Assaying
advertisement

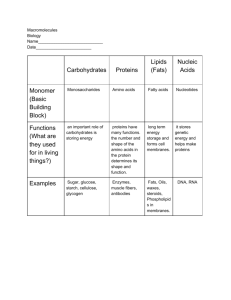
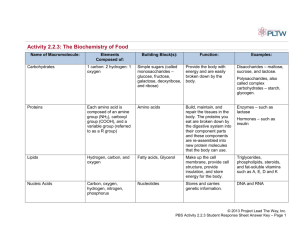
Course is an Introduction to Biotechnology with emphasis on lab methods Unit 1 Cells used and growth of cells Unit 2- DNA and DNA techniques Unit 3- Protein and Protein techniques Why are these things important in biotechnology -e.g. 1. To get a new protein product (a drug?) formed in cell have to be able to • grow cell, • recognise contaminants, • measure growth, • isolate new DNA to insert and perhaps cut or amplify it, • insert new DNA into cloning vector like a plasmid, • transform a bacterial cell • grow cell, monitor for protein production and then isolate protein -e.g. 2 Use all techniques to ask questions about how cells work at a molecular level -e.g. 3. Use techniques to develop diagnostic techniques Assaying Amino acids, and Proteins 1.Amino acids Structural building blocks of proteins 20+ amino acids, each differing only in the composition of the R groups. An R group could be a sulfydrl, another methyl, a string of methyls, rings of carbons, and several other organic groups. The general formula for an amino acid is represented in the following ways Parts of the Amino Acid: a) Amino group (NH2) b) Carboxyl group (COOH) c) R-group: variable- 2. Proteins Amino acids linked together sometimes with a non amino acid component Peptide is a short stretch (~50aa) of aa. Peptide bonds: Bond formed when 2 amino acids bond. peptide bond (See vertical arrow). Dipeptide- 2 amino acids joined by peptide bond. Polypeptide- many amino acids bonded together. Primary structure: specific amino acid sequence Secondary structure: H bonding to side groups, forms folds or coils Tertiary structure: Three dimensional shape. Final folded shape driven by hydrophobic interactions Quaternary structure: two or more polypeptide subunits linked Protein assays Protein quantitation is often necessary before processing protein samples for isolation, separation and analysis by chromatographic, electrophoretic and immunochemical methods. The criteria for choice of a protein assay are usually based on: • convenience, • availability of protein for assay, • presence or absence of interfering agents, • need for accuracy Assays can be quantitative or qualitative Need to be concerned with • Accuracy-proximity to true value • Precision- agreement between relicate data • Concentration range- range within which the method is accurate • Detection limit-minimum concentration that can be detected with a particular confidence level • Selectivity- extent method is free from interference due to other substances in the sample • Sensitivity- ability to discriminate between small differences in analyte concentration • Validation- process where accuracy and precision are checked in relation to specific standards Assays • Some labs have strict validation rules e.g. forensic labs. These include • adherence SOPs • calibration of assays using certified reference material • effective quality assurance and control systems • detailed record keeping Absorbance assays Monitors the absorbance of aromatic amino acids, tyrosine and tryptophan or if the wavelength is lowered, the absorbance of the peptide bond. Higher order structure in the proteins will influence the absorption Absorbance at 280 nm Range: 20 micrograms to 3 mg Accuracy: Fair Major interfering agents: Detergents, nucleic acids, particulates, lipid droplets Advantages Quick Sample can be recovered Useful for estimation of protein before using a more accurate method Well suited for identifying protein in column fractions Disadvantages Major interfering agents: Detergents, nucleic acids, particulates, lipid droplets Highly susceptible to contamination by buffers, biological materials and salts Protein amino acid composition is extremely important, thus the choice of a standard is very difficult, especially for purified proteins Absorbance is heavily influence by pH and ionic strength of the solution. This is often used to estimate protein concentration prior to a more sensitive method so the protein can be diluted to the correct range Colorimetric assays 1. Modified Lowry • The first step is a Biuret reaction which reduces Cu+2 to Cu+1 • The second reaction uses Cu+1 to reduce the Folin-Ciocalteu reagent (phosphomolybdate and phosphotungstate). This is detectable in the range of 500 to 750 nm – – – – Range: 2 to 100 micrograms Accuracy: Good Convenience: Fair Major interfering agents: Strong acids, ammonium sulfate Colorimetric assays: Modified Lowry Advantages • Sensitive over a wide range • The most commonly referenced procedure for protein determination • Can be performed at room temperature • 10-20 times more sensitive than UV detection • Can be performed in a microplate format Disadvantages • Many substances interfere with the assay • Alkaline copper reagent is laborious to prepare and will develop carbonate scales over storage which interfere with optical activity, thus it must be prepared fresh daily • Takes a considerable amount of time to perform • The assay is photosensitive, so illumination during the assay must be kept consistent for all samples • Amount of color varies with different proteins • Since reduced copper is detected in the procedure, make sure that the distilled water used in the procedure is fed from plastic lines and not copper lines Colorimetric assays 2. . Bradford assay – Range: 1 to 20 micrograms (micro assay); 20 to 200micrograms (macro assay) – Volume: 1 ml (micro); 5.5 ml (macro) – Accuracy: Good – Convenience: Excellent – Major interfering agents: None • Absorbance shift in Coomassie Brilliant Blue G250 (CBBG) when bound to arginine and aromatic residues • The anionic (bound form) has absorbance maximum at 595 nm whereas the cationic form (unbound form) has and absorbance maximum at 470 nm • 1-20 µg (micro assay) 20-200 µg (macro assay) Bradford assay cont. Advantages • Fast and inexpensive • Highly specific for protein • Very sensitive • Compatible with a wide range of substances • Extinction co-efficient for the dye-protein complex is stable over 10 orders of magnitude (assessed in albumin) • Dye reagent is complex is stable for approximately one hour Disadvantages • Absorbance spectra of the two Coomassie Brilliant Blue G-250 species partially overlap making the standard curve very important • Non-linear standard curve over wide ranges • Response to different proteins can vary widely, choice of standard is very important • The dye binds to quartz cuvettes so it is usually better to use glass or plastic cuvettes • The dye reagent is usually more convenient to purchase than to make, due to the use of phosphoric acid Colorimetric assays 3. BCA (Bicinchoninic Acid ) • The first step is a Biuret reaction which reduces Cu+2 to Cu+1 • In the second step BCA forms a complex with Cu+1 which it purple colored and is detectable at 562 nm • Range:0.2-50 µg • Accuracy: Good • Convenience: Good • Major interfering agents: Ammonium salts • • • • • • • Advantages Less susceptible to interference from common buffer substances Very sensitive and rapid if you use elevated temperatures Compatible with many detergents Working reagent is stable Very little variation in response between different proteins Broad linear working range • • Disadvantages The reaction does not go to completion when performed at room temperature or 37oC. This can be a problem if you are assaying a large number of proteins Dilution is often necessary for concentrated protein samples • Assaying Enzymes • When assaying enzymes want to know how much active enzyme you have. • 2 types of assay 1) fixed time assays, 2) kinetic • Fixed time assays are based on a single measurement and the relationship is known to be linear • Kinetic assays are based on product formation per unit time e.g grams per minute. Linearity has to be established. • Enzyme activity measured in Units Example Lactate + NAD+ LDH ↔ Pyruvate + NADH + H+ LDH = enzyme lactate dehydrogenase • The activity of LDH can be measured by measuring the rate of formation of the product NADH which can be measured by a change in absorbtion at 340nm. • The rate of formation of the product is dependent on the concentration of substrates and the concentration of products and the activity of the enzyme. • If start with lots of substrate and active enzyme the reaction will go forward and produce products • As the product concentration increases the forward reaction will slow down • Build up of product pushes the reaction backwards • To get a good measure of the enzyme activity need to measure it in the beginning of the reaction before the concentration of the products builds up enough to have an influence. • Before the concentration of products builds up the relationship between enzyme activity and product formation is linear. As the product accumulates this linear relationship often breaks down. • When you do an enzyme assay need to ensure it is in the linear range so need to plot the measurement (absorbance usually) versus time to see whether linear. • Use only the linear range.for calculation of number of units Enzyme activity is calculated by measuring it over short time periods to determine the initial velocity. Data from the linear portion of the curve only must be used Units of activity are calculated as below: Units = change in absorb/change in time (min) x 10 6uM/M x volume *6220 M -1 cm-1 = umol/min *6220 M -1 cm-1 = extinction coefficient for NADH, used to convert absorbance to moles. This would differ depending on the product.