Rate Laws
Chemical Reaction Engineering I
Booker T. Washington
“Success is measured not so
much by the position one
has reached in life, as by
the
obstacles
one
has
overcome while trying to
succeed”
An African-American Scholar and Inventor
Reactor Size
Design Equations
Batch
t N A0
CSTR
V
PFR
PBR
PFR
X
0
Levenspiel plot
dX
rAV
FA0 X
rA
V FA0
dX
rA
X
0
W FA0
X
0
V FA0
X
0
Graphical
method
dX
rA
dX
rA
rA f ( X )
FA0
For rA vs. X, the volume of a
CSTR and the volume of a PFR
can be represented as the shaded
areas in the Levenspiel plots.
If we know the molar flow rate to the
reactor and the reaction rate as a
function of conversion, then we can
calculate the reactor volume necessary
to achieve a specific conversion.
Design Isothermal Reactor
CHAPTER 2
CHAPTER 4
-rA=f(X)
CA = f(X)
CHAPTER 3
Rate Laws
rA kC C
a
A
b
B
stoichiometry
NA = f(X)
V= f(X)
Seoul National University
3.1.1 Relative Rates of Reactions
If the rate law depends on more than one species, we MUST relate the
concentrations of different species to each other. A stoichiometric table presents
the stoichiometric relationships between reacting molecules for a single reaction.
aA + bB
cC + dD
(2-1)
In formulating our stoichiometric table, we shall take species A as our basis of
calculation (i.e., limiting reactant) and then divide through by the stoichiometric
coefficient of A. In order to put everything on a basis of “per mole of A.”
A
b
c
d
B
C D
a
a
a
(2-2)
The relationship can be expressed directly from the stoichiometry of the reaction.
(3-1)
3.1.1 Relative Rates of Reactions
2 NO + O2
rNO
2
2 NO2
rO2
1
rNO2
2
aA +bB
cC + dD
r
rA
r
r
B C D
a b c
d
If rNO2= 4 mol/m3/s, (formation of NO2)
Then
rNO= -4 mol/m3/s (disappearance of NO)
rO2= -2 mol/m3/s (disappearance of O2)
3.2 Reaction Order and Rate Law
Let’s take A as the basis of calculation
a species A is one of the reactants that is disappearing as a result of
the reaction. The limiting reactant is usually chosen as our basis
for calculation.
The rate of disappearance of A, -rA, depends on temperature and
concentration and it can be written as the product of the reaction
constant k and
rA (T , C ) k A (T ) f (C A , CB ...)
Rate raw (Kinetic expression) : the algebraic equation that relates –rA to the species
concentration
3.2.1 Power Law Models and Elementary Rate Laws
The dependence of the reaction rate –rA on the concentration of
the species is almost without exception determined by
experimental observation.
The order of a reaction refers to the powers to which the
concentrations are raised in the kinetic rate law.
rA k AC ACB
(3-3)
order with respect to reactant A
order with respect to reactant B
n (=) : the overall order of the reaction
(concentration vs. activity)
ai (ai=iCi)
rA k Aa A aB
rA k A a A aB k A AC A B CB
k A A B C A CB k AC A CB
rA k AC ACB
kline and Fogler, JCIS, 82, 93 (1981), JCIS, 82, 103 (1981)
(3-3)
(concentration vs. activity)
Unit of Specific Reaction Rate
The unit of the specific reaction rate, kA, vary with the order of the reaction.
A products
k=
(Concentration)1-n
Time
Unit of Specific Reaction Rate
The unit of the specific reaction rate, kA, vary with the order of the reaction.
A products
0.
Zero - order :
rA k A
{k}
mol
(dm) 3 s
(3-4)
1.
First - order :
rA k AC A
{k}
1
s
(3-5)
2.
Second - order : rA k AC
(dm) 3
{k}
mol s
(3-6)
(dm 3 / mol ) 2
{k}
s
(3-7)
3.
Third - order :
2
A
rA k AC
3
A
Elementary and Non-elementary Reaction
Kinetic rate raw
“Elementary reaction”
“Non-elementary reaction”
O + CH3OH CH3O + OH
CO + Cl2 COCl2
-rO = k CO CCH3OH
3/ 2
rCO kCCOCCl
2
1st order w.r.t. atomic oxygen
1st order w.r.t. carbon monoxide
1st order w.r.t. methanol
3/2 order w.r.t. chorine
overall is 2nd order reaction
overall is 5/2 order reaction
In general, first- and second-order reactions are more commonly observed.
Elementary Reaction
An elementary reaction is one that evolves a single step such as the
bimolecular reaction between oxygen and methanol
O + CH3OH
CH3O + OH
The stoichiometric coefficients in this reaction are identical to the
powers in the rate law.
-r O = k CO C
The reaction is 1st order in oxygen free radical and 1st order in
methanol; therefore, we say both the reaction and rate law are
elementary.
Apparent Reaction Order
Sometimes reactions have complex rate expressions that cannot be separated into
solely temperature-dependent and concentration-dependent portions.
Pt
2N2O
rN 2O
2N2 + O2
k N 2O C N 2O
CO2
1 k NO
Kinetic rate raw
Limiting conditions: depending on oxygen concentration
(1) 1 k CO2 : rN2O k N2OCN2O
(2) 1 k CO2 : rN 2O
k N 2O C N 2O
k CO2
“apparent” 1st-order w.r.t. N2O
1st order overall
“apparent” reaction-order
-1 w.r.t. O2 , +1 w.r.t. N2O,
overall apparent zero order
3.2.2 Non-elementary Rate Laws and Reactions
H2 + Br2
2HBr
Non-elementary reaction (free radical)
rH 2
1/ 2
k1CH 2 CBr
2
k 2 CHBr / CBr2
(3-8)
CH3CHO
CH4 + CO
Non-elementary reaction (free radical)
gas-phase decomposition of acetaldehyde
@500oC
3/ 2
rCH3CHO kCCH
3CHO
3.2.2 Nonelementary Rate Laws and Reactions
In many gas-solid catalyzed reactions, it is sometimes preferable to write the
rate law in terms of partial pressures rather than concentrations.
C6H5CH(CH3)2
cumene
C6H6 + C3H6
benzene
C
propylene
B + P
k ( PC PB PP / K P )
rC
1 K C PC K B PB
KP = the pressure equilibrium constant [atm]
KB = the adsorption constants [atm-1]
k = the specific reaction rate [mol/kg cat·s ·atm]
LangmuirHinshelwood
kinetics
3.2.2 Nonelementary Rate Laws and Reactions
At equilibrium, -r’C =0; the rate law for the reversible reaction is indeed
thermodynamically consistent:
rC 0
k ( PC PB PP / K P )
1 K C PC K B PB
Solving for Kp yields
PBe PPe
KP
PCe
which is identical to the expression obtained from thermodynamics.
To express the rate of decomposition of cumene, -r’C as a function of conversion,
replace the partial pressure with concentration, using the ideal gas law:
pC CC RT
and then express concentration in terms of conversion.
Determination of Reaction Rate Law
It is important to remember that the rate laws are determined by
experimental observation! They cannot be deduced from reaction
stoichiometry.
They are function of the reaction chemistry and not the type of
reactor in which the reactions occur.
Even though a number of reactions follow elementary rate laws, at
least as many reactions do not. One must determine the reaction
order from the experiments or from literature.
Where do you find rate laws?
The activation energy, frequency factor, and reaction order
Floppy disks and CDROMs by National Institute of Standards and Technology (NIST)
Standard Reference Data 221/A320 Gaithersburg, MD 20899
Tables of Chemical Kinetics: Homogeneous Reaction,
National Bureau of Standards Circular 510 (Sept. 1951)
Suppl. 1 (Nov. 14, 1956), Suppl. 2 (Aug. 5, 1960), Suppl. 3 (Sept. 15, 1961)
Washington, D.C., U.S. Government Printing Office
Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling,
Evaluate No. 10, JPL Publication 92-20, Aug. 15, 1992, Jet Propulsion
Laboratories, Pasadena, CA, USA
International Journal of Chemical Kinetics, Journal of Physical Chemistry
Journal of Catalysis, Journal of Applied Catalysis
AIChE Journal, Chemical Engineering Science, Korean Journal of Chemical Engineering
Chemical Engineering Communications
Industrial and Engineering Chemistry Research
Example of Rate Law
Example of Rate Law
Example of Rate Law
A. First Order Reaction
B. Second Order Reaction
3.2.3 Reversible Reactions
All rate raws for reversible reactions must reduce to the
thermodynamic relationship relating the reacting species
concentrations at equilibrium. At equilibrium, the rate of reaction is
identically zero for all species (i.e., -rA=0). For the general reaction
aA + bB
cC + dD
The concentrations at equilibrium are related by the thermodynamic
relationship
c
d
CCe
CDe
KC a b
C Ae CBe
[(mol / dm3 ) d c b a ]
3.2.3 Reversible Reactions
2C6H6
C12H10 +H2
2B
D +H2
The rate of disappearance of benzene
2C6 H6
kB
C12 H10 H2
The rate of formation of benzene
B
C12 H10 H 2 k
2C6 H 6
rB ,reverse k B C D CH 2
rB , forward kB CB2
The net rate of formation of benzene
rB rB ,net rB , forward rB ,reverse
rB k B C k B CDCH 2
2
B
3.2.3 Reversible Reactions
The rate law for the rate of disappearance of benzene
rB k B C k B C D C H 2
2
B
2 k B
k B C B
C D C H 2
kB
2 CDCH 2
rB k B C B
KC
k BB
K C concentrat ion equilibriu m constant
k B-B
thermodynamically consistent with the equilibrium constant
2B
D +H2
We need to check
2 CDCH 2
rB k B CB
KC
KC
CDeCH 2e
2
CBe
whether the above rate law is thermodynamically consistent at equilibrium.
At equilibrium, -rB=0,
2 CDeCH 2e
rB 0 k B CBe
KC
Rearranging, we obtain
KC
CDeCH 2e
2
CBe
identical
Thermodynamically Consistent with Equilibrium Constant
2B
D +H2
The rate of formation of diphenyl is
2 CDCH 2
rD k D CB
KC
(3-15)
Using the relationship
rC rD
rA
rB
a b c
d
We can obtain the relationship between the various specific reaction rates,
kB, kD:
rD
rB
k B 2 CDCH 2
CB
1 2 2
KC
kB
kD
2
(3-16)
Temperature Dependence of Concentration Equilibrium Constant
H Rx
K C (T ) K C (T1 ) exp
R
Appendix C, Eq (C-9)
KC
Exothermic
reaction
T
1 1
T1 T
KC
Endothermic
reaction
T
3.3
k : The specific Reaction Rate (the Rate Constant)
A
The specific reaction rate is always referred to that species in the
reactions and normally should be subscripted w.r.t. that species. The
reaction rate constant k is not truly a constant, but is merely
independent of the concentrations of the species involved in the
reaction.
The quantity k is also referred to as the specific reaction rate (constant).
It is almost always strongly dependent on temperature. In gas-phase
reactions, it depends on the catalyst and total pressure. In liquid
systems, it depends on the total pressure, ionic strength and choice of
solvent.
These other variables normally exhibit much less effect on the specific
reaction rate than temperature does with the exception of supercritical
solvents, such as supercritical water.
In this text, it will be assumed that kA depends only on temperature.
Arrhenius equation
Activation energy, J/mol or cal/mol
Specific reaction rate (constant)
k A (T ) Ae
E
RT
Absolute
Temperature, K
frequency factor
or
pre-exponential
factor
mathematical number
e=2.71828…
Gas constant
8.314 J/mol · K
1.987 cal/mol · K
8.314 kPa · dm3/mol · K
Arrhenius equation
k A (T ) Ae
A
E
RT
high E
kA
kA
Low E
Slope= -E/R
T (K)
T 0, kA 0
T , kA A
1/T (K-1)
SVANTE AUGUST ARRHENIUS
1884 (25) : Ph.D. “ionic theory”
1887 (27) : work with Boltzmann
1888 (28): work with Ostwald and Van't Hoff
1889 (30) : introduced the concept of activation
energy as the critical energy that
chemicals need to react
1903(44) : Nobel Prize in chemistry for ionic theory
prove the influence of the electrolytic dissociation on the
osmotic pressure, the lowering of the freezing point and
increase of the boiling point of solutions containing
electrolytes. He also pointed out the existence of a
"greenhouse effect" in which small changes in the
concentration of carbon dioxide in the atmosphere could
considerably alter the average temperature of a planet.
1859 -1927
SVANTE AUGUST ARRHENIUS
Svante August Arrhenius was born in Vik, Sweden in 1859. At age 25 he turned in his PhD thesis at
the University of Uppsala, Sweden. His PhD examining committee did not think very highly of his
thesis and rated it 4th class. His oral thesis defense did not fair much better as they rated it as only
3rd class. Arrhenius left Sweden for five years to work with Oswald, Boltzmann and van't Hoff. In
1889 his interpretation of temperature-dependent equation by van't Hoff led to the universal
accepted Arrhenius equation for kinetic rate laws in chemistry. He received the Nobel Prize in 1903.
From 1905 until his death in 1927 he was director of Physical Chemistry at the Nobel Institute
k A (T ) Ae
E
RT
Activation energy
Activation energy E :
a minimum energy that must be possessed by reacting
molecules before the reaction will occur.
e
E
RT
The fraction of the collisions between
molecules that together have this minimum
energy E (the kinetic theory of gases)
Activation energy E is determined experimentally by
carrying out the reaction at several different
temperature.
E1
ln k A ln A
R T
Activation energy
A+BC
A-B-C
AB+C
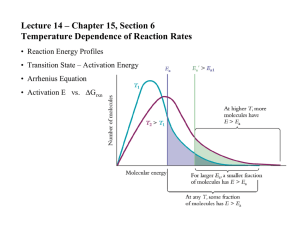
Energy distribution of reacting molecules
f(E,T)
EA
Energy distribution function
for kinetic energies
of the reacting molecules
Fraction of collisions that
have energy EA or great
Energy distribution of reacting molecules
Fraction of collisions at T2
that have energy EA or greater
Fraction of collisions at T1
that have energy EA or greater
Activation energy
Azo Dye
0~5oC
NO2
HNO3
3H2
NH2
Pd/C*
H2SO4
N=N-Cl
HNO3
HCl
Benzene
diazonium
Aniline
bp=184oC
0~5oC
0~5oC
OH
OH
OH
-N=N-N=N-
-OH
Yellow
(4-hydroxyl phenol) azobenzene
Bright red
mp=132oC
Ex. 3-1 Determination of the Activation energy
Decomposition of benzene diazonium chloride to give chlorobenzene and N2
Cl
Cl
N=N
+ N2
Calculate the activation energy using following information for this first-order
E 1
log k A log A
2.3R T
T(K)
313
319
323
328
333
-1
k (s )
0.00043
0.00103
0.00180
0.00355
0.00717
-1
1/T (K )
0.003195
0.003135
0.003096
0.003049
0.003003
-1
k (s )
0.00043
0.00103
0.00180
0.00355
0.00717
Finding the activation energy
Plot (log k) vs (1/T)
k (sec-1)
0.01
0.001
Slope = -E/R
0.0001
0.0030
0.0031
1/T (K-1)
0.0032
Finding the activation energy
Plot (log k) vs (1/T)
E 1
log k1 log A
2.3R T1
When k1=0.005 : 1/T1=0.003025
When k2=0.0005 : 1/T2=0.00319
E 1
log k 2 log A
2.3R T2
Therefore,
k2
E 1 1
log
k1
2.3R T2 T1
(2.3)( R ) log( k 2 / k1 )
E
1 / T2 1 / T1
(2.3)( R) log( k 2 / k1 )
E
1 / T2 1 / T1
(2.3)(8.314 J / mol K )(1)
(0.00319 0.003025) / K
116.5 kJ / mol 28.7 kcal / mol
To use the decade method,
choose 1/T1 and 1/T2 so that k2=0.1 k1.
ln A 37.12
Then, log(k1/k2)=1
14017 K
k 1.32 1016 EXP
T
A 1.32 1016 s 1
Finding the activation energy
Plot (ln k) vs (1/T)
0.01
k (sec-1)
0.005
0.001
0.0005
14017 K
k 1.32 1016 EXP
T
0.0001
0.0030
0.003025
0.0031
1/T (K-1)
0.0032
0.00319
Activation energy
The larger the activation energy,
the more temperature-sensitive
is the rate of reaction.
Frequency factor ~ 1013 s-1
Activation energy ~ 300 kJ/mol
k (sec-1)
Typical values of 1st order gasphase reaction
0.01
0.001
Low E
High E
0.0001
0.0030
0.0031
1/T (K-1)
0.0032
Specific reaction rate
k Ae
k0 Ae
E
RT
E
RT0
Taking the ratio
E 1 1
k k 0exp
R T0 T
3.4 Present Status of Our Approach to Reactor Sizing and Design
Design Equations
Differential
form
Batch
N A0
dX
rAV
dt
PBR
Integral
form
t N A0
dX
rAV
X
0
FA0 X
V
rA
CSTR
PFR
Algebraic
form
FA0
dX
rA
dV
FA0
dX
rA
dW
V FA0
X
0
W FA0
X
0
dX
rA
dX
rA
Reactor Size
Design Equations
Batch
t N A0
CSTR
V
PFR
PBR
PFR
X
0
Levenspiel plot
dX
rAV
FA0 X
rA
V FA0
dX
rA
X
0
W FA0
X
0
V FA0
X
0
Graphical
method
dX
rA
dX
rA
rA f ( X )
FA0
For rA vs. X, the volume of a
CSTR and the volume of a PFR
can be represented as the shaded
areas in the Levenspiel plots.
If we know the molar flow rate to the
reactor and the reaction rate as a
function of conversion, then we can
calculate the reactor volume necessary
to achieve a specific conversion.
Design Isothermal Reactor
CHAPTER 2
CHAPTER 4
-rA=f(X)
CA = f(X)
CHAPTER 3
Rate Laws
rA kC C
a
A
b
B
stoichiometry
NA = f(X)
V= f(X)
• 3.1.Rate Laws and Stoich_CHAPTER 3
FINAL.pptx
• 3.2.Nonelementary rxns_Bab 7.pptx