Presentation #13
advertisement

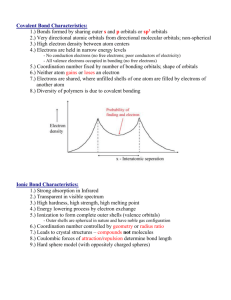
MODULE 11 Diatomic Molecules and Beyond Our next task is to extend the procedures and ideas that we have developed for H2+ to larger molecules. The track we follow is the customary one, that is, start with homonuclear diatomics, of which H2 is the paradigm, then consider heteronuclear diatomics, and finally polyatomic molecules. The overall procedure is to construct sets of MOs and then put in the electrons according to the Pauli principle analogous to the aufbau method for atoms The result is similar because we end up with the electronic configurations of molecules. MODULE 11 The Hydrogen Molecule 1 A 2 H2 has an additional electron over its molecule-ion and we can imagine a scheme such as shown in Figure. B The electron configuration follows the same rule as for the He atom, where the added electron was placed into the lowest energy 1s orbital with its spin opposed (Pauli). Thus the added electron in H2 is placed into the bonding orbital defined for the 1sA + 1sB combination. MODULE 11 The extra electron adds the distances r12, r2B, and r2A. This adds complexity in that an electron-electron repulsion term appears, as well as two more electron-nucleon potential energy terms. The hamiltonian for the hydrogen molecule in the BornOppenheimer approximation and in atomic units is 1 1 1 1 1 1 1 2 2 ˆ H (1 2 ) 2 r1 A r1B r2 A r2 B r12 R MODULE 11 1 y b (1) y b (2) y 2! y b (1) y b (2) where yb is the bonding orbital (1s). This can be rearranged to 1 y y b (1)y b (2) (1) (2) (2) (1) 2 Since the hamiltonian is independent of the spin terms, we calculate the energy using only the spatial part of equation. Thus our molecular wavefunction yMO is given by y MO 1 1sA (1) 1sB (1)1sA (2) 1sB (2) 2(1 S ) Where the first term on the RHS is the normalization constant (it is the square of the normalization constant for y. MODULE 11 Thus our molecular wavefunction is a product of molecular orbitals both of which are linear combinations of atomic orbitals. The procedure for constructing molecular wavefunctions is known as the Linear Combination of Atomic Orbitals-Molecular Orbitals, or LCAO-MO, method. This is a commonly used procedure for a variety of molecules. The ground state energy of H2 can then be calculated as before using EMO y MO (1, 2) Hˆ y MO (1, 2) Where the hamiltonian and the wavefunction are as above MODULE 11 The results of the integration are shown in figure. As for the molecule ion the results lack accuracy, but the form is clearly shown and the results can be improved using a larger basis set. The ground state configuration of H2 is classified as 1Sg, which echoes the term symbols for atoms (Module 9A). The superscript 1 is the multiplicity of the state (here the 2 electrons are paired, S = 0). The S (analogous to atomic S) indicates that the total OAM around the inter-nuclear axis is zero because both electrons are in s-type AOs MODULE 11 Whereas for atoms we used L = l1+l2, etc to compute the total OAM, in molecules we use L =l1 + l2 to compute the total OAM In H2, L = 0, hence S. The subscript g indicates the overall parity of the state and we calculate this from the individual values for the electrons by the products g x g = g; g x u = u; u x u = g In H2 both electrons occupy the sg orbital, hence the overall parity is g. Had one occupied a 2su orbital the overall parity would have been u. MODULE 11 Homonuclear Diatomic Molecules Beyond Hydrogen The MOs for homonuclear diatomics beyond H2 are formed from pairs of AOs of many-electron atoms Therefore we must to use orbitals beyond 1s (higher energy). Nevertheless the 1s AOs are involved and we can start there and the first pair of MOs is given by y 1sA 1sB In Module 10 we saw that the two MOs have cylindrical symmetry with respect to the inter-nuclear axis. For this reason they are termed s orbitals. MODULE 11 Because they are constructed from a pair of 1s orbitals they are conventionally labeled s1s, indicating they are s orbitals formed from 1s AOs, and nothing else. The positive combination, y, builds up electron density between the nucleons and is the bonding orbital, The negative combination, y, excludes electron density from that region, and is the antibonding orbital, often written as s*1s. As we saw in Module 10 the bonding orbital has gerade inversion symmetry, hence it is symbolized as sg1s, and the antibonding orbital is u, therefore su1s. We do not include the asterisk in the symbol since the subscript gives the antibonding designation. MODULE 11 Next consider combinations (sum and diff) of a pair of 2s AOs. Both AOs have the same energy and the same symmetry and these factors are important in the LCAO procedure. The two MOs resulting from 2sA+/-2sB are sg2s and su2s, one bonding and the other antibonding. These MOs have similar shape to those shown in the figure above, except they are larger because the composite AOs are larger. By the same token the energy of the 2s-based orbitals are higher than those based on 1s. The energies can be calculated in an analogous way to that outlined above for the 1s combination. MODULE 11 The relative energies are in the sequence s g 1s s u 1s s g 2s s u 2s In the hydrogenic ions the 2s and 2p orbitals are degenerate, but this degeneracy is lifted in manyelectron atoms because of differences in the nuclear screening, thus E2s < E2p In Figure the way in which the atomic 2p orbitals combine is indicated. 2pz AOs are oriented along the inter-nuclear axis. Both u and g combinations of the 2pz orbitals are symmetric around the axis and are therefore of s-type. MODULE 11 The 2px and 2py combinations overlap “sideways” and the result is that the four MOs generated do not have cylindrical symmetry. This change in symmetry leads to the designation of p-bonds. [An easy way of determining whether a MO is s or p is to “view” the orbital along the inter-nuclear axis. If you see a circle of electron density centered on the nucleus (like an s-AO), then that orbital is s. If what you see looks like a p-AO (two circles separated by a nodal plane) then you are dealing with a p-orbital.] The negative combination 2pzA-2pzB is the one that yields the bonding MO; for the other combinations the positive ones are bonding. The bonding orbital resulting from the 2pz combination transforms as g on inversion, whereas the opposite is true for the bonding orbitals resulting from the 2px and 2py pair combinations. MODULE 11 From figure we see that the energies depend on Z. Moreover the sg2pz orbital changes so much with Z that between N2 and O2 it switches with the degenerate x and y-pairs. Also Figure shows the electron occupancy of the MOs built up by the LCAOMO procedure, with aufbau. Not shown are H2 and He2. The former we have already considered, it has two electrons of opposed spins in the sg1s MO and its configuration is (sg1s)2. MODULE 11 He2 has a configuration (sg1s)2(su1s)2 in which there are an equal number of bonding and antibonding electrons. The two sets cancel such that there is no net bond, according to the simple version of MO theory used here (He2 has been detected spectrometrically in the gas phase at T~0.001 K). The molecular ion He2+ is a stable, but reactive, species. Some properties of hydrogen and helium molecules are in the Table MODULE 11 bond order = (nb-na)/2 where na and nb are the number of electrons in bonding and antibonding orbitals, respectively. Note that bond order can be a half-integer. Diatomic lithium has an electron configuration of (sg1s)2(su1s)2(sg2s)2 and thus the bond order is one. Li2 exists in Li vapor and its bond energy is 105 kJmol-1. Electron density contour maps, generated by computer solutions of the Schrödinger equation are presented in the figure over page MODULE 11 The important thing to note about these diagrams is that the sg1s and su1s electrons are found close to the nuclei and play virtually no part in the bonding interaction, which is mostly due to the electrons in the sg2s MO. This leads to an alternative way of writing molecular electronic configuration for Li2 as KK(sg2s)2 in which K represents the filled n = 1 shell of the Li atom. MODULE 11 This becomes even more accurate as Z increases and the 1s electrons are held progressively more tightly than in the Li case. At this level of approximation only the valence electrons need to be considered for the homonuclear diatomics beyond He2. Returning to the orbital energy diagram and focusing on B2 and O2, we see that each molecule has a degenerate pair of orbitals (pu2px,y in the case of B and pg2px.y in the case of O). Hund’s rule informs us that we place electrons into degenerate sets of orbitals one at a time in order to maximize the spin multiplicity. Both molecular boron and molecular oxygen have been shown to have paramagnetic (triplet) ground states. MODULE 11 In the Table are shown the ground state electron configurations of the 2nd row homonuclear diatomics MODULE 11 The tabulated data are plotted here, showing the inverse correlation between bond energy and bond length. MODULE 11 Experimental Demonstration of Orbitals The concepts of atomic and molecular orbitals have been arrived at through theoretical manipulations, and one might wonder that there is any reality in the ideas. In fact, there are experimental ways to support the actual existence of orbitals. One is by the use of photoelectron spectrometry. This is a technique that measures the energy of electrons ejected from gaseous molecules by vacuum uv/soft x-ray radiation. This provides the ionization energy of the molecule, which depends on the MO from which the ionized electron originated. MODULE 11 The figure shows the PE spectrum of N2 gas. Peaks occur that can be assigned to all the occupied orbitals listed for N2 in the Table MODULE 11 Heteronuclear Diatomic Molecules Examples are HF, HI, CO, NO, CN-. Earlier we stated that for LCAO-MOs we need to use AOs that are close in energy. In HF and HI the energies of the atomic orbitals are very different. In CO, NO, CN- the energies are not very different (atoms are close in terms of Z). It is useful to look closely at one from each group. First consider CO. MODULE 11 The PE spectrum of CO Peaks that are characteristic atomic 1s orbitals on C and O are visible The high energies of s1s and s*1s are indicative of these electrons being closely associated with the respective nuclei and supports the contention that K electrons play no part in the bonding process after Z = 3. Moreover the energies of these orbitals are very similar to the energies of the constituent O1s and C1s AOs respectively MODULE 11 The spectrum is consistent with the electronic configuration of CO being KK (s 2s)2 (s *2s)2 (p 2 px )2 (p 2 py ) 2 (s 2 pz ) 2 Thus CO has a bond order of 3. Note that there are no gerade/ungerade subscripts because there is no inversion symmetry in heteronuclear diatomics. MODULE 11 In HF, the valence electrons are in different shells. In F, E2s = -1.477 au, E2p = -0.684 au. In H, E1s = -0.5 au. Thus it is more favorable for the H1s and F2p orbitals to form the combination. Now we have to consider the symmetry of the situation. Figure shows clearly why only the 2pz AO on F has the proper symmetry for bonding with the H1s AO. The coupling of H1s and F2px,y leads to no bond since the positive and negative overlaps cancel out. MODULE 11 The MO for HF will be approximated by y c11sH c2 2 pzF the bond has cylindrical symmetry and hence is s. An energy level diagram for HF is shown (less the 1sF and the 2sF orbitals). H has 1 valence electron and F has 7, so the total of 8 occupy the four orbitals shown according to the Pauli principle. The 2sF (not shown), 2pxF, and 2pyF orbitals are non-bonding MOs (2sF )2 (s b )2 (2 pxF )2 (2 pyF )2 The bond order is 1 MODULE 11 Most of what we have said so far about diatomics has concerned MOs that have been formed from a single AO from two contributing atoms. When we were considering many-electron atoms in Module 10 we found that we could lower our computed value of Emin by using linear combinations of orbitals (e.g. STOs) for the trial function and using the Hartree-Fock SCF procedure to generate the HartreeFock limit. The molecular case uses similar procedures. For example we could use a trial function of the form y c1 (1s A 1sB ) c2 (2s A 2sB ) c3 (2 pZ , A 2 pZ , B ) ... and use variation theory to find the relative contributions of the different LCAOs by determining the variational coefficients, ci. MODULE 11 One outcome of using the larger MO basis set is that it becomes no longer possible to identify an MO as arising from a single AO. Thus our sg1s, etc designations need to be modified. Thus we find designations such as 1sg, indicating the first (lowest energy) s orbital, and 1pu, indicating the first (lowest energy) p orbital, and so on. These days MOs are determined by Hartree-Fock SCF computations employing basis sets of a large number of linear combinations, and the notation above is the one you will find in the literature.



![6) cobalt [Ar] 4s 2 3d 7](http://s2.studylib.net/store/data/009918562_1-1950b3428f2f6bf78209e86f923b4abf-300x300.png)