Cell organelles and functions
advertisement

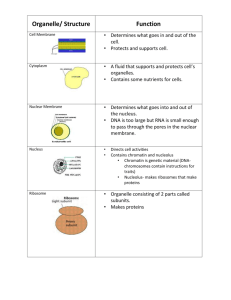
CELL ORGANELLES AND FUNCTIONS By: Hemangi Atalia INTRODUCTION Cell is the structural and functional unit of life. Cells Tissue Organ Organ system. Cells contain a variety of Internal Structures called ORGANELLES. An organelle is a cell component that performs specific functions for the cell. NUCLEUS Spherical in shape. Covered by nuclear membrane. The nuclear membrane encloses nucleoplasm and nucleolus. Nuclear membrane is double layered and porous in nature. This allows the nucleoplasm to communicate (exchange of material) with the cytoplasm. Nucleoplasm is a gel like substance that contains large quantities of DNA, which forms the gene. One or more nucleoli are present in each nucleus. The nucleolus synthesizes ribosomes, which inturn build proteins. FUNCTIONS 1. 2. 3. 4. Control of all the activites of the cell. Control the cell division through genes. Storage of hereditary information. Sending genetic instruction to the cytoplasm for protein synthesis through the mRNA. RIBOSOMES Granular and small dot like structures. Some are attached to RER while others are present as free ribosomes in the cytoplasm. Ribosomes attached with RER are involved in the synthesis of proteins like the hormonal proteins or the proteins of the cell membrane. The free ribosomes are responsible for the synthesis of proteins of hemoglobin, and mitochondria. MAIN FUNCTION SYNTHESIS OF PROTEINS. ROUGH ENDOPLASMIC RETICULUM (RER) Tubular in structure. Also known as “granular endoplasmic reticulum.” due to the present of ribosomes attached to its outer surface. FUNCTION 1. Synthesis of proteins in the cell. INSULIN. 2. Degradation of the worn out organelles and later digested by lysosomal enzymes. SMOOTH ENDOPLASMIC RETICULUM (SER) “Agranular reticulum.” Smooth appearance FUNCTION 1. Synthesis of lipids and steroids. 2. Storage and metabolism of calcium. GOLGI APPARATUS “Golgi body” or “Golgi Complex” Membraneous tubules Situated near the nucleus Also called “post office of the cell.” Site for lysosomes FUNCTION 1. Processing and delivery of proteins and other molecules lipids 2. Labeling and sort out the processed and packed materials for distribution to their proper destinations. “Shipping department of the cell.” CYTOSKELETON Complex network of structures of various sizes present throughout the cytoplasm. Consists of three protein components Microtubule Intermediate filaments Microfilaments FUNCTION 1. Determination of shape of the cell. 2. Stability of cell shape. 3. Cellular movements. MITOCHONDRION Globular in structure. Bilayered membranous organelle. “Power house of the cell” Outer membrane is smooth and contains enzymes responsible for biological oxidation. Inner membrane is folded like inward projection called CRISTAE and it covers the inner matrix space. The cristae contain many enzymes which are involved in RESPIRATION and synthesis of ATP. Matrix Enzymes HEAD BASEPIECE STALK Capable of reproducing themselves Contain their own DNA. FUNCTION 1. Production of energy 2. Synthesis of ATP LYSOSOME Membrane bound vesicular. Formed by Golgi Apparatus. Contain enzymes called Hydrolytic enzymes. “Garbage System” their degradation activity. Autolysis FUNCTION 1. Degradation of macromolecules like bacteria. 2. Degradation of worn out organelles 3. Secretory function PEROXISOME Similar in structure as lysosomes. Pinched off from ER Contains oxidative enzymes such as 1. 2. 3. catalase, urate oxidase D-amino acid oxidase. FUNCTION 1. 2. 3. 4. Degradation of toxic substances Hydrogen peroxide Oxygen utilization Breakdown of excess fatty acids Role in formation of myelin and bile acids. CENTRIOLE 2 cylindrical structure visible during cell division. Responsible for the movement of chromosomes during cell division. Arranged perpendicular to each other and is made up of proteins. LYSOSOMAL STORAGE DISEASES Hereditary disorders caused by a deficency in specific lysosomal acid hydrolases. Therefore, lysosomes are unable to degrade certain compounds, which accumulates and interfere with cell function. TAY-SACHS DISEASE TAY-SACHS DISEASE autosomal recessive disease caused by mutations in both alleles of a gene (HEXA) on chromosome 15. HEXA codes for the alpha subunit of the enzyme βhexosaminidase A. This enzyme is found in lysosomes, Normally, βhexosaminidase A helps to degrade a lipid called GM2 ganglioside, but in this case, the enzyme is absent allowing excessive accumulation of the GM2 ganglioside in the lysosomes of neurons. The large buildup of gangliosides in the neurons of the brain results in marked degenerative changes in the CNS, & death usually occurs before age 4 years. Symptoms: seizures, vision and hearing loss, mental retardation, and paralysis. No cure or treatment. PEROXISOMAL DISEASES ZELLWEGER SYNDROME Is a genetic disease in which normal peroxisomes are absent. Infants with this syndrome have profound neurological, liver, and kidney problems and usually die within a few months. There is no cure nor is there a standard course of treatment. ADRENOLEUKODYSTROPHY Is caused by the inability of peroxisomes to metabolize fatty acids. Therefore, lipids accumulate in the nervous system and adrenal glands, impairing their function. X-linked disease and affects males. Symptoms normally start between the ages of 4 and 10 and include loss of previously acquired neurologic abilities, seizures, ataxia, as well as degeneration of visual and auditory function. No cure for this therefore, infants die at young age. REFERENCE LIST http://my.clevelandclinic.org/disorders/Mitochondr ial_Disease/hic_Mitochondrial_Disease.aspx http://www.ninds.nih.gov/disorders/taysachs/taysa chs.htm http://rarediseases.about.com/cs/mitochondrialdis/ a/062103.htm http://en.wikipedia.org/wiki/Adrenoleukodystrophy