive of various
epitaxial relationships, is tabulated in Table 3.7 for both completeness and convenience. The discussion here details the intricacies of lattice mismatch as affected by
the epitaxial relationship driven by strain minimization brought about in the first
place by the lattice-mismatched substrates.
j377
a-Al2O3
ð2 1 1 0Þ=ð0 1 1 2Þ
ð0 0 0 1Þ=ð2 1 1 0Þ
ð0 1 1 3Þ=ð0 1 1 0Þ
(0 0 0 1)/(0 0 0 1)
[0 0 0 1]//[0 0 0 1]
ð1 1 2 0Þ=ð1 1 2 0Þ
Zinc blende (a ¼ 4.33)
Hexagonal (a ¼ 4.7589, c ¼ 12.991)
½1 0 1 0==½1 0 1 0
(0 0 0 1)/(0 0 0 1)
Wurtzite (a ¼ 3.1129, c ¼ 4.9819)
AlN
—
Zinc blende (a ¼ 4.511)
½0 0 0 1==½0 1 1 1
½0 1 1 0==½2 1 1 0
½2 1 1 0==½0 0 0 1
½0 1 1 0==½0 1 1 0
½2 1 1 0==½0 0 0 1
½0 3 3 2==½2 1 1 0
½0 1 1 0==½2 1 1 0
½21 10==½0110
—
—
—
0.18
0.06
—
—
Lattice misfit (%)
Thermal strain (%)
(growth temperature) (1000–25 C)
aGaN aAlN
¼ 2:41
2.35
aAlN
c GaN c AlN
¼ 4:08
c AlN pffiffiffi
2aGaN 3aAl2 O3
pffiffiffi
¼ 22:65 22.83
pffiffiffi 3aAl2 O3
3aGaN aAl2 O3
¼ 16:02
aAl2 O3
xaGaN c Al2 O3
¼ 6:09;
c Al2 O3 qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
pffiffiffi
where x ¼ c 2 þ ð2 3aÞ2
4aGaN c Al2 O3
¼ 1:85
pffiffiffi c Al2 O3 pffiffiffi
3aGaN 3aAl2 O3
pffiffiffi
¼ 33:01
3aAl2 O3
pffiffiffi
3aGaN aAl2 O3
¼ 16:02
aAl2 O3
4aGaN c Al2 O3
¼ 1:85
c Al2 O3
3cGaN x Al2 O3
¼ 1:19;
x Al2 O3 qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
pffiffiffi
where x ¼ c2 þ ð2 3aÞ2
—
—
—
Wurtzite (a ¼ 3.1878, c ¼ 5.185)
GaN
—
In-plane direction Lattice misfit (%)
Epitaxial relationship (GaN || substrate) (room temperature)
Lattice parameter (Å)
Crystal
substrates inclusive of various epitaxial relationships between GaN and substrates mentioned.
Table 3.7 Room-temperature lattice mismatch of GaN with other III-N compounds and
378
j 3 Growth and Growth Methods for Nitride Semiconductors
Zinc blende (a ¼ 4.35997)
Hexagonal (a ¼ 3.07997, c ¼ 10.083) (0 0 0 1)/(0 0 0 1)
3C-SiC
4H-SiC
Orthorhombic
(0 0 0 1)/(0 0 1)
(a ¼ 5.4063, b ¼ 6.3786, c ¼ 5.0129)
Orthorhombic
(a ¼ 5.428, b ¼ 5.498, c ¼ 7.710)
Zinc blende (a ¼ 5.4309)
Zinc blende (a ¼ 5.652)
b-LiGaO2
NdGaO3
Si
GaAs
(0 0 1)/(0 0 1)
(0 0 0 1)/(1 1 1)
(0 0 1)/(0 0 1)
(0 0 1)/(1 1 1)
(0 0 0 1)/(1 1 1)
(0 0 0 1)/(1 0 1)
(0 0 0 1)/(0 1 1)
(0 0 0 1)/(0 0 1)
Tetragonal (a ¼ 5.169, c ¼ 6.282)
g-LiAlO2
ð1 1 0 0Þ=ð1 0 0Þ
(0 0 1)/(0 0 1)
Hexagonal (a ¼ 3.0806, c ¼ 15.1173) (0 0 0 1)/(0 0 0 1)
6H-SiC
[0 1 0]//[0 1 0]
aGaN a6H-SiC
¼ 3:48
½1 0 1 0==½1 0 1 0
a6H- Sic
aGaN a3C-SiC
[0 1 0]//[0 1 0]
¼ 3:46
a3C-SiC
aGaN a4H--SiC
¼ 3:50
½1 1 2 0==½1 1 2 0
a4H--SiC
c GaN aLiAlO2
[0 0 0 1]//[0 1 0]
¼ 0:31
pffiffiffi aLiAlO2
3aGaN aLiAlO2
¼ 6:8
½1 0 1 0==½0 1 0
pffiffiffi aLiAlO2
3aGaN - aLiGaO2
½1 0 1 0==½1 0 0
¼ 0:13
aLiGaO2
ða-directionÞ
p
ffiffiffi
3aGaN bLiGaO2
¼ 13:43
½1 0 1 0==½0 1 0
bLiGaO2
ðb-directionÞ
p
ffiffiffi
3aGaN aNdGaO3
½1 0 1 0==½1 0 0
¼ 1:72
pffiffiffi aNdGaO3
b
3
a
GaN
NdGaO3
¼ 0:43
½1 0 1 0==½0 1 0
bNdGaO
pffiffiffi3
2a
2
a
GaN
Si
pffiffiffi
¼ 16:99
½1 0 2 0==½1 1 0
2aSi
a
a
GaN
Si
½110==½101
¼ 16:93
aSi pffiffiffi
2aGaN 2aGaAs
pffiffiffi
[0 1 0]//[0 1 0]
¼ 20:19
2aGaAsi
aGaN aGaAs
¼ 20:19
½1 0 2 0==½1 1 0
aGaAs
In-plane direction Lattice misfit (%)
Epitaxial relationship (GaN || substrate) (room temperature)
Lattice parameter (Å)
Crystal
20.26
16.8
1.06
15.1 (b-direction)
1.59 (a-direction)
0.1
3.53
3.55
3.49
(Continued)
0.07
0.19
0.66
1.67 (b-direction)
0.54 (a-direction)
0.41
0.03
0.09
0.01
Lattice misfit (%)
Thermal strain (%)
(growth temperature) (1000–25 C)
3.3 GaN Epitaxial Relationship to Substrates
j379
Zinc blende (a ¼ 4.216)
Wurtzite (a ¼ 3.252, c ¼ 5.213)
MgO
ZnO
(0 0 0 1)/(1 1 1)
Mixed perovskite (a ¼ 7.730)
LSAT
aGaN aMgO
¼ 6:99
pffiffiffi aMgO pffiffiffi
3aGaN 2aMgO
pffiffiffi
½1 0 1 0==½1 1 0
¼ 7:99
2aMgO
a
a
GaN
ZnO
¼ 1:97
½1 0 2 0==½1 0 2 0
aZnO
aGaN - aScAlMgO4
½1 0 2 0==½1 0 2 0
¼ 1:49
aScAlMgO
pffiffi4ffi
4a
2
a
GaN
MgAl
2 O4
pffiffiffi
½1 0 2 0==½1 1 0
¼ 11:55
2ap
ffiffiffi 2 O4
MgAl
4a
2
a
GaN
LSAT
pffiffiffi
½1 0 2 0==½1 1 0
¼ 16:64
2aLSAT
[0 1 0]/[0 1 0]
5.8
16.68
11.61
1.64
0.04
0.06
0.15
0.21
1.19
Lattice misfit (%)
Thermal strain (%)
(growth temperature) (1000–25 C)
2.18
The growth temperature for each of the cases is tabulated in Table 2.36. Courtesy of J. Chaudhuri, Wichita State University.
(0 0 0 1)/(1 1 1)
Spinel (a ¼ 8.083)
(0 0 0 1)/(0 0 0 1)
(0 0 0 1)/(0 0 0 1)
(0 0 0 1)/(1 1 1)
(0 0 1)/(0 0 1)
In-plane direction Lattice misfit (%)
Epitaxial relationship (GaN || substrate) (room temperature)
MgAl2O4
ScAlMgO4 Hexagonal (a ¼ 3.236, c ¼ 25.15)
Lattice parameter (Å)
Crystal
Table 3.7 (Continued)
380
j 3 Growth and Growth Methods for Nitride Semiconductors
3.3 GaN Epitaxial Relationship to Substrates
3.3.2
Epitaxial Relationship of GaN and AlN with SiC
Both AlN and GaN deposit on hexagonal SiC substrates in a simple epitaxial
relationship, that is, [0 0 0 2]III-N//[0 0 0 6]SiC and ½1 1 2 0III-N ==½1 1 2 0SiC . Using
ð1 1 0 0Þ 6H-SiC substrates (not readily available commercially), Horino et al. [97]
established the epitaxial relationship as ð1 1 0 0ÞGaN==ð1 1 0 0ÞSiC and ½1 1 2 0
III-N==½1 1 2 0SiC.
3.3.3
Epitaxial Relationship of GaN and AlN with Si
The epitaxial relationship of GaN on Si is GaN h0 0 0 1i||Sih1 1 1i and h2 1
1 0i
GaNjjh0 1 1iSi.
If the wurtzitic phase is produced on (0 0 1) Si, then the epitaxial relationship is
(0 0 0 1) GaN || (0 0 1) Si and ½1 1 2 0 GaN || [1 1 0] Si. AlN on Si(1 1 1) showed two
epitaxial relationships [13,98] namely AlN(0 0 0 1) ½2 1
1 0 || Si(1 1 1) ½0 2
2 for
deposition temperatures greater than 650 C and AlN(0 0 0 1) ½1 1 0 0 || Si(1 1 1)
½0 2 2. These two relationships correspond to the (4 4) and (7 7) lattices,
respectively. AlN grown at low temperatures of 400–600 C by MBE shows the
relationship of AlN(0 0 0 1) ½0 1 1 0 || Si(1 1 1) ½1 1 2 [99]. These results are given in
Table 3.8 and the atomic distance mismatches in Table 3.9. Figure 3.36 shows the
orientational relationship between wurtzite AlN and the underlying Si (0 0 1) substrate. GaN deposited on Si(1 1 1) with an AlN buffer layer shows Ga polarity, and GaN
grown directly on Si(1 1 1) shows N polarity. For details, refer to Refs [13,100,101],
assuming the epitaxial relationship between AlN and Si to be the same as that between
GaN and Si.
3.3.4
Epitaxial Relationship of GaN with ZnO
Epitaxial relationships between GaN and ZnO are expected to be [0 0 0 1] of GaN being
parallel to [0 0 0 1] of ZnO. Because the lattice mismatch between GaN and ZnO in the
Table 3.8 Epitaxial relationship of Wz GaN and AlN grown on Si
when buffer conditions are such that wurtzitic form of GaN or AlN
is produced.
Si crystal plane Miller
indices (h j k l) or (h k l)
GaN plane || Si plane in
the first column
GaN direction
|| Si direction
(1 1 1)
(0 0 1) if Wz GaN is produced
(1 1 1) if Wz AlN is produced (Ts > 650 C)
(0 0 0 1)
(0 0 0 1)
(0 0 0 1)
(0 0 0 1)
(0 0 0 1)
½2 1 1 0 jj ½0 1 1
½1 1 2 0 jj ½1 1 0
½2 1 1 0jj½0 2 2
½1 0 1 0jj½0 2 2
½0 1 1 0jj½1 1 2
(1 1 1) if Wz AlN is produced (Ts ¼ 400–600 C)
j381
j 3 Growth and Growth Methods for Nitride Semiconductors
382
Table 3.9 Epitaxial relationship of GaN grown on Si with the
atomic distance mismatch (ADM) and the extended atomic
distance mismatch (EADM).
Epilayer/substrate
ð0 0 1Þh-GaN/(1 0 0)Si
(1 0 0)c-GaN/(1 0 0)Si
(0 0 1)h-GaN/(1 1 1)Si
Atomic distance (Å) of
epilayer/substrate
[0 1 0]Ga–Ga/[0 1 0]
Si–Si
3.186
5.431
[2 1 0]Ga–Ga/[1 0 0]
Si–Si
5.518
5.431
[0 1 0]Ga–Ga/[0 1 0]
Si–Si
4.500
5.431
[0 1 0]Ga–Ga/[1 1 0]
Si–Si
3.186
3.84
ADM (%)
EADM (%) of
epilayer/substrate
41.3
(Ga–Ga) · 5/(Si–Si) · 3
1.6
2.5
(Ga–Ga) · 1/(Si–Si) · 1
17.1
1.6
(Ga–Ga) · 6/(Si–Si) · 5
17.0
0.571
(Ga–Ga) · 6/(Si–Si) · 5
0.438
For details, refer to Refs [13,100,101].
c-plane is very small, under 2%, no discernible in-plane rotation for strain reduction
is expected. However, if ZnO is deposited on sapphire followed by GaN deposition,
the sapphire substrate and both epitaxial layers are oriented toward each other by a
30 rotation of the unit cell. That is, the in-plane epitaxial layer and substrate are in the
2 0, GaN ½1 1 0 0k Al2O3½1 1 2 1 0, GaN or
form of ZnO, GaN ½1 0 1 0 k Al2O3½1 1 ZnO ½2 1 1 0 k Al2O3½1 1 0 0, all of which indicates a 30 rotation of the epitaxial layer
with respect to sapphire substrate.
3.3.5
Epitaxial Relationship of GaN with LiGaO2 and LiAlO2 and Perovskites
Some oxides have also been explored because of the small lattice misfit with GaN they
provide, with LiGaO2 and LiAlO2 being the ones, particularly the latter, generating a
good deal of interest. The epitaxial relationships between GaN and LiGaO2 are
expected to be ½1 1 2 0 GaN || [0 1 0] LiGaO2 and [0 0 0 1] GaN || [0 0 1] LiGaO2. On the
contrary, the epitaxial relationships between GaN and LiAlO2 are expected to
1
1 0 GaN//[0 0 1] LiAlO2, ð1 1 0 0Þ GaN//
be ð0 1 1 0Þ GaN//(1 0 0) LiAlO2 with ½2 2 0GaNjj½0 0 1LiAlO2 .
g-LiAlO2 (1 0 0) with ½1 1 0 0GaNjj½1 0 0LiAlO2 and ½1 1 1 0 0Þ=ð1 0 0Þ and along
The lattice misfit strain between GaN and LiAlO2 with ð1 the [0 0 0 1]//[0 1 0] in-plane is 0.41%. The lattice structure of LiGaO2 is similar to
the wurtzite structure. However, owing to Li and Ga atoms having different ionic
radii, the crystal has orthorhombic structure [102]. Figure 3.37 shows the transformation of the hexagonal unit cell of GaN to an orthorhombic cell that has lattice
parameters close to that of LiGaO2 (Table 3.10).
Perovskite oxides have also been employed in the growth of GaN in an effort to
attain a better match compared to more conventional substrates or for applications
3.3 GaN Epitaxial Relationship to Substrates
1
2
Dangling
bonds
Dangling
bonds
Si top layer
Si first underlayer
[0 1 0]
2
〈2 1 1 0〉
Si second underlayer
Al or N sublattice
[1 0 0]
[0 0 1]//[ 0 0 0 1]
〈1 1 1 0〉
Figure 3.36 Atomic arrangement for the heteroepitaxial
nucleation of 2H-AlN on the Si(0 0 1) surface. The two AlN
domains with a 30 rotation are formed on neighboring terraces
(1) and (2), separated by a single atomic step boundary according
to the Si dangling bond directions. Refs [13,101].
where the properties of nitride semiconductors and electro-optic and nonlinear
optics properties of perovskites could be offered in the same template/stack. Among
the perovskite is NdGaO3 with its orthorhombic unit cell. The lattice mismatch of
GaN with NdGaO3 has been calculated [102] by assuming a perovskite cell of NdGaO3
with lattice parameters a, b, and c each equal to 3.86 Å. This is followed by creating a
new unit cell with a0 and b0 where a0 and b0 are the diagonals of the old perovskite cell,
as shown in Figure 3.38. The c0 -axis of the new cell is naturally parallel to the c-axis of
the perovskite cell but its length is doubled. Basically, this operation transforms a
perovskite unit cell to a tetragonal unit cell representing GaN, as tabulated in
j383
j 3 Growth and Growth Methods for Nitride Semiconductors
384
GaN
b
a
a'
b'
Figure 3.37 Transformation of a hexagonal unit cell to an
orthorhombic unit cell. Courtesy of J. Chaudhuri and Ref. [102].
Table 3.11. Accordingly, (1 0 0) plane becomes ð1 1 0Þ plane and (0 0 1) becomes
(0 0 1). The corresponding lattice misfit is 1.72% and misfit strain is 0.66%.
3.4
Nitride Growth Techniques
HVPE, OMVPE (inorganic VPE), RMBE, and bulk crystal growth from Ga solution
are the main growth methods used for nitrides. By far the most frequently used
methods are the variants of VPE methods. Although HVPE is used to produce thick
GaN layers, including those thick enough to be self-supporting once peeled from the
sapphire substrate, OMVPE produces sharp heterojunctions for devices. With the
exception of FETs, OMVPE is the primary method employed in the investigation and
production of optoelectronic devices, such as LEDs and lasers, albeit the quality of
MBE films grown on HVPE buffers is slightly better than of those grown by OMVPE.
Inorganic VPE was the first method used to grow epitaxial III-N semiconductors, but
was nearly abandoned. The technique, however, was revived recently by growing very
high quality and thick buffer layers and templates for the growth of device structures
by MBE and OMVPE.
Table 3.10 Transformation of GaN hexagonal unit cell to an orthorhombic unit cell [102].
GaN original hexagonal
cell with a and b lattice
parameters
GaN transformed
orthorhombic cell with
lattice parameters a0 and b0
LiGaO2 orthorhombic cell
a ¼ 3.189 Å
b ¼ 3.189 Å
c ¼ 5.185 Å
pffiffiffi
a ¼ 3a ¼ 5:52 Å
0
b ¼ 2b ¼ 6.38 Å
c0 ¼ 5.185 · 1 Å
a ¼ 5.4063 Å
b ¼ 6.3786 Å
c ¼ 5.0129 Å
0
3.4 Nitride Growth Techniques
a
a'
b'
b
Figure 3.38 Transformation of a perovskite unit cell to a
tetragonal unit cell. Courtesy of J. Chaudhuri and Ref. [102].
3.4.1
Vapor Phase Epitaxy
VPE has long been employed for the growth of many semiconductor structures. With
ongoing source developments and improved reactor designs, this technique has
become very powerful, particularly for GaN and related materials. Growth from the
vapor phase is categorized on the basis of the sources used. If the sources are inorganic
in nature, the term inorganic vapor phase epitaxy is used. This too can be subdivided
on the basis of the sources used. For example, if a hydride source is used for the group
V element, the term hydride vapor phase epitaxy is applied. If at least some of the
sources are organic in nature, the terms organometallic vapor phase eptixay, organometallic chemical vapor deposition, metalorganic chemical vapor deposition, or
metalorganic vapor phase epitaxy are employed.
3.4.1.1 Hydride Vapor Phase Epitaxy
The genesis of the HVPE growth method can be traced to its wide use in silicon and
conventional III–V semiconductors. In the early 1960s, the development of the halide
precursor techniques applied to Si and Ge provided the foundation for their
subsequent application to the growth of GaAs, which was coming to eminence.
The HVPE method has since played an important role in the growth of III–V
Table 3.11 Transformation of a perovskite unit cell to a tetragonal unit cell.
Lattice parameters
a
b
c
Perovskite unit cell
New unit cell
Final unit cell
3.86 Å
3.86 Å
3.86 Å
pffiffiffi
a= ¼ 3:86pffiffi2ffi
=
b ¼ 3:86 2
c0 ¼ 3.86 · 2
a0 ¼ 5.43 Å
b0 ¼ 5.43 Å
c0 ¼ 7.72 Å
Courtesy of J. Chaudhuri [102].
j385
j 3 Growth and Growth Methods for Nitride Semiconductors
386
semiconductors. In fact, it has the unique distinction of being the first method to
produce AlN [103] and, as reported by Maruska et al. [17], to produce single crystalline
GaN with quality sufficient to launch the first stages of GaN technology that gained so
much prominence in the 1980s. Typical thicknesses for these deposits were in the
range of 50–150 mm. The advantage of this technique is that it is conducive to the
growth of thick buffer layers at high growth rates on any available substrates to be
used as templates for OMVPE and MBE growth, particularly the latter, of high-quality
heterostructures with relatively low defect concentrations. A comprehensive treatment of this method can be found in Ref. [104]. A brief historical treatment and a
succinct review will be given.
After the report of Maruska et al. [17], Wickenden et al. [105] reported on GaN
deposition on a-SiC and a-Al2O3, meaning wurtzitic varieties, in 1971, and Ilegems [106] obtained 100–200 mm thick single-crystalline GaN layers on sapphire
substrates in 1972. Continuing on, Shintani et al. [107] investigated in 1974 the effects
of the important growth parameters, such as the position of the substrate in view of
the gas flow dynamics in the reactor, the reactant gas flow rate, and the substrate
temperature, on the epitaxial growth rate of GaN on (0 0 0 1) sapphire substrates.
Sano et al. [108] considered the influence of the surface sapphire orientation on the
growth rate in 1976. In 1977, Madar et al. [109] and Jacob et al. [110] achieved doped
GaN with n-type conductivity on sapphire substrates. A study on the growth rate of
GaN in hydrogen as well as in inert gas ambience was undertaken by Seifert et al. [111]
in 1981, which resulted in growth rates up to 800 mm h1.
Owing to difficulties associated with uniform seeding of GaN on sapphire, coalescence of islands in a timely manner and resultant high n-type background doping
(typically 1019 cm3), large defect concentrations, and inability to produce p-type
GaN for light emitters, this technique was largely abandoned in the early 1980s,
although Maruska et al. [112] later showed that Zn and Mg doping could be achieved by
the simultaneous evaporation of the dopant source in the HCl stream. Since the first
report of Maruska and Tietjen [17] many reports on GaN growth [113–124] became
available in the literature. Further, several others [103,125,126] have extended this
method to the growth of high-quality AlN.
But when low-temperature nucleation buffer layers were employed in the context
of OMVPE, as discussed in Section 3.5.5.1, followed shortly thereafter by reports of
p-type conductivity in GaN, the HVPE method reappeared [127–129] because, in part,
of its improved ability to grow thick GaN films with relatively low defect concentration
and new techniques for nucleation layers (NLs). Further improvements resulted
from lateral epitaxial overgrowth on patterned SiO2 masks [130–132], the concept of
which is discussed in Section 3.5.5.2. Freestanding GaN substrates have been
prepared by Kim et al. [133] and Melnik et al. [134], which eventually culminated
in the production of very high quality freestanding GaN templates prepared at
Samsung Advanced Institute of Technology [135], the detailed characteristics of
which are discussed in Section 3.5.1.2. Despite these remarkable achievements, the
nitride materials still suffer from a very high defect density because of the lattice
mismatch between the nitrides and all the available foreign substrates. Recent twostep processes employing low-temperature GaN buffer layers [136] and techniques
3.4 Nitride Growth Techniques
for substrate removal [135,137] have shown good-quality materials with very promising characteristics. The development of the HVPE technique combined with other
techniques for producing GaN templates may be the key to resolve the high density
defect issue in the III nitride device technology.
In HVPE, the group III precursors are chlorides formed by flowing hydrogen
chloride gas over the liquid metal in a quartz tube. The group V precursors are
hydrides fed into the reaction chamber by a separate quartz line in order to avoid
premature reaction with molten source metal. For GaN growth, the chloride and
hydride precursors are GaCl, which is formed by reacting Cl from HCl gas with
molten Ga, and NH3, respectively. The GaCl in vapor phase is transported to the
deposition zone by a carrier gas, which can be hydrogen, and/or an inert gas. The
pressure inside the reaction chamber is kept at the atmospheric pressure. The reactor
walls are made of high-purity quartz tube. For high-quality semiconductor films to
result, the gases are of electronic quality with purity better than 1 ppm for contaminants. For nitrogen or hydrogen, each impurity is in concentration below 1 ppm.
For the other gases, the total concentration of all the impurities is below 1 ppm. The
metallic sources employed are of 7N, meaning they have a purity of 99.99999%. A
multiple zone furnace is used as the metal source zone and substrate temperatures
are different. In particular, the zone containing the metallic gallium, the central zone
where the gases are homogeneously mixed, and the zone where the substrate resides
and the deposition takes place are all kept at different temperatures, as shown
schematically in Figure 3.39. The three-temperature zone allows one to independently set the partial pressure of each species, such as the chlorides and hydrides, and
explore optimum growth conditions systematically. The influence of parametric
variations in the vapor phase composition on the growth rate becomes relatively
easier. The same applies to physical processes taking place during a growth. In brief,
the vapor phase composition depends on the metallic source efficiency, the ammonia
decomposition, and the flow of various gases introduced into the reactor. The vapor
phase composition, the partial pressures of the various reactive gaseous species, and
the temperature of the three zones in the reactor determine the growth rate and the
solid composition of the epitaxial layer if ternaries are attempted. Obviously,
the growth takes place in a thermodynamical equilibrium and a wide range of
conditions can be applied. If a large flow of HCl is not introduced into the reactor, the
Source zone
Mixing zone
Deposition zone
NH3
NH3
N2/H2+HCl
N2/H2+HCl
GaCl
N2/H2+HCl add
Substrate
GaCl HCl H2/N2/NH3
Ga source
Bypass line
Bypass
Three-zone furnace
Figure 3.39 Schematic diagram of a three-zone HVPE reactor,
which utilizes Ga and ammonia sources, used for nitride growth.
Patterned after Ref. [104].
Exhaust
j387
j 3 Growth and Growth Methods for Nitride Semiconductors
388
use of NH3 leads to initial conditions far from thermodynamical equilibrium. The
growth rates from a low of 1 mm h1 to a high of more than 100 mm h1 can be
achieved [104].
In HVPE as employed by Maruska and Tietjen [17] for GaN, HCl vapor flowing over
a Ga melt causes the formation of GaCl, which is transported downstream. On the
substrate surface, the GaCl reacts with NH3 and leads to GaN through the following
chemical reaction:
2GaðlÞ þ 2HClðgÞ ! 2GaClðgÞ þ H2 ðgÞ;
GaClðgÞ þ NH3 ðgÞ ! GaNðsÞ þ HClðgÞ þ H2 ðgÞ;
ð3:9Þ
where g, l, and s depict gaseous, liquid, and solid species, respectively. The efficiency
of the first reaction had been estimated by Ban [138] to be about 99.5%.
Using the GaAs analogy [139], two thermodynamic reaction pathways leading to
the deposition of GaN [140] can be forwarded:
GaClðgÞ þNH3 ðgÞ()GaN þ HClðgÞ þ H2 ðgÞ;
3GaClðgÞ þ 2NH3 ðgÞ()2GaN þ GaCl3 ðgÞ þ 3H2 ðgÞ:
ð3:10Þ
The gaseous species in the reactor are GaCl, GaCl3, HCl, NH3, H2, and either H2 or
some other inert gas is used as carrier gas. Barin [141] used thermodynamical data to
calculate the reactions of Equations 3.9 and 3.10. For additional details, see Ref. [104].
The supply of GaCl is controlled by the Ga boat temperature and the flow rates of
the HCl gas and the carrier gas. The reactor in this method is heated by a two-zone
resistance furnace with the region containing the Ga boat being kept at a different
temperature than the region housing the substrate for reaction, as shown in
Figure 3.39. The Ga zone temperature impacts the growth rate a great deal. The
Ga source is held at a constant temperature between 850 and 900 C. The reaction
efficiency of HCl with Ga is near unity. Although dependent on the reactor itself, the
typical flow rates are about tens of sccm for HCl, 1 l min1 for NH3 and 2 l min1 for
the carrier gas. GaN films can be grown with rates up to 1 mm h1 on Al2O3
substrates at atmospheric pressure. However, those high growth rates deplete the
Ga source material rather quickly and lead to very rough surfaces with columnar
growth. The substrate zone temperature varies between 1050 and 1200 C. At lower
substrate zone temperatures, the growth rate decreases exponentially owing to the
decreasing pyrolysis efficiency of the GaCl and NH3. On the contrary, at higher
substrate zone temperatures, thermally induced decomposition of reactants reduces
the growth rate. The hydrogen ambience also aids this reduction in that competing
processes such as GaHx would take place.
The main issue of concern associated with HVPE and other nitride growth
methods is that the initial nucleation layer on sapphire substrates determines to a
larger extent the material properties of the subsequent epitaxial layer than in other
methods [142–144]. The nucleation or the prelayer is typically deposited using a GaCl
or NH3 pretreatment consisting of flowing GaCl or NH3 over the sapphire surface
prior to the initiation of growth at high temperatures. In other cases, a ZnO wetting
layer is used [127,145,146]. Nitridation of sapphire has been mentioned as a means to
improve HVPE materials by several groups [147,148].
3.4 Nitride Growth Techniques
Thermal dissociation of group V species during HVPE of GaAs leads to the
formation of As2 or As4 molecules, which remain volatile and chemically reactive and
thus participate in the film growth. However, in the case of GaN, by-products of
decomposed NH3 are N2 and H2, and N2 molecules that are stable and unreactive at
the temperature of interest. Other forms of N species such as NCl3 are not
considered, as they are explosive. Moreover, there is a strong thermodynamic driving
force for forming parasitic gas-phase reactions, which cause deposition on the walls,
making the growth mechanism difficult to unravel. The process also tends to produce
large amounts of NH4Cl, GaCl3, and GaCl3-NH3, which condense and clog the
exhaust lines unless they are heated to sufficiently high temperatures (>150 C).
Exchange reactions with the hot quartz walls of the reactor make it difficult to use
HVPE for aluminum- and magnesium-bearing compounds required for AlGaN
growth and efficient p-doping, respectively.
Endemic to MBE and not considered that important in the case of vapor phase
deposition techniques until the advent of GaN is the concept of kinetics, involving
adsorption and desorption discussed in detail for OMVPE in Section 3.4.1.3 and for
MBE in Section 3.4.2. Adsorption and desorption processes depend on the kinetics of
the gaseous species on the substrate surface and the diffusion kinetics of the adatoms
or admolecules before arriving at the incorporation sites dubbed half-crystal or K sites
(see Figure 3.40) [104]. To put it simply, kinetics consists of complex but simplified
approaches that can be considered to get insight into the growth resulting from two
superficial diffusion flows, namely, the NGa admolecules and the other from the
NGaCl admolecules. Near the step edges, only the NGa flow directly leads to
incorporation through desorption of chlorine from the NGaCl. Because GaN growth
relies on GaCl, which is identical to that in GaAs, much of the GaN work benefited
from systematic investigations performed for GaAs.
Among the studies of GaAs growth were those reported by Shaw [149,150] in a
series of articles including a systematic measure of the growth rate of GaAs on {0 0 1},
{1 1 1}A, {1 1 1}B, and {1 1 0} surfaces (using hydrogen as carrier gas and the chloride
Vapor phase
AB
AB+B
Vapor phase
followed by
diffusion
Adsorption
followed by
diffusion
B*
A
AB*
Dissociation A
A*
Su bstra
A
te
* Adsorbed species
Surface diffusion
Figure 3.40 Kinetics processes occurring from the growth by
vapor phase. Patterned after Ref. [104].
j389
j 3 Growth and Growth Methods for Nitride Semiconductors
390
method) as a function of temperature and the GaCl partial pressure. The nitride
growth rate in HVPE exhibits an increase followed by a decrease in the growth rate
with decrease in substrate temperature. The latter was found consistent with
Langmuir GaCl adsorption isotherm together with a kinetic model based on GaCl
adsorption [151,152]. In this treatment, the lateral interaction between GaCl adsorbed
molecules and the Cl desorption by hydrogen of adsorbed GaCl molecules (dubbed
the H2 mechanism) are invoked to account for the slight decrease in the GaAs growth
rate at low temperatures when the GaCl partial pressure is increased. Hollan and
Schiller [153,154] and Hollan et al. computed surface diffusion, a component of
kinetics at play, by fitting the experimental data and computed values as a function of
the substrate orientation, and of Gentner [155] on 6 off {0 0 1} GaAs substrates. In
the latter study, both atmospheric and reduced pressures in hydrogen and helium
carrier gases were considered. These studies were also expanded to include AsCl3
instead of HCl gas. A desorption mechanism for two adsorbed chlorine atoms by
GaCl in GaCl3 was, therefore, considered with an intermediate GaCl3 adsorption
step [139]. This mechanism is called the GaCl3 mechanism.
Cadoret [156] developed a model involving adsorption and desorption of species on
the surface of the substrates. Among the adsorbing species considered are NH3
molecules, adsorption of N atoms resulting from NH3 decomposition, and adsorption of GaCl molecules on N atoms. They follow the reactions:
V þ NH3 ðgÞ()NH3 ;
3
NH3 ()N þ H2 ðgÞ;
2
ð3:11Þ
N þ GaClðgÞ()NGaCl;
where (g) depicts the gas-phase species and V is a vacant site.
Cadoret also considered two desorption mechanisms of chlorine: that is, desorption in HCl vapor molecules following a surface reaction with H2 and desorption in
GaCl3 vapor molecules following adsorption of a GaCl molecule on two GaCl
underlying molecules, which is schematically shown in Figure 3.41. The representation is based on the premise that the substrate surface, which is sapphire, is
terminated with Al atoms on which N atoms bond leading to a Ga polarity sample. As
in the GaAs model [139], the processes follow the reactions:
2NGaCl þ H2 ðgÞ()2NGa þ 2ClH;
NGa ClH()NGa þ HClðgÞ:
ð3:12Þ
The two mechanisms are labeled H2 and GaCl3 mechanisms. They can be treated by
means of a one-monolayer model of adsorption on a (0 0 0 1) Ga or Al surface. The
adsorbed species are NH3 molecules, N atoms, NGaCl, NGa–ClH, and 2N Ga–GaCl3
molecules. The one-monolayer adsorption model and the Bragg–Williams approximation are used to simplify the problem. The number of activated molecules involved
in the reactions described by Equations 3.11 and 3.12 as well as possible intermediate
states of hydrogen desorption from NH3 are neglected. GaCl adsorption on a Ga
adatom [157,158] is assumed to be negligible because it would lead to antisite positions
following chlorine desorption or would act as a simple inhibitor of the deposition
3.4 Nitride Growth Techniques
Ga
N
H
H
Cl
Cl
Ga
Ga
N
N
Cl
Ga
NH3
N
N
N
V
(a)
Cl
Ga
Ga
N
N
Cl
Cl
Ga
Ga
N
Cl
Ga
N
NH 3
N
N
V
(b)
Figure 3.41 Schematic steps of adsorption and desorption
processes involved in the (a) H2 mechanism and (b) GaCl3
mechanism. Patterned after Ref. [104].
process that is not necessarily observed in GaAs. The two overall reactions corresponding to the H2 and GaCl3 growth mechanisms can be written as
V þ NH3 ðgÞ þ GaClðgÞ()NGa þ HClðgÞ þ H2 ðgÞ;
2V þ 2NH3 ðgÞ þ 3GaClðgÞ()2NGa þ GaCl3 ðgÞ þ 3H2 ðgÞ:
ð3:13Þ
The adsorption and desorption processes including their flux, potential barriers
associated with their activation barriers, are somewhat involved and are beyond the
scope of the present treatment. An in-depth treatment of the topic can be found in
Ref. [104]. In addition to adsorption and desorption, quantification dealing with
growth by HVPE also involves mass transport, which is then followed by growth. In the
mass transport case, partial pressures of reactant species are treated that, depending
on local conditions and the substrate area, can lead to production or depletion of
species. Coupled with the treatment of mass transport, the growth phase, which
involves equilibrium between the vapor and substrate surface, must be treated. To get a
handle on the problem, superficial flow of diffusive molecules toward step edges,
which are assumed to be monomolecular in height, and surface coverage of vacant
sites, their distribution over the substrate surface (assumed uniform), are considered.
So is whether the growth is based on GaCl3 or H2 growth mechanisms. When the
terrace width is small enough to not hinder surface diffusion, the parameters of
importance are the adsorption energies of GaCl(g), HCl(g), and GaCl3(g). Further
details can be found in Ref. [104]. Those who are more focused on the growth- and
properties-related issues involving GaN by MBE are referred to Ref. [159].
j391
j 3 Growth and Growth Methods for Nitride Semiconductors
392
Summarizing, the growth process of GaN by HVPE could be analyzed by a
combination of thermodynamical and kinetic considerations. The ample experimental data available in the literature regarding the growth rate on exact and misoriented
{0 0 1} surfaces including those measured by Seifert [111] in He and H2 environments on 3 off (0 0.1), in the hkl configuration and (0 0 0 1) in the hjkl configuration,
GaN by HVPE allowed for modeling and understanding the physical processes
involved in growth. Relative kinetics considerations would indicate that the mass
transfer is larger for GaN than for GaAs. Additionally, the high supersaturation, in
effect, generally leads to important parasitic GaN deposition before the substrate
zone, or the deposition zone, that reduces and even could potentially negate the
relative supersaturation. The accuracy with which the growth rate is measured and
controlled is the key to the study of the thermodynamics of the system.
The variants of HVPE have also been explored and used for growth of GaN and its
ternaries with AlN. Among them is a modified VPE process, dubbed the sublimation
sandwich method (SSM), which was reported by Wetzel et al. [160] and Fischer
et al. [161] (Figure 3.42). Initially, the GaN films were grown from metallic Ga and
ammonia on (0 0 0 1) 6H-SiC, using a modification of the sandwich method described
previously by Vodakov et al. [162]. In this approach, the quartz reactor contains a Ga cell
for each substrate for multiwafer processing with one ammonia stream only. The gap
between the substrate and the Ga source is typically about 5 mm. The ammonia flow
rate through the gap is very high, 25–50 l m1 at atmospheric pressure. Under these
conditions, there is an effective mass transport of Ga vapor and nitrogen to the surface
of the substrates. At growth temperatures between 1170 and 1270 C, GaN layers
were obtained at growth rates of up to 0.3 mm h1.
Many other variations of this approach, including [163–171] gaseous sources such
as GaCl, GaCl2, GaCl3, Ga(C2H5)2Cl, GaCl2NH3, AlC3, AlBr3, InCl3, and GaBr3 for
group III element(s) as reactants for NH3, have been used. Pastrnak et al. [172] chose
to react N2 with GaCl3, AlCl3, and InCl3 in their CVD process. Dryburgh [173] grew
RF Coils
Water
Graphite
Substrate
NH3
ΔT
Ga
Graphite
Water
Figure 3.42 Schematic diagram of a proximity HVPE vessel for the
growth of GaN at very high growth rates, approaching
0.3 mm h1. It is dubbed the SSM.
Quartz
3.4 Nitride Growth Techniques
AlN from AlSe and N2. By introducing PH3, Igarashi et al. [174] achieved several
percent P incorporation in GaN. The technique became popular because of the highquality buffer layers on the freestanding GaN templates for epitaxy by heterostructure
deposition systems such as OMVPE and MBE [175]. The transport and optical
properties of HVPE-grown films are discussed in Volume 2, Chapters 3 and 5,
respectively.
3.4.1.2 Organometalic Vapor Phase Epitaxy
High-quality epitaxial III-N films and heterostructures for devices have been accomplished by OMVPE technique. Manasevit et al. [176] applied this technique to the
deposition of GaN and AlN in 1971. Using triethylgallium (TEG) and ammonia
(NH3) as source gases for group III and V species, respectively, the authors obtained
c-axis oriented films on sapphire (0 0 0 1) and on 6H-SiC(0 0 0 1) substrates.
 0
0
advertisement
![Structural and electronic properties of GaN [001] nanowires by using](http://s3.studylib.net/store/data/007592263_2-097e6f635887ae5b303613d8f900ab21-300x300.png)
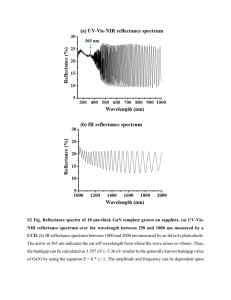



Download
advertisement
Add this document to collection(s)
You can add this document to your study collection(s)
Sign in Available only to authorized usersAdd this document to saved
You can add this document to your saved list
Sign in Available only to authorized users