Practical Guide to the ENCODE project
advertisement

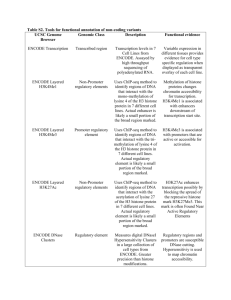
Practical Guide to the (mod)ENCODE project February 27 2013 Fundamental Goals • Improve comprehensiveness and accuracy of gene annotation • Define novel protein coding and noncoding gene products, including variants • Define noncoding regulatory elements, including both sequence and epigenetic features • Begin to measure the extent of tissue-specific deployment of functional elements Rationale for the Consortium • Synergistic expertise of large groups • Coordinated sample and data collection procedures • Systematic data analysis • Rapid release of the data to the public • Common data repository History and Relationship of ENCODE Projects U. S. National Human Genome Research Institute 2007-2012 modENCODE (100% of genome) 2003-2007 pilot human ENCODE (1% of genome) C. elegans Waterston/Celniker (transcribed elements) Piano/Lai (3’ UTR elements) 2007-20?? human ENCODE scale-up (100% of genome) Drosophila Snyder/White (TF binding sites) Lieb/Karpen (chromatin function) Henikoff (histone replacement) Model organism advantages… • Compact, well-annotated “simpler” genome • Functional elements can be identified in vivo • Experimental advantages for both generating and interpreting genomic data …and disadvantages • Not human • Most studies performed in whole animals modENCODE Publications of the “half-way point” in Science Dec 2010: 237 C. elegans datasets and >700 Drosophila datasets Verified data available at http://www.modencode.org Defining the transcriptome Extract total RNA, mRNA, and small RNAs from samples taken at distinct developmental stages and conditions L4 male adult hermaphrodite early embryo L4 dauer L3 late embryo L1 L2 C. elegans transcriptome features and alternative splicing increase in splice junction confirmation fractional differences in isoform composition for 12,875 genes in pair-wise comparison across seven developmental stages M B Gerstein et al. Science 2010;330:1775-1787 stage-specific isoforms stage-specific pseudogene expression Drosophila coding and noncoding genes and structures malespecific expression combine RNAseq data with conserved structures novel miRNA found in protein coding exon Roy et al. Science 2010;330:1787-1797 Tagging (worm) vs endogenous (fly) TF-ChIP Generate antibodies to proteins of interest Create GFP-tagged transcription factor fosmids by recombineering Generate transgenic lines by microparticle bombardment Characterize sensitivity and specificity Characterize expression and culture large scale preps culture large scale preps Perform ChIP-seq define binding sites and analyze data 10 C. elegans Highly Occupied Target (HOT) Regions 22TFs -> 304 HOT regions with 15+ TFs M B Gerstein et al. Science 2010;330:1775-1787 tend to be at the promoters of broadly expressed genes Discovery and characterization of chromatin states and their functional enrichments in Drosophila 30 discrete -> 9 continuous chromatin states Roy et al. Science 2010;330:1787-1797 Statistical models predicting TF-binding and gene expression from chromatin features in C. elegans an example color represents accuracy of statistical model in which a chromatin feature(s) acts as a predictor for TF binding/HOT regions Chromatin based predictions for expression of both coding genes (top) and miRNAs (bottom) Spearman correlation coefficient of each chromatin feature with expression levels M B Gerstein et al. Science 2010;330:1775-1787 Predictive models of regulator, region, and gene activity in Drosophila predicting cell type specific regulators of chromatin activity Roy et al. Science 2010;330:1787-1797 predicting target gene expression from regulator expression DREM: Dynamic Regulatory Events Miner Human (and mouse) ENCODE PLoS Biol 9:e1001046, 2011 ENCODE methods and organization PLoS Biol 9:e1001046, 2011 PLoS Biol 9:e1001046, 2011 Selected cell lines Standardized data collection and processing • • • • • • cell growth conditions antibody characterization requirements for controls requirements for replicates assessment of reproducibility data submission formats Caveats • • • • assays on unsynchronized cell populations several of the cell lines are karyotypically unstable some Tier 3 lines could be of heterogenous composition mappability in the human genome is variable and repetitive sequences (~15% of the genome) are not included currently • variable confidence regarding assigned function for the different types of elements • data types lacking focal enrichment (spread over broad regions) could have variation across the enriched domain Programs utilized for data analysis PLoS Biol 9:e1001046, 2011 Location of data sources PLoS Biol 9:e1001046, 2011 Exploring the ENCODE analysis http://www.nature.com/encode/#/threads Companion Papers In the same issue of Nature (6 September 2012): Landscape of transcription in human cells Djebali, S., Davis, C.A. et al. The accessible chromatin landscape of the human genome Thurman, R.E., Rynes, E., Humbert , R. et al. An expansive human regulatory lexicon encoded in transcription factor footprints Neph, S., Vierstra, J., Stergachis, A.B., Reynolds, A.P. et al. Architecture of the human regulatory network derived from ENCODE data Gerstein, M.B., Kundaje, A., Hariharan, M., Landt, S.G., Yan, K.K. et al. The long-range interaction landscape of gene promoters Sanyal, A., Lajoie, B.R. et al. In Genome Biology (6 September 2012): Analysis of variation at transcription factor binding sites in Drosophila and humans Spivakov, M. et al. Genome Biol. Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3 Frietze, S. et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription related factors Yip, K.Y. et al. Functional analysis of transcription factor binding sites in human promoters Whitfield, T.W. et al. Analysis of variation at transcription factor binding sites in Drosophila and humans Spivakov, M. et al. Modeling gene expression using chromatin features in various cellular contexts Dong, X. et al. The GENCODE pseudogene resource Pei, B. et al. Companion Papers In Genome Research (6 September 2012): Annotation of functional variation in personal genomes using RegulomeDB. Boyle, A.P. et al. ChIP-seq guidelines and practices used by the ENCODE and modENCODE consortia. Landt, S.G. et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs Tilgner, H. et al. Discovery of hundreds of mirtrons in mouse and human small RNA data Ladewig, E. et al. GENCODE: The reference human genome annotation for the ENCODE project Harrow, J. et al. Linking disease associations with regulatory information in the human genome. Schaub, M.A. et al. Long noncoding RNAs are rarely translated in two human cell lines Bánfai, B. et al. Sequence and chromatin determinants of cell-type–specific transcription factor binding. Arvey, A. et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors Wang, J. et al Combining RT-PCR-seq and RNA-seq to catalog all genic elements encoded in the human genome Howald, C. et al. Personal and population genomics of human regulatory variation. Vernot, B. et al. Predicting cell-type–specific gene expression from regions of open chromatin. Natarajan, A. et al. RNA editing in the human ENCODE RNA-seq data Park, E. et al. GENCODE Harrow et al., 2012 • GENCODE is a manual/automated curation of genes • annotation is verified by RT-PCR and RACE experiments • v7: 20,687 protein-coding genes with, on average, 6.3 alternatively spliced transcripts (3.9 different protein-coding transcripts) per locus Frankish et al., Genome Research 2012 TF mapping by ChIP-seq across 72 cell lines data is organized in “Factorbook” www.factorbook.org Encode Project Consortium, Nature 489: 57-74, 2012 Chromatin accessibility mapping • 2.89 million unique, non-overlapping DNase I hypersensitive sites (DHSs) by DNase-seq in 125 cell types • 4.8 million sites across 25 cell types that displayed reduced nucleosomal crosslinking by FAIRE, many of which coincide with DHSs • DNA methylation by RRBS [average of 1.2 million CpGs in each of 82 cell lines and tissues (8.6% of non-repetitive genomic CpGs), including CpGs in intergenic regions, proximal promoters and intragenic regions (gene bodies)] Encode Project Consortium, Nature 489: 57-74, 2012 Histone modification mapping 12 histone modifications and variants in 46 cell types, including a complete matrix of eight modifications across tier 1 and tier 2. Modelling transcription levels from histone modification and transcription-factor-binding patterns histone modifications TFs Encode Project Consortium, Nature 489: 57-74, 2012 Patterns and asymmetry of chromatin modification at transcription-factor-binding sites histone modifications show asymmetric patterns across TFBS Encode Project Consortium, Nature 489: 57-74, 2012 Co-association between transcription factors Encode Project Consortium, Nature 489: 57-74, 2012 Integration of ENCODE data by genome-wide segmentation Label CTCF E PF R TSS T WE Description CTCF-enriched element Predicted enhancer Predicted promoter flanking region Predicted repressed or low-activity region Predicted promoter region including TSS Predicted transcribed region Predicted weak enhancer or open chromatin cis-regulatory element Encode Project Consortium, Nature 489: 57-74, 2012 High-resolution segmentation of ENCODE data by selforganizing maps (SOM) Encode Project Consortium, Nature 489: 57-74, 2012 Allele-specific ENCODE elements Chrom HMM segments single genes Encode Project Consortium, Nature 489: 57-74, 2012 Examining ENCODE elements on a per individual basis in the normal and cancer genome Comparison of genome-wide-association-study-identified loci with ENCODE data UCSC broswer Browser interface http://encodeproject.org -> Genome Browser link both hg18 and hg19 genome versions are available and worth viewing – hg18 has the “Integrated Regulation Track” on by default, while hg19 has newer and more datasets PLoS Biol 9:e1001046, 2011 UCSC browser visualization of ENCODE data novel independent transcript in the first intron of TP53 session includes proteogenomics data in conjunction with ENCODE gene, transcriptome and regulatory data sets Roadmap Epigenomics Project next-generation sequencing technologies to map DNA methylation, histone modifications, chromatin accessibility and small RNA transcripts in stem cells and primary ex vivo tissues selected to represent the normal counterparts of tissues and organ systems frequently involved in human disease rapid release of raw sequence data, profiles of epigenomics features and higher-level integrated maps to the scientific community development, standardization and dissemination of protocols, reagents and analytical tools to enable the research community to utilize, integrate and expand upon this body of data Epigenomics Data www.roadmapepigenomics.org/data Epigenomics Data www.roadmapepigenomics.org/data Databases, data visualization, and access modENCODE: http://www.modencode.org http://www.intermine.modencode.org http://www.modencode.org/publications/worm_2010pubs/ http://www.wormbase.org http://www.flybase.org ENCODE: http://www.encodeproject.org http://www.genome.ucsc.edu/ENCODE/ http://www.genome.ucsc.edu/ENCODE/downloads.html http://www.factorbook.org Epigenomics RoadMap: http://nihroadmap.nih.gov/epigenomics http://ncbi.nlm.nih.gov/epigenomics http://www.epigenomebrowser.org http://genomebrowser.wustl.edu/ http://epigenomegateway.wustl.edu/