FIGURE 5. Plot of peptide charge state ratios. Quality Control Concept
advertisement

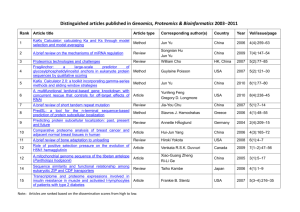
Rawputator – A New Tool to Combine Proteomic Data Mining with Method Development, Result Validation and Quality Control PP443 Martin Zeller, Bernard Delanghe, Christoph Henrich, Torsten Ueckert Thermo Fisher Scientific, Bremen, Germany The big advantage of having all that information in the search output is the additional connection to the search score. This can be used to evaluate the system performance. Overview Purpose: FIGURE 5. Plot of peptide charge state ratios. date 11/6/10 0:22 11/6/10 2:33 11/6/10 4:43 11/6/10 6:44 11/6/10 8:44 11/6/10 10:45 11/6/10 12:30 11/6/10 14:16 11/6/10 16:02 11/6/10 17:32 11/6/10 19:03 11/6/10 20:34 Implementation of proteomics data evaluation for quality control and method optimization. FIGURE 2. Rawputator node in Proteome Discoverer software. Methods: Hybrid linear ion trap-OrbitrapTM mass spectrometer with specialized proteomics software. Results: Rawputator tool delivers quantitative system performance assessment, method optimization and result verification. Introduction One of the biggest recurring questions in proteomics in a high-throughput environment is whether or not all parameters in the acquisition method are set optimally and the acquired data meets your quality criteria. Good practice therefore is to include quality controls in between the samples to assess the system performance. Figure 1 shows the system in a proteomic workflow which comprises sample preparation as well as LC, MS and data analysis. Why quality control? • Optimum performance of the analytical system for the reproducible analysis of complex samples • Identification of the source of the sub-optimal performance and variability Extraction of the dynamic parameter: - Precursor S/N - TIC - Ion inject time - Elapsed scan time - Effective collision energy [eV] - Lockmass correction Additional „costs“: - ~ 6% .msf file size increase - < 3 min per search job (complex cell lysate sample, 13665 MS/MS) dataset proteins 90 min 1 676 90 min 2 672 90 min 3 675 75 min 1 634 75 min 2 599 75 min 3 644 60 min 1 601 60 min 2 601 60 min 3 638 45 min 1 553 45 min 2 552 45 min 3 550 Quality Control Concept Lockmass Correction One example is to plot the lockmass correction over time (see Figure 3). Low (internal) lockmass correction values indicate on the one hand a valid external mass calibration and on the other hand indicate appropriate external mass calibration intervals. FIGURE 6. Outlook on a quality control system within Xcalibur / Proteome Discoverer software. FIGURE 3. Lockmass correction over time. date 11/6/10 0:22 11/6/10 2:33 11/6/10 4:43 11/6/10 6:44 11/6/10 8:44 11/6/10 10:45 11/6/10 12:30 11/6/10 14:16 11/6/10 16:02 11/6/10 17:32 11/6/10 19:03 11/6/10 20:34 • Evaluation of the impact of the changes or improvements in the analytical system • Documentation of the system performance metrics to support the assessment of the proteomic differences between biologically interesting samples Figure 6 shows a concept for the implementation of quality control as system suitability test as a system check before samples are analyzed and a concept for including a quality control sample in the sample queue. After the data acquisition of the quality control sample is finished, a quality control check is done automatically and the system is stopped if it does not fulfill minimum quality requirements. dataset proteins 90 min 1 676 90 min 2 672 90 min 3 675 75 min 1 634 75 min 2 599 75 min 3 644 60 min 1 601 60 min 2 601 60 min 3 638 45 min 1 553 45 min 2 552 45 min 3 550 1. System suitability test QC sample and QC analysis, QC sample re-injection after corrective actions if system shows low performance QC sample Methods QC ok? QC analysis YES real samples NO: Restore system performance Sample Preparation Different enzymatic degradation preparations of complex proteomes were prepared using standard protocols. 2. Quality control sample in sample sequence Liquid Chromatography QC sample in sequence (and sequence pause if QC shows low system performance) All samples were separated by a Thermo Scientific EASY-nLC II liquid chromatograph using a peptide trap (C18 BioBasic, 100 μm inner diameter, 2 cm length) and a C18 analytical column (C18 BioBasic, 75 μm inner diameter, 10 cm length, both NanoSeparations, NL), at a flow rate of 300 nL/min using standard data-dependent acquisition methods. Mass Spectrometry Proteomic data sets were acquired using a Thermo Scientific LTQ Orbitrap Velos hybrid mass spectrometer. Data Analysis Data analysis was done using Thermo Scientific Proteome Discoverer software evaluation version 1.3. FIGURE 1. Proteomic workflow. Sample preparation Proteome Mass spectrometry Data analysis Charge state ratios One example for a digestion quality check is the plot of the charge state ratios of all ions triggered for MS/MS. Figure 4 shows a plot of all precursor ions triggered for MS/MS per charge state and identified peptides per charge state for a single run. If the same enzyme is used for digestion and other instrument parameters are not changed then the ratio of the triggered precursor ions (2+/3+, 2+/4+, 3+/4+, ...) should be on the same level (see Figure 5). This plot also allows the identification of possible outliers. In this example the same sample is analyzed in triplicates with different gradient lengths. The number of identified proteins is for the 90-min and 45-min runs at the same level but for the 75-min and 60-min runs one outlier per triplicate can be spotted. For 75-min run 2 and for 60-min run 3 shows deviation from the normal distribution for the precursor ion charge states as well as in the number of identified proteins. FIGURE 4. Number of precursor ions triggered for MS/MS and identified peptides per charge state. QC sample real samples QC analysis QC ok? NO: Pause sequence, restore system performance and reinject samples Conclusion New Rawputator node allows Extraction of additional dynamic precursor ion information Diagnosis of instrument performance (LC + MS) and sample integrity Identification of outliers Method development and optimization References Ionization (ESI) Precursor ions Identified peptides 1. Rudnick PA et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol Cell Proteomics. 2010 Feb;9(2):225-41. 12000 10000 Digest YES: Continue with sample injections Full MS 8000 6000 2. Köcher T, Pichler P, Swart R, Mechtler K. Quality control in LC-MS/MS. Proteomics. 2011 Mar;11(6):1026-30. MS/MS 4000 2000 0 Identification Quantitation 2+ 3+ 4+ charge state Results Rawputator Node in Proteome Discoverer Software The Rawputator node can be easily added to any database search workflow within Proteome DiscovererTM software and extracts from the raw file additional dynamic precursor ion parameter per MS/MS spectrum as shown in Figure 2. The extracted values are stored in the .msf search output format. The additional time for the extraction is almost neglectable as well as the file size increase of the .msf output. 5+ 6+ All trademarks are the property of Thermo Fisher Scientific and its subsidiaries. This information is not intended to encourage use of these products in any manners that might infringe the intellectual property rights of others.