Pediatric Leukodystrophy

Leukodystrophy
Tyler Reimschisel, MD
September 6, 2013
Clinical Presentations of IMD
• Intoxication
– Urea cycle defects
• Energy Failure
– Mitochondrial disease
– Glycogen storage disease
• Complex Molecule
– Lysosomal storage disease
– Glycogen storage disease
– Peroxisomal storage disorders
Diseases that Cause
Leukodystrophy
Some examples
• Adrenoleukodystrophy
• Metachromatic leukodystrophy
• Tay-Sachs
• Krabbe
• Canavan
• Mitochondrial
Clinical Presentation of
Leukodystrophy
• Developmental delay: relentless regression
• Seizures
• UMN signs
• Failure to thrive (less common)
• +/- dysmorphisms
Testing for Leukodystrophy
• Lysosomal enzyme profile
• VLCFA (very long chain fatty acids)
• Urine organic acids
• Lactate
• Pyruvate: not clinically useful lab due to timing; in equilibrium with alanine
• Alanine (order via Plasma amino acids)
Pelizaeus-Merzbacher
• Xq22 mutation in proteolipid protein 1 (PLP1)
• Onset in first few months of life with rotary head movements, rotary nystagmus, & motor delay
• Then ataxia, tremor, choreoathetosis, spasticity
• Seizures
• Optic atrophy and ocular impairments
• MRI: Reversal of gray-white signal due to diffuse dymyelination
Pelizaeus-Merzbacher
Gal-GalNAc
Nana-Gal-Glc-Cer
GM
GalNAc
1
(
- Galactosidase)
Nana-Gal-Glc-Cer
GM
2
(
-Hexosaminidase A)
Nana-Gal-Glc-Cer
GalNAc-Gal-Gal-Glc-Cer
Sandhoff
(
-Hexosaminidase
A & B)
Gal-Gal-Glc-Cer
Fabry (
-Galactosidase)
Neuraminidase
Gal-Glc-Cer
SO
3
Glc-Cer
Gaucher (
-Glucosidase)
H-Gal-Cer Gal-Cer Ceramide
Phosphorylcholine-Cer
(Sphingomyelin)
Farber (Ceraminidase)
Sphingosine
Krabbe
• AR defect of galactocerebroside-betagalactosidase on chromosome 14
• Pure neurologic condition
• Onset at 3-8 months of age
• Irritability, intermittent fevers, heightened startle reflex, feeding problems
• Develop seizures, opisthotonus
• Deafness and blindness by 9 months
• MRI:
KRABBE DISEASE
Metachromatic Leukodystrophy
• AR defect of arylsulfatase-A
• Leukodystrophy as well as disease of adrenal glands, kidneys, pancreas, liver
Metachromatic Leukodystrophy
• 3 Presentations
– Late infantile (18-24 months)
• Gait disturbance, hypotonia to hypertonia, regression, involuntary movements, neuropathy, cherry red spot
– Juvenile (4-10 years)
• Bradykinesia, poor school performance, ataxia, movement disorder, neuropathy, slower progression
– Adult
• After puberty get personality and mental changes, cortical and cerebellar regression to frank dementia in third to fourth decade
Metachromatic Leukodystrophy
L.B.
• 4-year-old girl with GDD, hypotonia, & worsening ataxia
– Development at 12-18 month level
– Hyperactivity, inattention and aggression (Tenex)
• Family history
– Maternal cousin with chromosome deletion
– Paternal half-sister with B12 deficiency (?)
• Labs
– CMA, karyotype, FRX, purine/pyrimidines, biotinidase,
MECP2, AS/PWS, EEG, brain MRI (9/2010)
First Visit
• Labs: PAA, acylcarnitine profile, vitamin
B12, homocysteine, MMA level, creatine metabolites
• Repeat brain MRI consistent with MLD
Second Visit
• Lysosomal enzyme panel, VLCFA, coenzyme Q10 level
– Arylsulfatase A level 1.5 (low)
– GM1, mannosidosis, fucosidosis, Krabbe,
Tay-sachs normal
Follow Up Testing
• No mutations in arylsulfatase A gene
• Parental testing showed normal arylsulfatase A enzyme activity
Additional Testing
• Arylsulfatase B enzyme activity at 4-5% normal
• Huge peak of sulfatides in patient
• Multiple sulfatase deficiency diagnosed
• Molecular testing pending
Multiple Sulfatase Deficiency
• AR, mutations in sulfatase-modifying factor-1 gene
(SUMF1) on 3p26
• Austria: 1 in 1.4 million individuals
• Affects 12 sulfatase enzymes
– Post-translation modification defect in which cystein residue of enzyme is not activated
– Defect in enzyme that causes oxidation of a thiol group in cysteine to generate an alpha-formylglycine residue
– Alpha-formylglycine residue may accept the sulfate during sulfate ester cleavage by hydrolysis
– Examples: arylsulfatase, steroid sulfatase, heparan sulfatase, N-acetylglucosamine-6-sulfatase
Multiple Sulfatase Deficiency
• 3 phenotypes
– Neonatal MSD: severe mucopolysaccharidosis
– Late infantile MSD: late-onset MLD
– Juvenile MSD
• Combined features of MLD, Hunter,
Sanfilippo A, Morquio, Maroteaux-Lamy, Xlinked ichthyosis
Canavan
• AR deficiency of asparto-acylase
• Macrocephaly , lack of head control, and developmental delays by the age of three to five months
• Develop severe hypotonia and failure to achieve independent sitting, ambulation, or speech
• Hypotonia eventually changes to spasticity
• Life expectancy is usually into the teens
• Diagnosis of Canavan disease relies upon demonstration of very high concentration of N-acetyl aspartic acid (NAA) in the urine
Canavan disease
Courtesy Dr Isabelle Desguerre, Paris Necker Hospital
Canavan disease
NAA
Courtesy Dr. Ralph Lachman
L-2-Hydroxyglutaric Aciduria
• Underlying defect unknown
• Clinical
– Normal to mild delays in infancy and early childhood
– Slowly progressive encephalopathy
– Variable rate of progressive ataxia, seizures, pyramidal signs, movement disorder (dystonia, tremor, choreoathetosis), dementia
– 50% with macrocephaly
• Laboratory: no metabolic decompensation, increased plasma lysine, elevated 2hydroxyglutaric acid in urine
Brain MRI
L-2-Hydroxyglutaric Aciduria
• Neuroimaging
– Severe cerebellar atrophy
– Mildly swollen white matter with gyral effacement
– Leukoencephalpathy more prominent closer to cerebral cortex
– Increased signal intensity in dentate and striatum
• Differential Diagnosis
– D-2-hydroxyglutaric aciduria presents earlier
– GAII causes elevations of D-2-hydroxyglutaric acid
• Treatment - none
Alexander Disease
• AD mutation in GFAP at 17q21.31
• Onset at around 6 months (birth – 2 yrs)
• Psychomotor regression, spasticity and seizures
• Juvenile patients have ataxia and spasticity
• Adult patients have MS-like presentation
• Diffuse demyelination, especially in frontal lobes
Alexander Disease
Brain MRI: Leigh Syndrome
From Osborn. Neuroradiology , 2000
Brain MRI
From Osborn. Neuroradiology , 2000
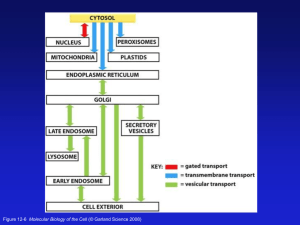
Peroxisome Function
• Synthesis
– Plasmologens (ether-phospholipids)
– Bile acid from mevalonate
• Catabolism
–
-oxidize very long chain fatty acids (esp C24:0 and C26:0), pristanic acid and bile acid intermediates
–
-oxidize phytanic acid (chlorophyll derivative) to pristanic acid
– Lysine via pipecolic acid and glutaric acid
– Glyoxylate to prevent conversion to oxalate
Peroxisomal Disorders
• 16 disorders
– 15 are autosomal recessive
– 1 is X-linked (adrenoleukodystrophy)
• Predominant features
– Dysmorphisms
– Neurologic dysfunction
– Liver disease
Peroxisomal Disorders
• Biosynthesis Defects
– Zellweger spectrum disorders (ZD, IRD, NR)
– Rhizomelia chondrodysplasia punctata
• Single Peroxisomal Enzyme Deficiencies
– Adrenoleukodystrophy (ABCD1 on Xq28)
– RCDP type 2 (GNPAT on 1q42.1-42.3)
– RCDP type 3 (AGPS on 2q33)
– Refsum (PHYH/PAHX on 10p15-p14)
– Glutaric aciduria type 3 (?)
– Mulibrey nanism (TRIM on 17q22-23)
– 9 others
Peroxisomal Biogenesis
• Peroxisomes multiply by division
• Proteins carried from free polyribosomes to peroxisomes by peroxisomal targeting signals (PTS)
• PTS1
– Last 3 carboxy terminal amino acids
– PTS1 receptor encoded by PEX5
• PTS2
– Stretch of 9 amino acids
– PTS2 receptor encoded by PEX7
Peroxisomal Biogenesis
• PTS receptors deliver proteins to peroxisomal protein import machinery
• Import machinery transports proteins across membrane
• Transporter complex has at least 15 peroxins ( PEX1, 2, 3, 5, 6, 10, 12, 13,
14, 16, 19, 26)
Zellweger Spectrum Disorders
• CZ, NALD, and IRD
• Genetic heterogeneity
• Dysmorphism
(large fontanelle, high forehead, abn ears, micrognathia, low/broad nose, redundant skin folds)
• Neuronal migration disorders and delayed myelination
• Seizures
• Hypotonia
• Sensorineural deafness
• Ocular abnormalities
(retinopathy, cataracts, ON atrophy)
• Liver disease
(hepatomegaly, cholestasis, hyperbilirubinemia)
• Failure to thrive
• Death in first year of life
Zellweger Syndrome
From Google Images
ZELLWEGER SYNDROME
Zellweger Spectrum Disorders
• Classic Zellweger (CZ)
• Neonatal adrenoleukodystrophy (NALD)
– Somewhat less severe than CZ
– May lack dysmorphisms altogether
– Neonatal or infantile onset of seizures, hypotonia, and progressive leukodystrophy
– May have pachypolymicrogyr ia
• Infantile Refsum disease (IRD)
– Least severe phenotype, regression over time
– May be asymptomatic at birth
– No progressive leukodystrophy
– Variable expressivity of cognitive dysfunction
– Deafness and vision changes (retinopathy)
– May survive to adulthood
Adrenoleukodystrophy/
Adrenomyeloneuropathy
• Most common peroxisomal disorder (1/20,000)
• Mutation in ABCD on Xq28 leads to defect in peroxisomal uptake of VLCFA
• ALD: progressive neurologic disorder that begins at 5-
12 years
– Boys with new onset school difficulties & ADHD
– Visuo-spatial deficits and hearing loss
– Spasticity, ataxia, maybe seizures
– Hypoglycemia, salt losing, hyperpigmentation
– Rx: steroids, presymptomatic stem cell transplant, Lorenzo ’ s oil ineffective (oleic and erucic acids )
• AMN: early adulthood progressive spastic paraparesis, cerebral demyelination (males)