ELECTROCHEMICAL ANALYSES AND REACTIONS OF MALONONITRILE DERIVATIVES AND PHENYL AZIDE
advertisement

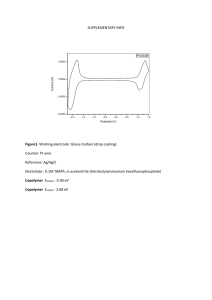
ELECTROCHEMICAL ANALYSES AND REACTIONS OF MALONONITRILE DERIVATIVES AND PHENYL AZIDE A RESEARCH PAPER SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE MASTERS OF ART BY JINYU LIU DR. CHONG ‐ ADVISOR BALL STATE UNIVERSITY MUNCIE, INDIANA JULY 2013 CONTENT Abstract 1 Introduction 1 Material 3 4-methoxybenzalmalonoitrile 3 Phenyl azide: 4 Set up 6 Experiment 6 1. 4-methoxybenzalmalonoitrile 6 a. Low concentration experiment to check and determine the potential of 4-methoxybenzalmalononitrile. b. High concentration experiment to confirm the reaction and the product. c. Product isolation. 6 8 10 2. Electronic effect for different substituted group 12 3. Phenyl azide reaction. 15 Result 16 Acknowledgement 18 Reference 19 Abstract: The versatile and relatively inexpensive (low energy cost) synthesis of new organic molecules is important for pharmaceutical production, polymerization, and other industrially applicable facets of chemistry. Electrochemical reduction of unsaturated organic compounds at room temperature to new organic products offers a benign and ambient method for the production of such molecules. The compounds, (p-methoxybenzal)malononitrile, 1 , E pc = -1.64 V vs. Ferrocene/Ferrocenium (Cp 2 Fe0/+), its derivatives and phenyl azide, 2 , E pc = -2.43 V vs. Cp 2 Fe0/+, were investigated for their redox activities with and without alkylation agents, R-X, via scanning and pulse voltammetric techniques in acetonitrile, acetonitrile/water and tetrahydrofuran as solvents, using 0.1 M [NBu 4 ][PF 6 ] as the supporting electrolyte. The redox potentials were measured at glassy carbon and platinum disk electrodes. Controlled potential electrolysis (CPE) of 1 at E appl = -1.75 V vs. Cp 2 Fe0/+ was yielding p-methoxybenzaldehyde (> 90%). CPE of 2 at E appl = -2.54 V vs. Cp 2 Fe0/+ in air gave aniline in good yield. Bulk cathodic electrolyses of 1 and 2 were exhausted in < 0.5 - 1 h, following the passage of 1 F/mol of analyte. Upon completing bulk reductions at each applied potential, products were characterized by 1H-NMR and GC-MS data analyses. Introduction: Nowadays, the C-C bonds cleavage and the C-C bonds coupling are top prior in the organic field. However, in the traditional way, the synthesis of aldehyde from olefin is an oxidation reaction. In the presence of PdCl 2 (MeCN) 2 , 1, 4-benzoquinone, and t-BuOH, aryl-substituted olefins can selectively be oxidized to aldehydes. The reaction was as followed. [1] 1 The use of PhI(OAc) 2 in dichloromethane enables a clean oxidative cleavage of 1,2-diols to aldehydes. Using OsO 4 as catalyst, NMO and 2,6-lutidine, olefinic bonds can be cleaved in acetone/water to yield the corresponding carbonyl compounds. [2] Based on the both catalytic reaction, in the reaction, expensive catalyst was used. One reaction is in the high temperament and another one need a long time to get a good yield. Also, the hydration can be the first step, and followed by the oxidation to get the aldehyde from olefin. The olefin was hydrated to alcohol. Then the alcohol was oxidized to aldehyde. H+ + H2O OH [O] OH O From the above reaction mechanism, after hydration of olefin, only ketone produced, and it could not cleave the C=C double bond. Depend on the traditional organic synthesis reaction, it will spends lots of time and energy especially in the conjugated structures, like the compound in this project, the olefin double bonds conjugated with the phenyl group which stabilized the C=C bond. In the previous research, to make the 2 p-methoxybenzalmalononitrile, p-methoxybenzaldehyde and malononitrile are reactants, but no research shows the inverse reactions. However, after checking the potential of p-methoxybenzaldehyde, one cathodic peak is shown in the cyclic voltammetry graph that means some reduction reactions can happen on this compound by using bulk electrolysis on Pt electrode under the room temperature with less than 1h. There is no previous research to indicate the cathodic reduction induced hydration of olefin. Based on the reaction of hydration of benzalmalononitrile, the CS-gas which is in the similar family can be easy to decompose into nontoxic compounds. In the organic synthesis, it is an important reaction to reducing azide to amines. The methods for converting the azide to amines contain catalytic hydrogenation [3], the phosphine-based Staudinger reduction [4] and the metal hydride reduction [5]. However, for these methods, they have significant limitation. [6] In electrochemistry, voltammetries are important tools to show the nature of the compound based on different solvents. The solvent effect can affect the behavior of compounds depended on the donor number and the dielectric constant. [7] The voltammetries can be related to the thermodynamic and kinetic properties of the compounds. Bulky electrolysis is a normal tool in electrochemistry to make the reaction happen. Materials: 4-methoxybenzalmalonoitrile: Light yellow solid 0.0745g; b.p. less than 220 oC IR (acetonitrile, cm-1) 3621 (w, hydrocarbon), 2225 (s, nitrile), 1514(s, aromatic double 3 bond), 1038 (m, C-O group) 1 H-NMR (400MHz, acetonitrile) δ 7.95 (singlet, 1H [A]), 7.94 (doublet, 1H [B], J=8.8Hz), 7.10 (doublet, 1H [C], J= 9.16), 3.88 (singlet, 3H [D]) [M+1]=185. B C H H NC D CN H3CO H C H B A H It is a family member as the malononitrile, which also has the cyanocarbon group. It can be synthesized by the Knoevenagel condensation. [8] Take the CS gas as an example: The reaction is catalysed with weak base like piperidine or pyridine. The production method has not changed since the substance was discovered by Corson and Stoughton. Other bases, solvent free methods and microwave promotion have been suggested to improve the production of the substance. Phenyl azide: It is an organic compound with the formula C 6 H 5 N 3 . It is one of the prototypical 4 organic azide. It has a pungent odor. The structure consists of a linear azide substituent bound to a phenyl group. Molecular formula C6H5N3 Molar mass 119.12 g/mol Appearance Pale yellow, oily liquid Boiling point 49 °C at 5mm Hg Main hazards Explosive N3 Fig 1: MS spectrum for phenyl azide. [9] 5 Setup: The photo above is a sample of the e-chem cell which had been set up. Working electrode as 2mm Pt or 2mm glassy carbon electrode was placed in the middle which was C, the reference as Ag/ AgCl was placed in part A, and the counter electrode as Pt was placed in part B. Though the change of the potential and current by using the potentialstat, it shows the graph for the voltammetry to get and check the potential of the analysts and using that potential to do the electrolysis to make some unknown reaction in the e-chem cell. Experiment: 1. 4-methoxybenzalmalonoitrile reaction. a. Low concentration experiment to check and determine the potential of 6 4-methoxybenzalmalononitrile. a E pc = -1.70V 5μA i ca background pre-electrolysis i an post-electrolysis b 2 1 0 -1 E (V vs -2 -3 Cp2Fe+/0) Fig2. CV scan for 2m M of 1 in acetonitrile with 0.1M [NBu 4 ][PF 6 ] as electrolyte at room temperature, the scan rate was 200mV/s on 2mmPt electrode. From a which is pre-electrolysis, it showed the peak potential of sample was 1.7V vs ferrocene on Pt electrode which has a huge peak, and post-electrolysis (b), when it ran in the same potential range, the huge peak disappeared, which may mean the starting material was reacted during the bulk electrolysis. i ca post-electrolysis i ca i an 2μA 0.5 0 E (V vs-0.5 Cp2Fe+/0) -1 -1.5 7 Fig3. CV scan for 2mM 1 post-electrolysis at room temperature in acetonitrile with 0.1M [NBu 4 ][PF 6 ] as electrolyte, the scan rate was 200 mV/s on 2mm Pt electrode. Post electrolysis, two new peaks showed up at -0.33V vs. ferrocene and 0.107V vs. ferrocene that it did not show up in the pre-electrolysis graph. It means that some unknown reaction happened and some new stuff came up. If all of the product can dissolve in the acetonitrile, it may two products there. Also, after electrolysis, the color of the solution in the working electrode turned to orange like the honey. SWV 1μA Half Width=130mV -1 -1.2 -1.4 -1.6 -1.8 E (V vs Cp2Fe+/0) -2 -2.2 -2.4 Fig4. Square wave voltammetry for 2mM 1 at room temperature with 0.1M [TBA][PF 6 ] as electrolyte on 2mm Pt electrode. This graph showed that the half potential of the sample was -1.65V vs. ferrocene, and by calculating the half width of the peak, it indicated that the reaction in the cell was one-electron transfer reaction whose half width was larger than 90mV. Seeing the SWV, it is found that the half width was 90mV, it is likely a one-electron transfer reaction. b. High concentration experiment to confirm the reaction and the product. 8 50 μA background i ca molanonitrile post-electrolysis i an 2 1 0 -1 E (V vs Cp2Fe0/+) -2 -3 Fig5: 30mM 1 with 0.109M [TBA][PF 6 ] in acetonitrile in the room temperature on 2mm Pt electrode. The scan rate was 200mV/s. For the high concentration, a little difference appeared by comparing the low concentration, at the end of range (about 1V vs. ferrocene) a reversible peak showed up. So the concentration may have some unknown effect on the reaction. And also at -2.5V vs. ferrocene, there was a new peak there. Also the two old peaks were at the simile place, because of the high concentration, the peak potential shifted a little. It showed the potential more clear in the graph blow. 9 i ca i an 10μA 1.5 1 0.5 0 -0.5 -1 E (V vs Cp2Fe0/+) Fig6: 30mM 1 post-electrolysis with 0.109M [TBA][PF 6 ] in acetonitrile in the room temperature on 2mm Pt electrode. The scan rate was 200mV/s. Two peaks potentials were -0.169V, 0.182V and the half potential for the last peak was 7.75V vs. ferrocene. Compared with the background, it means that after electrolysis, some unknown compound formed in the solution. c. Product isolation. After the solution was collected in a small vessel, use nitrogen gas to evaporate the solvent, stirring and keep the evaporation speed slowed to prevent to lose some of organic product which has a low boiling point. Then mix benzene (1st experiment) or ethyl ether (2nd experiment) with the residual. As we know, the electrolyte could not dissolve in both solvent. After mix very well, filter the solution through the silicon gel. What’s more, remove the solvent again to get the major product. By using the NMR and GC-MS, a major was fingered out which was 10 4-mehtoxybenzaldehyde. 4-methoxybenzaldehyde: light yellow liquid 0.0236 (yield 31.7%); b.p. 248 ℃ IR spectrum (dichloromethane, cm-1) 2926 (m, alkane), 1606 (m, CO double bond), 1464 (m, aromatic double bonds) 1 H-NMR (400MHz, chloroform) δ 9.886 (singlet, 1H [A]) 7.851 (doublet, 1H [B] J=8.8Hz) 7.015 (doublet, 1H [C] J=8.8Hz) 3.847 (singlet, 3H[d]) [M]=136 c H H b O d H3CO H a H c H b Fig7: MS spectrum for the isolated product. The molar mass is 136g/mol. 11 Fig8: NMR spectrum for the isolated product. The two spectrums above for NMR and GC-MS, show the evidence to prove the structure for my one major product. After calculation, the yield is around 80%. 2. Electronic effect for different substituted group. After the cathodic reduction of p-methoxybenzalmalononitrile, the methoxybenzaldehyde was formed in the solution with the potential (-1.64V vs. Ferrocene). However, in the benzalmalononitrile family, series of organic compounds with different substituted groups was shown the different voltammetry activity. So the electric effect would be investigated by using some various substituted groups in two different solvents by using two different electrodes (glassy carbon electrode and Pt electrode) Based on the different effects of the substituted group on the phenyl group, both activated and deactivated groups were investigated in three weeks. Also the position of substituted groups can stabilize or destabilize the conjugated C=C double bonds. 12 The structures of the five compounds were followed: CN NC NC CN MeO MeO OMe OMe p-methoxybenzalmalononitrile 3,4,5-trimethoxybenzalmalononitrile NC CN NC CN MeO OMe Cl m-chlorobenzalmalononitrile NC 3,4-dimethoxybenzalmalononitrile CN MeO p-methoxybenzalmalononitrile 13 b a 3.00E-05 3.00E-05 2.00E-05 2.00E-05 1.00E-05 1.00E-05 I (A) I(A) 0.00E+00 -1.00E-05 0.00E+00 -2.00E-05 -1.00E-05 -3.00E-05 -2.00E-05 -4.00E-05 -5.00E-05 2 1.5 0.5 1 0 -0.5 -1 -1.5 -2 -3.00E-05 -2.5 2 E (V vs. Ferrocene) 1.5 1 0.5 0 -0.5 -1 -1.5 -2 -2.5 -3 E ( V vs. Ferrocene) c 2.50E-05 2.00E-05 1.50E-05 I (A) 1.00E-05 5.00E-06 0.00E+00 -5.00E-06 -1.00E-05 2 1.5 1 0.5 0 -0.5 -1 -1.5 -2 -2.5 E ( V vs. ferrocene) Fig 9: CV scan for (a) 1mM 3,4,5-trimethoxybenzalmalononitrile (b) 1Mm 3,4-dimethoxymalononitrile (c) 1mM m-methoxybenzalmalononitrile. 0.1M [TBA][PF 6 ] was used as electrolyte in the solvent acetonitrile on 2mm GC electrode. The scan rates are 200mV/s. I checked the peak potentials and half potentials by using CVs, LSVs and DPVs for five different compounds by using the acetonitrile as the solvent and the [TBA][PF 6 ] as supporting electrolyte. 14 The data of the potentials as followed: Glassy Carbon Electrode Compound Platinum Electrode Epc E1/2. Half Peak Width Epc E1/2 Half Peak Width (V) (V) (V) (V) (V) (V) m-chlorobenzalmalononitrile -1.38 -1.32 0.0991 -1.42 -1.33 0.1023 p-methoxybenzalmalononitrile -1.62 -1.55 0.082 -1.72 -1.68 0.123 m-methoxybenzalmalononitrile -1.50 -1.43 0.086 -1.60 -1.49 0.16 3,4,5-trimethoxybenzalmalononitrile -1.53 -1.49 0.096 -1.53 -1.50 0.135 3,4-dimethoxybenzalmalononitrile -1.61 -1.56 0.087 -1.65 -1.56 0.099 Table1: voltammetry data for different substituted groups on benzalmalononitrile on two different electrodes. The data of potential was converted to use the ferrocene as a standard. 3. Phenyl azide reaction. 8.00E-05 6.00E-05 i ( A) 4.00E-05 2.00E-05 0.00E+00 -2.00E-05 -4.00E-05 2 1 0 -1 -2 -3 -4 E ( V vs ferrocene) Fig10: CV scan for 5mM phenyl azide in THF with 0.1M [TBA][PF6] as electrolyte on glassy carbon electrode. The scan rate was 200mV/s. In order to get the half potential of phenyl azide, which has an unsaturated group and may have some reaction on that group based on gaining electrons, the CV scan was used to check the half potential of this compound in THF. The cathodic peak potential is -3.01V vs. ferrocence. Then based on the same condition, LSV was used to check the half potential which was -2.4V vs. ferrocence. 15 2.50E-05 2.00E-05 i ( A) 1.50E-05 1.00E-05 E1/2 = -2.4V 5.00E-06 0.00E+00 -1 -1.5 -2 -2.5 -3 -3.5 -4 E ( V vs ferrocene) Fig 11: LSV for 5mM phenyl azide in THF with 0.1M [TBA][PF 6 ] as electrolyte on glassy carbon electrode. The scan rate was 1mV/s. However, the peak potential shifted a lot when the Glassy Carbon electrode was replaced by Pt electrode. The peak potential shifted to -3.31V vs. ferrocene After measuring the half potential of the phenyl azide, controlled potential electrolysis was running. Aniline was detected by GC-MS which was the product from one electron reaction, and another product was N 2 which escaped during the process of electrolysis. Result: Combining all the voltammetry graphs and the spectrums, for my starting material, it has some reaction in the cell, and one of the products, 4-methoxybenzalmalononitrile, formed. However, depended on some unknown reason, only one product can be detected. So the equation for the reaction is: 16 NC CN O CN e MeO - H2O + CN MeO Also, for the electronic effect of different substituted group on this family compound, the chloro is weakly deactivated group and the methoxy is strongly activated group. Based on the table, the compound with chloro group has a low half potential. However, for the chloro, the deactivated group is meta- directors, and the methoxy is ortho or para directors. The substituted group was used to stabilize the negative charge on the аC to make the starting material easy to get an electrons. So seeing from the methoxy group, the lowest one is m-methoxy because it only has one methoxy group on the phenyl group and the rest is on the meta-position. The largest one is p-methoxy because it has a activated group, but the rest was connect to the para-position. It decreases the resonance of the phenyl group and the olefin double bond to increase the energy which is need to adding the electrons into the bond. For the two other methoxy compounds, the meta position was decreasing the potential. The otho and para position was increasing the potential. Also, in these compounds, numbers of methoxy group has some effect on the potential shift. It can offset the part of the other position’s effect. After this, more substituted groups will be investigated and electrolysis will be used to investigate the electronic effect on the reaction product. For cathodic reduction reaction of phenyl azide, the aniline was detected in the product. The mechanism was followed. The phenyl azide gains one electron to form the 17 intermediate and release the nitrogen gas. Then gain two protons to form the aniline. [10] During the reaction, the [PhN]-. anion was formed. In the future, methyl iodide will be used to do the electrolysis with phenyl azide to try to make N-C bond. 18 Reference: 1. P. Teo, Z. K. Wickens, G. Dong, R. H. Grubbs, Org. Lett., 2012, 14, 3237-3239. 2. K. C. Nicolaou, V. A. Adsool, C. R. H. Hale, Org. Lett., 2010, 12, 1552-1555. 3. Johnstone RAW, Wilby AH, Entwistle ID. Heterogeneous catalytic transfer hydrogenation and itsrelation to other methods for reduction of organic-compounds. Chem. Rev. 1985; 85:129–170. 4. Gololobov YG, Kasukhin LF. Recent advances in the Staudinger reaction. Tetrahedron. 1992; 48:1353–1406 5. Scriven EFV, Turnbull K. Azides - their preparation and synthetic uses. Chem. Rev. 1988; 88:297–368. 6. Chen Y, Kamlet AS, Steinman JB, Liu, DR. A Biomolecule-Compatible Visible Light-Induce Azide Reduction from a DNA-Encoded Reaction Discovery System. Nat Chem. 2011, 3(2): 146-153. 7. Chong D, Slote J, Geiger WE. The Role of Solvent in Stepwise Electrochemical Oxidation of Nickelocene to the Nickelocenium Dication. J. Electroanalytical Chemistry 630 (2009) 28-.4 8. Pande A, Ganesan K, Jain AK, Gupta PK, Malhotr RC. Novel Eco-Friendly Process For the Synthesis of 2-Chlorobenzylidenemalononitrile and ITS Analogues Using Water as A Solvent. Org Proc Res Develop , 2005, 9(2): 133-136 9. NIST Chemistry Webbook (http://webbook .nist.gove/chemistry). 10. Takeuchi H, Koyama K. Photolysis and Thermolysis of Phenyl Azide in Acetic Acid. J.C.S.Chem. Comm., 1981, 202-204 19