PowerPoint bemutató
advertisement

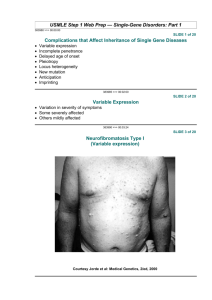
Autosomal monogenic inheritance Dr. habil. Kohidai Laszlo Department of Genetics, Cell- and Immunobiology Semmelweis University Budapest /2014/ Autosomal - Dominant Minimum one of the parents is affected Phenotype of homozygotes is more severe than heterozygotes Male and female are affected equally Male and female transmit evenly Affected x Non affected results 50%<affetced (sick) phenotype Vertical pedigree Frequency of the mutations shows correlation to the age of father AD mutations influence receptor, structural or carrier proteins Variable expressivity and penetrance Dominant autosomal ~ 2200 known dominant trait frequency 0.1-3/1000/birth most frequently affected organs: skeleton central nerve system 4p16.3 Achondroplasia Frequency 1:25000 FGFR3 gene mutation (fibroblast-growth factor receptor 3) Longitudinal growth of tubular bones is affected Limbs are affected forehead is dominant, middle part of the face is less developed Achondroplasia Rhinoceros unicornis Teleoceras fossiger sheeps FGFR3 gene locus: 4p16.3 DNA: 16.5 Kb; 19 exon; exon 1 is not known in human RNA: 4.0 Kb mRNS; alternative splicing exons 7 and 8: two mRNA isoforms IIIb and IIIc Expressed in: brain, cartilage, liver, kidney, inner ear The protein 806 aa; 115 kDa function: tyrosin kinase receptor structure: extracellular part 3 Ig-like loops (I, II, III) strongly hydrophobe TM domain (22 aa) -TM intracellular domain with ttyrosine kinase activity -TK Mutationsa of the FGFR3 gene 3 diseases are associated to the mutations of FGFR3 Arachnodactylia – Marfan syndrome Antoine Bernard-Jean Marfan (1896) Gabrielle Arachnodactylia – Marfan syndrome Tutankhamen pharaoh Ehnaton pharaoh Mary of Scotland Abraham Lincoln Marfan syndrome – Symptomes affected bones and joints height chest long fingers hyperflexibility Marfan szindróma - Symptomes Eye and vision myopia (short sight) axis of the eye is longer position of the lens is abnormál Heart and circulation valve prolapse aorta aneurysm hypotension Frequency of mutations is increasing by age Marfan syndrome Fibrillin gene (FBN1) 15q21.1 There are several mutations of fibrillin gene (see green bands) Fibrillin protein ~ 60 domain binds 47 Ca2+ similar to epidermal growth factor (EGF) Osteogenesis imperfecta I. blue sclera Penetrance 100% extremely fragile bones Deafness or loss of hearing (penetrance is less than 100%) Level of pleiotropy is high Osteogenesis imperfecta COL1A1 gene 17q21.31-q22 COL1A1 - 18 kb 52 exon ( 6 – 49: alpha helical domain) short exons: 45 bp, 54 bp or repeats of these two RNA: 2 RNA: 5.8 kb and 4.8 kb difference in 3’ UTR Protein : 140 kDa Structure of collagen fibre Healthy Osteogenesis imp. Type I. Central helical domain: - 338 x repeat of Gly-X-Y triplet - X and Y amino acids are frequently prolins (Pro) Osteogenesis Imperfecta: Mutation map of collagen Osteogenesis imperfecta Familiar hypercholesterinaemy Main clinical symptoms: - early onset of cardial and circulatory system diseases (myocardial innfarction, vascular diseases of brain and peripherial blood vessels) - xanthoma - diseases of the eye Familiar hypercholesterinaemy (FH) LDL lifespan in the body healthy: 2.5 days FH: 4.5 days LDL-level in sera is increased Reasons: - Mutation of LDL-receptor - ApoB defect LDL 19p13.1-13.3 Familiar hypercholesterinaemia Mutations of LDL-receptor Heterozygtes: 1:500-1000 Homozygotes: 1:1.000.000 Most frequent mutation: 9. exon 408 kodon CTG → CTA Val → Met Familiar hypercholesterinaemy Outcomes of LDL-receptor mutation Ligand kötő domain EGFP domain O-linked szénh.dom. Citopl. domain Membrán Joseph Goldstein, Michael Brown (Nobel Prize 1985) Trinucleotide-repeat diseases Huntington chorea Starts in age 35-44 Complex disease of locomotor, cognitive and psychiatric symptomes CAG trinucleotide repeats Number of CAG repeats: Normal - >26 Transient 27-35 Low penetrance 36-39 High penetrance above 40 Huntington chorea Huntington chorea - (CAGn) 4 Gain-of-function mutation The function of the Huntingtin gene in human is not known Huntington chorea Huntington chorea – CNS parts affected Huntington chorea Effects of huntingtin on gene level Inhibited expression of Dopamine D2 receptor gene Huntington chorea Effects on cytoskeleton level BDNF - brain-derived neurotrophic factor Transport of vesicles containing neurotransmitters via microtubular system: Huntingtin – huntingtin-related-protein (HAP) – dynactin - dynein Correlation between the ‘CAG’ repeat-number and the age of onset George Huntington (1850-1916) Grandfather and father were farmer doctors – their anamnestic files supported Huntington to describe the disease The disease was described in 1872 Medical and Surgical Reporter of Philadelphia chorea = maniac dance Trinucleotide-repeat CAG Anticipation – Trinucleotide repeat The disease is expressed in gradually more severe levels and earlyier in the offspring generations Sickle cell anemia Co-dominant Haemoglobinopathy HBB gene – 11 chrs. b-globin chain mutation HbS variant Haemoglobin structural change HbA HbS Due to the irregular structure of HbS: The membrane of RBC is damaged The affected RBCs are eliminated in great numbers in the peripheral organs, (e.g. kidney/spleen) The O2 transport is disturbed Malaria – spreading cycle Plasmodium falciparum Plasmodium vivax Selective advantage of heterozygotes: Sickle cell anemia Malaria Selective advantage of heterozygotes: Sickle cell anaemia Malaria BUT: The ratio is decreasing by changing the environment E.g. Cyprus – frequency of thalasszemia is decreasing Autosomal Recessive Parents of the affected person usually not expressing the trait, they are heterozygotes (Aa) Expression rate in male:female is 1:1 Transferred by male and females Co-saguinity of parents is frequent Risk to have an affected individual in the offspring is 25% (Aa x Aa) Horizontal pedigree Recessive autosomal 1700 known human recessive traits More than 15% is enzymopathy (e.g. phenylalanine hydroxylase, hexose aminidase) Mutations of haemoglobin Multiplex allelism – several mutations of one gene are responsible for the development of a symptome Complex heterozygote – an individual who possess two diverse, mutant allels of a gene mutáns allélját hordozza E.g. Cystic fibrosis – 850 different mutations Frequency of recessive diseases There is a significant difference in diverse ethnic groups REASON: reproductive advantage of heterozygotes to homozygotes ENVIRONMENTAL FACTORS Selective advantages of heterozygotes Other reasons: directed marriages in some ethnic groups b-Thalassemia (thalassa = sea) b-Thalassemia (thalassa = sea) haemoglobin b chain mutations short life span of RBCs O2 transporter capacity is decreased Cystic fibrosis Cystic fibrosis chief symptome is the obliteration of tubular organs affected organs: lung, pamcreas, gonads laboratory: sweat Cl- ion conc. increased deafness Selective advantage: Cholera – results high loss of Cl- ions Aa heterozygotes (aa – CF sick, AA – cholera infected, Aa CF–infected BUT no loss of Cl-ions) Cystic fibrosis deletion of F508 (Phe) deletion hits an ABC transporter role in Cl- ion release from the cell Cystic fibrosis chloride-channel mutation 1/25 frequency 850 mutation Development of clinical symptoms in cystic fibrosis Tay-Sachs disease GM2 gangliosid metabolism affected HAXA gene b-hexose aminidase enzyme is affected (lysosomes in neurones) CNS diseases: - paralysis - demency - blindness - early death GM2 gangliosid deposites in the brain (LM and TEM) Tay-Sachs disease Lysosome membrane phospholipid GM2 GM2 activator GM3 GM2 Hex A Tay-Sachs disease phosphorylation of Man is failed on HA-a subunit enzym is not transported into lysosomes Tay-Sachs disease Carrier frequency: 1:300 Askenazi Jewish population: 1:30 15q23-q24 Environmental effect: Galicia – bad hygenic conditions - high mortality due to TBC – AA homozygotes - high mortality due to Tay-Sachs – aa homozygotes Aa heterozygotes have reproductive advantage Sandhoff disease Both hexose aminidase A and B are affected GM2 ganglioside storage – toxic effects infantile, juvenile and adult forms mental ret., muscular probl., eye, organiomegaly Saskatchewan, Christian Maronits (non Ashkenazi Jewish origin) 5q12-q13 Other monogenic recessive autosomal diseases Other recessive autosomal diseases 12 Phenylketonuria – deficient phenylalanin hydroxylase 11, 15, 9, X Albinism - mental retardation - slow development - Tyr -> melanin synthesis is failed - Enzyme def.: failed tyrosinase enzyme - affected organs skin and eye - more known forms Galaktosemia Albino and normal kangaroos Albino Barking Deer Albino crab Albino dingo Effect of ethnic diferences on the expression of inherited diseases Factors complicating determination of inheritance • • • • • • • • New mutations Germ line mosaicism Late onset Decreased penetrance Variable expressivity Pleiotropy and heterogeny Genomic imprinting Anticipation Expressivity - level of expression of the inherited trait (severity of disease) Penetrancy – level of transmission of the trait from generation to generation Pleiotropy gene Phenylalanin-hydroxylase gene mutation Symptome < Syndrome Trait 1 Trait2 Pleiotropy: Stickler syndrome Collagen 2A1 mutation Symptomes: - Retina-ablation - Myopy - Cleft lip - Cleft palate - Size of mandibula is small - Problems with joints Heterogeny Forms: Gene 1 Gene 2 trait Phenotype ~ (clinical) – mutations of one gene –div. phenotype Allel ~ – allels of the same gene – phenotype is similar Locus ~ – genes are different – phenotype is similar Deafness Forms: Retinitis pigmentosa •Aut.Dom. •Aut. Rec. •X-linked Hirshprung dis. – Phenotype - Allel - Locus Cystic fibrosis Genotype Phenotype Environment ~ Phenocopy Retinoic acid-defic. 22q11 deletion - Aorta def. - Small ears - IQ decr. - Cleft palate