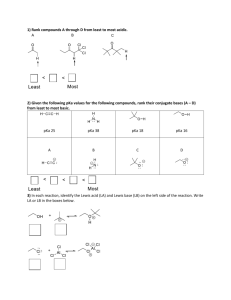
weak acids and bases
advertisement

Introduction to Biochemistry
Aim: to understand life on a molecular level
3 principle areas:
structure/function relationships
metabolism-chemical reactions
genetics
Biochemistry is interdisciplinary and is more
experim ental than theoretical
biochemist needs to understand:
techniques based on physics
basic chemical elem ents & structure of
biological compounds including:
-stoichiom etry
-m echanisms of reactions
-therm odynamics
Outline of first two lectures:
•size scales- biochemical events
depend on the structure of large
molecules and assemblies of
molecules
•weak interactions- the structures
depend on weak interactions (ie
weaker than those usually considered
in organic chemistry)
•ionic equilibria
•thermodynamics in biology- 2nd law
vs order in biological systems
Many important biologic al molecules are
polymers:
joining of prefabricated monomers
monomers of a given polym er have limited
diversity
monomers polymerized by identic al
mechanism to form covalent bonds
monomer
polymer
glucose
cellulose- homopolymer
Polysaccharides serve as
structural component & store
energy
nucleotides
(4 types)
nucleic acids- heteropolymers
RN A & DNA- storage &
transmission of genetic info
amino acids
(20)
Lipids
proteins or peptides-hetero
Greater structural & functional
diversity
-chemically diverse group
-low solubility in aqueous solution and
amphipathic nature results in formation of
particular structures
-major structural element in cell membranes
Range of Object Sizes of interest to Biochemists
and Techniques used to study them
Covalent vs Non-covalent bonds
some covalent bond energies (kJ/mole):
C-H
C-C
C C
C
C
C-O
O2
O-H
415
345
615
810
350
499
463
This is the energy required to break one mole of
chemical bonds of a particular type in the gas state.
In many cases, it is not very dependent on the
molecule.
Conjugated double bonds are an exception. These
bonds are more stable than those molecules
containing isolated C-C and C C bonds.
Example: For benzene, it takes ~ 158 kJ/mole more
energy than expected to dissociate due to resonance
energy which stabilizes the molecule.
Non covalent interactions
~ 10-100 times weaker than covalent
essential that they can be broken and reformed
consider Energy available in a typical biological
system (aqueous environment) to break/reform
non covalent interactions
RT =
the thermal energy of the “bath”
surrounding any molecule;
R = ideal gas constant (8.314 J/mol K)
T = 273 + 37 = 310 K
~ 2.5 kJ/mole
(not usually enough to break covalent bonds)
THUS, biochemical function has evolved to center
around “weak” or non covalent interactions
Weak interactions are fundamentally electrostatic
in nature:
depend on the forces that electrical charges or
dipoles exert on one another
Classification of Non Covalent Interactions
1. Charge-charge (ion-ion)
Coulomb’s Law: F = k q 1 q 2 / r 2
In vacuum
q = magnitude and sign of charge
r = distance between charges
k = proportionality constant (depends on units)
if charges are repulsive (++ or - -), F is positive
if charges are attractive (+ -), F is negative
in some medium: F = k q 1 q 2 / r 2
= dielectric constant of the medium
reflects the effect the medium has on separating
the charges
the medium shields the charges from each other
Relevant
’s:
vacuum
1
Hydrocarbon (organic solvent) 1-10
W ater
80
NOTE:
Force between 2 charges in non-polar environment;
for example, inside a biological membrane or inside a
protein (low
), can be much larger than in water
we are more concerned with changes in energy that
occur during interactions, integration yields...
energy of interaction: U(r) = k q 1 q 2 / r
the energy required to separate 2 charged particles
until r reaches infinity
when working in common units:
U(r) = 1389 q 1 q 2 / r (kJ/mole)
For: q in integral multiples of the electronic charge;
r in angstroms
eg. two electrons in vacuum separated by 1 have:
U = + 1389 kJ/mole (repulsion)
Moving on from:
to:
and:
1. charge-charge interactions
2. ion-dipole
3. dipole-dipole
what do we mean by dipole?
molecules that do not carry a net charge and have
an asymmetric internal distribution of charge are
called polar molecules and have a permanent
dipole moment ()
dipole mom ent-
µ = qx
the product of the magnitude of
the charge and the distance
separating the charges
where q = fractional (partial) charge
x = distance between the
charges (q- and q+) in the
molecule
eg. Carbon monoxide
q+
q-
C
O
is a vector, in your book directed towards the
partial + charge (most books directed to - charge)
for larger molecules, have to sum all individual µ’s
through-out the molecule to get the overall dipole
moment
µ x = qi xi (sum of all in x-direction)
similarly for y and z components
i
eg. water
H
i
O
H
overall
Units of : (Coulomb-meter) or Debye (D)
1 Debye = 3.34 X 10-30 Coulomb-meter
2. Ion-dipole interaction:
consider charge Q at a distance r from the center
of a polar molecule with dipole moment, ,
subtending an angle to the line joining the two
molecules
q+
r
Q+ ----------------------------------------------
q–
U(r) = – ( Q cos ) / r2
Simplified example:
Na+ ion near a water molecule (m = 1.85 D) requires
96 kJ/mole at 300o K to pull them apart
•Consider cos as an orientation term
•Important to note that U is inversely proportional to r2
where for charge-charge interactions it was inversely
proportional to r.
3. Dipole-dipole interaction:
2
1
1---------------------------------2
r12
U(r) = 1389 1 2 (cos 12 - 3cos1cos2) / r123
where: 12 = angle between 1 and 2
dipole-dipole interaction energy depends on the
inverse cube of the distance between the two
dipoles and their orientations
consider the following dipole orientations:
repulsion
attraction
no
interaction
attraction
repulsion
The magnitudes of dipole moments can be substantial.
HCl
Urea
Peptide bond
H
C i-1
1.04 D
4.56 D
3.70 D
Total
N
C
Ci
O
Ri
Peptide bond dipole moment
is parallel to N-H bond
(N is q-)
for amino acid: glycine (Ri is H, simplest)
16.7 D
glycylglycine
28.6 D
for large protein:hemoglobin
hundreds
QUESTION?
for the same r, which would have the largest interaction
energy?
A. a charge-charge interaction
B. an ion-dipole interaction
C. a dipole-dipole interaction
NEXT,
even if molecules have no permanent dipole moments,
there are forces between them! (induced dipoles)
So far we have seen weak interactions that are
electrostatic in nature, now we will consider
Polarization interactions (also weak)- arising from
dipole moments induced in atoms and molecules by
the electric fields of nearby charges or permanent
dipoles
4. Charge-induced dipole : proportional to 1/r 4
5. Dipole-induced dipole: proportional to 1/r 5
these are even shorter range than permanent dipole
interactions
other non covalent interactions include:
6. Dispersion forces (London forces) -all atoms attract each other
-results from instantaneous fluctuations in
charge distributions
-depends on the polarizability of the molecule (ie
the deformability of the electron cloud)
U(r) = A/r 6 where A > 0
-this force occurs between all atoms. It is small but
is summed over all species in a structure.
7. Steric repulsion (van der Waals repulsion)
-all atoms repel at short distances
-occurs when outer electronic orbitals overlap
U(r) = + B/r 12
where B > 0
Non covalent Interaction Energy
of two approaching particles
Energy of Interaction
Van der Waals
radii
One last important non covalent interaction :
Hydrogen bonds•stabilize and specify the structures of proteins and DNA
•determine the structure of liquid water
•an H atom that interacts simultaneously with two other
atoms is said to form H bond
•takes place between:
•an acidic hydrogen
• H atom that is covalently bound to a donor
group; like -O-H or N-H
• depends upon electronegativity of donor
• and a pair of non-bonded electrons on an acceptor
group; like O=C- or N
B
+
H-A
B ……H-A
•length of H-bond is nearly the same in all species
• approximately 0.3 nm
•defined as the distance between the center of the Hdonor atom (A) to the center of the acceptor atom (B)
•H-bonds are directional
•the H-donor atom (A) tends to point directly at acceptor
e- pair on B
•maximum stability when B …… H-A are co-linear
•bent H-bonds have reduced stability
H-bonds are extremely important in dictating protein
structure. Both a-helical and b-sheet structures are
stabilized by intramolecular H-bonds.
The nature of H-bonds is electrostatic
-attraction of one proton to two nuclei provides an
efficient path for moving charges
-the proton can easily move between nuclei
O-H …. O
O …. H-O
Hydrogen bonding in ice and in water
a: ice, space filling;
b: ice, skeletal
c: water, simulation
Hydration of ions in solution
As salt dissolves, non-covalent interaction between ions and water
produces a hydration shell. The energy gained helps overcome ion-ion
interactions that stabilize the crystal.
Amphipathic molecules are both hydrophobic
and hydrophilic
Hydrophilic: the ability of an atom or a molecule to engage in attractive
interactions with water molecules. Substances that are ionic or can
engage in hydrogen bonding are hydrophilic. Hydrophilic substances
are either soluble in water or, at least, wettable.
Hydrophobic: the molecular property of being unable to engage in
attractive interactions with water molecules. Hydrophobic
substances are nonionic and nonpolar; they are nonwettable and
do not readily dissolve in water.
SUMMARY
Typical magnitudes of Interaction Energies
•covalent
200-800 kJ/mole
•H-bonds
25
•ion-ion
20-250
•ion-dipole
15
•dipole-dipole
0.3-2
•dispersion
2
SUMMARY:
•Several weak interactions are present within or
between biomolecules. The energy sums up to an
impressive total.
•The energy is minimized for a particular
conformation.
•One central problem of modern Biochemistry is to
predict what the 3-D structure of a protein will be
given its primary sequence.
Important Properties of Water
•Water’s tendency to form H-bonds makes it unique
•causing boiling point, melting point, heat of
vaporization to be anomalously high
•Each water molecule can be H-bond donor and acceptor
simultaneously
•Dipolar nature can reduce effective electrostatic force
between two interacting ions:
the orientation of water dipoles between the two
charges acts as counterfield
•Hydrophilic molecules tend to form H-bonds with water
and readily dissolve
•Ions in aqueous solutions become hydrated as water
forms hydration shells around them (energetically
favorable, energy is released)
•Hydrophobic molecules have limited solubility in water
•clathrate structures form
•the ‘caged’ water structure is ordered which
decreases the entropy (randomness) of the mixture
•hydrophobic molecules tend to aggregate in water
with a single “cage” surrounding them
•Amphipathic molecules tend to form ordered structures
in water: monolayers, micelles, bilayers
Ionic Equilibria
Acids and Bases (proton donors and acceptors)
REVIEW:
strong acids- essentially complete dissociation
HCl
H+
+
Cl -
strong base- essentially complete ionization to yield
OH- ions; proton acceptors
weak acids and bases - important in biochemistry,
do not completely ionize at physiological pH, get partial
dissociation with equilibrium between weak acid and
conjugate base
Weak Acid
Conjugate base + proton
H3PO4
H2PO4– + H+
phosphoric acid
Dihydrogen phosphate ion
H2PO4–
HPO4 2– + H+
monohydrogen phosphate ion
HPO4 2–
PO4 3– + H+
phosphate ion
the stronger the acid, the weaker the conjugate base
(the conjugate base does not have a strong tendency to
accept a proton and reform the acid)
-which of the above is the strongest acid?
Equilibria involving water and pH
•water has a slight tendency to ionize
•it can act as acid and base
H 2O + H 2O
H 3 O+
+
OH–
often see written as:
H 2O
H+ + OH–
but proton never exists as free ion in solution, it is always
associated with other water molecules
equilibrium can be expressed as ion product of water:
Kw = [H+][OH–] = 1 x 10 –14 M2
[H+] and [OH-] do not vary independently
If [H+] is high, [OH-] must be ______?
For pure water at 25° C [H+] = [OH–] = 1 x 10 –7 (neutral)
to simplify, define
pH = – log [H+]
for neutral solution: pH = –log (1 x 10 –7) = 7
physiological pH range = 6.5 - 8.0
Weak Acid and Base Equilibria
Many weak acids are found in biological systems, for eg:
catalytic proteins have ionizable side chains whose state
of ionization is dependent on pH (catalytic activity occurs
only at a certain pH)
to describe weak acid strength consider:
dissociation constant, Ka, (equilibruim constant)
HA
H+ + A–
where Ka = [products]/[reactants] = [H+] [A–] / [HA]
(large Ka , greater tendency to dissociate, stronger acid)
pKa = – log Ka
also define
small pKa , stronger acid
H3PO4
H2PO4– + H+
pKa = 2.14
H2PO4–
HPO4 2
pKa = 6.86
HPO4 2–
PO4 3– + H+
+ H+
pKa = 12.4
Environment influences pKa values:
-hydration of proton favors dissociation
-electrostatic attraction between proton and conjugate
base opposes dissociation
-identical groups in different local environments (different
regions in a protein) will dissociate to differing degrees
-consider high dielectric constant vs. low dielectric
constant media
Buffers and the Henderson-Hasselbalch Equation
-many biological processes generate or use H+
- the pH of the medium would change dramatically if it were
not controlled (leading to unwanted effects)
--biological reactions occur in a buffered medium where
pH changes slightly upon addition of acid or base
-most biologically relevant experiments are run in buffers
how do buffered solutions maintain pH under varying
conditions?
to calculate the pH of a solution when acid/base ratio of
weak acid is varied: Henderson-Hasselbalch equation
comes from:
Ka = [H+] [A–] / [HA]
take (– log) of each side and rearrange, yields:
pH = pKa + log ( [A–] / [HA] )
some examples using HH equation:
what is the pH of a buffer that contains the following?
1 M acetic acid and 0.5 M sodium acetate
Titration example (similar one in text:)
Consider the titration of a 2 M formic acid solution with
NaOH.
1. What is the pH of a 2 M formic acid solution?
Ka = [H+] [A–] / [HA]
use
HCOOH
H+ + HCOO–
let x = [H+] = [HCOO–]
then
Ka = 1.78 x 10 –4 = x2 / (2 – x)
for an exact answer, need the quadratic equation but since
formic acid is a weak acid (Ka is small),
x <<< [HCOOH]
and equation becomes Ka = 1.78 x 10 –4 = x2 / 2
so
x = [H+] = [HCOO–] = 0.019
and pH = 1.7
2. Now start the titration. As NaOH is added, what
happens?
•NaOH is a strong base --- completely dissociates
•OH– is in equilibrium with H+ , Kw = [H+] [OH–] = 10–14 ,
•Kw is a very small number so virtually all [OH–] added
reacts with [H+] to form water
Titration continued:
- to satisfy the equilibrium relationship given by Ka
Ka = [H+] [HCOO–] / [HCOOH] = 1.78 x 10 -4
more HCOOH dissociates to replace the reacted [H+] and
-applying HH, see that [HCOO–] / [HCOOH] will increase
pH = pKa + log ( [HCOO–] / [HCOOH] )
-leading to a slow increase in pH as the titration proceeds
_______________________________________________
consider midpoint of titration where half of the HCOOH
has been neutralized by the NaOH
[HCOO–] / [HCOOH] = 1
HH becomes: pH = pKa + log 1 = pKa = 3.75 for HCOOH
Titration curve:
- within 1 pH unit of pKa
over most of curve
- so pKa defines the
range where buffering
capacity is maximum
- curve is reversible
Simple problem:
-have one liter of a weak acid (pKa = 5.00) at 0.1 M
-measure the initial pH of the solution, pH = 5.00
-so it follows that initially,
[A–] = [HA] where pH = pKa
-add 100mL of 0.1M NaOH, following occurs
HA + OH– = A–
0.01moles
+
H2 O
-so, 0.01 moles of HA reacted and
new [HA] = 0.1 – 0.01 = 0.09
new [A–] = 0.11
-use HH to get new pH = 5 + log (0.11 / 0.09) = 5.087
_______________________________________________
now consider,100mL of 0.1 M NaOH added to 1 L without
the weak acid to see how well the weak acid buffers
0.01 moles OH– / 1.1L = 9.09 x 10 -3 = [OH– ]
use Kw = [OH–] [H+] = 1 x 10 -14
to get pH = 11.96
_______________________________________________
what happens when 0.1 moles of base have been added?
what happens when the next 1 mL of base is added?
Known as overrunning the buffer
Sample Buffer Calculation (in text)
-want to study a reaction at pH 4.00
-so to prevent the pH from drifting during the reaction, use
weak acid with pKa close to 4.00 -- formic acid (3.75)
-can use a solution of weak acid and its conjugate base
-ratio of formate ion to formic acid required can be
calculated from the Henderson - Hasselbalch equation:
4.00 = 3.75 + log [HCOO–] / [HCOOH]
[HCOO–] / [HCOOH] = 10 0.25 = 1.78
-so can make a formate buffer at pH 4.0 by using equal
volumes of 0.1 M formic acid and 0.178 M sodium formate
-Alternatively, exactly the same solution could be prepared
by titrating a 0.1 M solution of formic acid to pH 4.00 with
sodium hydroxide.
_______________________________________________
some buffer systems controlling biological pH:
1. dihydrogen phosphate-monohydrogen phosphate
pKa = 6.86 - involved in intracellular pH
control where phosphate is abundant
2. carbonic acid-bicarbonate pKa = 6.37, blood pH control
3. Protein amino acid side chains with pKa near 7.0
Example of an ampholyte - molecule with both acidic
and basic groups
NH3+ – CH2 – COOH
net charge +1
pH 6
NH3+ – CH2 – COO–
zwitterion
net charge 0
pH 14
NH2 – CH2 – COO–
net charge –1
glycine: pH 1
pKa values
carboxylate group
amino group
2.3
9.6
can serve as good buffer in 2 different pH ranges
______________________________________________
use glycine to define an important property
isoelectric point (pI) - pH at which an ampholyte or
polyampholyte has a net charge of zero.
for glycine, pI is where:
[NH3+ – CH2 – COOH] = [NH2 – CH2 – COO– ]
can calculate pI by applying HH to both ionizing groups
and summing (see text) yields:
pI = {pK COOH + pK NH 3+ } / 2 = {2.3 + 9.6} / 2 = 5.95
pI is the simple average for two ionizable groups
polyampholytes are molecules that have more than 2
ionizable groups
lysine
NH3+- C- (CH2)4 - NH3+
COOH
titration of lysine shows 3 pKa’s:
• pH<2, exists in above form
•first pKa = 2.18, loss of carboxyl proton
•second at pH = 8.9
•third at pH = 10.28
•need model compounds to decide which amino group
loses a proton first
_____________________________________________
to determine pI experimentally use electrophoresis
(see end of Chapter 2)
1. Gel electrophoresis-electric field is applied to solution
of ions, positively charged ions migrate to cathode and
negatively charged to anode, at it’s pI an ampholyte does
not move because net charge = zero
2. Isoelectric focusing- charged species move through a
pH gradient, each resting at it’s own isoelectric point
_____________________________________________
Macromolecules with multiples of either only negatively or
only positively charged groups are called polyelectrolytes
polylysine is a weak polyelectrolyte - pKa of each group
influenced by ionization state of other groups
Solubility of macroions (polyelectrolytes and
polyampholytes, including nucleic acids and proteins)
depends on pH.
For polyampholytes:
•high or low pH leads to greater solubility (due to – or +
charges on proteins, respectively)
•At the isoelectric pH although net charge is zero, there are
+ and – charges and precipitation occurs due to:
- charge-charge intermolecular interaction
- van der Waals interaction
•to minimize the electrostatic interaction, small ions (salts)
are added to serve as counterions, they screen the
macroions from one another
Ionic Strength = I = ½ (Mi Zi2)
(sum over all small ions)
M is molarity
Z is charge
Consider the following 2 processes that can take place for
protein solutions:
1. Salting in: increasing ionic strength up to a point
(relatively low I), proteins go into solution
2. Salting out: at high salt, water that would normally
solvate the protein goes to solvate the ions and protein
solubility decreases.
Most experiments use buffers with NaCl or KCl