Thermodynamic Systems
advertisement

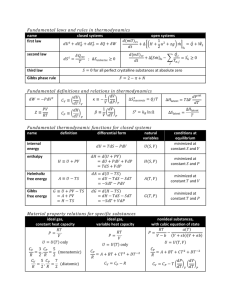
Learning Objectives and Fundamental Questions • What is thermodynamics and how are its concepts used in petrology? • How can heat and mass flux be predicted or interpreted using thermodynamic models? • How do we use phase diagrams to visualize thermodynamic stability? • How do kinetic effects affect our interpretations from thermodynamic models? What is Thermodynamics? • Thermodynamics: A set of of mathematical models and concepts that allow us to describe the way changes in the system state (temperature, pressure, and composition) affect equilibrium. – Can be used to predict how rock-forming systems will respond to changes in state – Invert observed chemical compositions of minerals and melts to infer the pressure and temperature conditions or origin Thermodynamic Systems - Definitions Isolated System: No matter or energy cross system boundaries. No work can be done on the system. Open System: Free exchange across system boundaries. Closed System: Energy can be exchanged but matter cannot. Adiabatic System: Special case where no heat can be exchanged but work can be done on the system (e.g. PV work). Thermodynamic State Properties • Extensive: These variables or properties depend on the amount of material present (e.g. mass or volume). • Intensive: These variables or properties DO NOT depend on the amount of material (e.g. density, pressure, and temperature). Idealized Thermodynamic Processes • Irreversible: Initial system state is unstable or metastable and spontaneous change in the system yields a system with a lower-energy final state. • Reversible: Both initial and final states are stable equilibrium states and the path between them is a continuous sequence of equilibrium states. NOT ACTUALLY REALIZED IN NATURE. Spontaneous Reaction Direction First Law of Thermodynamics The increase in internal energy as a result of heat absorbed is diminished by the amount of work done on the surroundings: dEi = dq - dw = dq - PdV By convention, heat added to the system, dq, is positive and work done by the system, dw, on its surroundings is negative. This is also called the Law of Conservation of Energy Definition of Enthalpy We can define a new state variable (one where the path to its current state does not affect its value) called enthalpy: H = Ei + PV Enthalpy = Internal Energy + PV Upon differentiation and combing with our earlier definition for internal energy: dH = dEi + PdV + VdP dEi = dq - PdV dH = dq + VdP Enthalpy, Melting, and Heat For isobaric (constant pressure) systems, dP = 0 and then the change in enthalpy is equal to the change in heat: dHp = dqp Three possible changes in a system may occur: 1) Chemical reactions (heterogeneous) 2) Change in state (e.g. melting) 3) Change in T with no state change Heat capacity is defined by the amount of heat that may be absorbed as a result of temperture change at constant pressure: Cp = (dH/dT)p Enthalpy of Melting Second Law of Thermodynamics • One statement defining the second law is that a spontaneous natural processes tend to even out the energy gradients in a isolated system. • Can be quantified based on the entropy of the system, S, such that S is at a maximum when energy is most uniform. Can also be viewed as a measure of disorder. DS = Sfinal - Sinitial > 0 Change in Entropy Relative Entropy Example: Ssteam > Sliquid water > Sice ISOLATED SYSTEM Third Law Entropies: All crystals become increasingly ordered as absolute zero is approached (0K = -273.15°C) and at 0K all atoms are fixed in space so that entropy is zero. Gibbs Free Energy Defined G = Ei + PV - TS dG = dEi + PdV + VdP - TdS - SdT dw = PdV and dq = TdS dG = VdP - SdT (for pure phases) At equilibrium: dGP,T = 0 Change in Gibbs Free Energy Gibbs Energy in Crystals vs. Liquid dGp = -SdT dGT = VdP Melting Relations for Selected Minerals dGc = dGl VcdP - ScdT = VldP - SldT (Vc - Vl)dP = (Sc - Sl)dT Clapeyron Equation dP (Sc Sl ) DS dT (Vc Vl ) DV Thermodynamics of Solutions • Phases: Part of a system that is chemically and physically homogeneous, bounded by a distinct interface with other phases and physically separable from other phases. • Components: Smallest number of chemical entities necessary to describe the composition of every phase in the system. • Solutions: Homogeneous mixture of two or more chemical components in which their concentrations may be freely varied within certain limits. Mole Fractions nA nA XA n (nA n B nC ) , where XA is called the “mole fraction” of component A in some phase. If the same component is used in more than one phase, Then we can define the mole fraction of component A in phase i as X Ai For a simple binary system, XA + XB = 1 Partial Molar Volumes & Mixing Temperature Dependence of Partial Molar Volumes Partial Molar Quantities • Defined because most solutions DO NOT mix ideally, but rather deviate from simple linear mixing as a result of atomic interactions of dissimilar ions or molecules within a phase. • Partial molar quantities are defined by the “true” mixing relations of a particular thermodynamic variable and can be calculated graphically by extrapolating the tangent at the mole fraction of interest back to the end-member composition. Partial Molar Gibbs Free Energy As noted earlier, the change in Gibbs free energy function determines the direction in which a reaction will proceed toward equilibrium. Because of its importance and frequent use, we designate a special label called the chemical potential, µ, for the partial molar Gibbs free energy. GA A X A P,T ,X B ,X C , We must define a reference state from which to calculate differences in chemical potential. The reference state is referred to as the standard state and can be arbitrarily selected to be the most convenient for calculation. The standard state is often assumed to be pure phases at standard atmospheric temperature and pressure (25°C and 1 bar). Thermodynamic data are tabulated for most phases of petrological interest and are designated with the superscript °, for example, G°, to avoid confusion. Chemical Thermodynamics MASTER EQUATION dG VdP SdT i dX i i dX i i A dX A B dX B C dX C n dX n i This equation demonstrates that changes in Gibbs free energy are dependent on: • changes in the chemical potential, µ, through the concentration of the components expressed as mole fractions of the various phases in the system • changes in molar volume of the system through dP • chnages in molar entropy of the system through dT Equilibrium and the Chemical Potential • Chemical potential is analogous to gravitational or electrical potentials: the most stable state is the one where the overall potential is lowest. • At equilibrium the chemical potentials for any specific component in ALL phases must be equal. This means that the system will change spontaneously to adjust by the Law of Mass Action to cause this state to be obtained. If melt H 2O melt H 2O melt CaO gas H2 O gas H2 O biotite H 2O gas CaO biotite CaO n H 2O n CaO then system will have to adjust the massmelt (concentration) to make them equal: H 2O gas H2 O Gibbs Free Energy of Mixing Activity - Composition Relations The activity of any component is always less than the corresponding Gibbs free energy of the pure phase, where the activity is equal to unity by definition (remember the choice of standard state). A A G ; B G B G RT ln a A i A a i A i A X i A For ideal solutions (remember dG of mixing is linear), such that the activity is equal to the mole fraction. A G RT ln X i A i A i A 1 i A P, T, X Stability of Crystals Equilibrium stability surface where Gl=Gc is defined by three variables: 1) Temperature 2) Pressure 3) Bulk Composition Changes in any of these variables can move the system from the liquid to crystal stability field Fugacity Defined For gaseous phases at fixed temperature: dGT = VdP - Assume Ideal Gas Law PV nRT;n 1 RT V P RT dGT VdP dP RT ln dP P PA = XAPtotal and the fugacity coefficient is defined as fA/PA, which Is analogous to the activity coefficient. As the gas component Becomes more ideal, fA goes to unity. A A G RT ln f A Equilibrium Constants Mg2SiO4 + SiO2 = 2MgSiO3 olivine melt opx DG = 2 opx MgSiO3 ol Mg 2 SiO4 0 mel t SiO2 mel t glass t SiO GSiO RT ln a mel SiO 2 ol Mg 2 SiO4 opx MgSiO3 2 2 ol Mg 2 SiO4 G G opx MgSiO3 RT ln a ol Mg 2 SiO4 RT ln a opx MgSiO3 Equilibrium Constants, con’t. opx MgSi O3 2G ol Mg 2 SiO4 glass SiO2 G G RT ln( a ) (a ) ol Mg 2 SiO4 opx 2 MgSi O3 melt SiO2 a F DG RT ln K eq where dG°F is referred to as the change in standard state Gibbs free energy of formation, which may be obtained from tabulated information K eq opx 2 (aMgSi ) O3 ol melt (aMg a ) SiO SiO 2 4 2 Silica Activity, Buffers, and Saturation Mg2SiO4 + SiO2 = 2MgSiO3 olivine melt opx NeAlSiO4 + SiO2 = NaAlSi3O8 nepheline melt albite Oxygen Buffers <--- Calculated fO2 from Fe-Ti oxides Fe2TiO4 + Fe2O3 = FeTiO3 + Fe3O4 Arrhenius Equation and Activation Energy Kinetic Rate = A exp -Ea/RT log D = log A - Ea/2.303RT y = b + m•x Slope = dy/dx = -Ea/2.303R Intercept = b = log A All processes that are thermally activated have similar form! Gibbs Free Energy - Temperature Relations Metastability for polymorphs A & B State A is stable for T > Te because GA < GB State B is stable for T < Te because GB < GA Undercooling allows metastability of phase A over B Irreversible Path SYSTEM STATE CHANGES YIELD REACTION OVERSTEPPING Silica Polymorph Free Energy Relations and Reaction Progress Ostwald’s Step Rule: In a change of state the kinetically most favored phase may form at an intermediate step rather than the most thermodynamically favored (lowest G) phase! Glass -> Qtz (favored) Glass -> Cristobalite or Tridymite