Good Clinical Practices - World Health Organization
advertisement

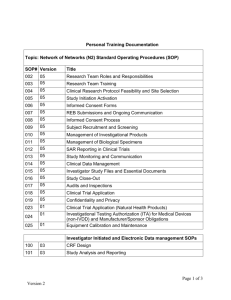
Good Clinical Practices Guilin, PRC Dr AJ van Zyl for Quality Assurance and Safety: Medicines Medicines Policy and Standards Health Technology and Pharmaceuticals Cluster World Health Organization Program • Thursday: •Presentation on guidelines: •GCP, GLP, CRO •Group sessions •Clinical and bio-analytical • Friday: •Presentation on GMP •Group sessions •Presentation on GMP •Group sessions Outline of presentation Bio-equivalence studies • Good Clinical Practices (GCP) • Good Practices for Quality Control Laboratories (GPQCL) • Good Laboratory Practices (GLP) • Good Practices for Contract Research Organizations (GPCRO) Guidelines GCP World Health Organization WHO Technical Report Series, No. 850, 1995, Annex 3 GLP UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR) HANDBOOK GOOD LABORATORY PRACTICE (GLP) CRO DRAFT ADDITIONAL GUIDANCE FOR ORGANIZATIONS PERFORMING IN VIVO BIOEQUIVALENCE STUDIES[1] [1] The present working document QAS/05.120 always Good Clinical Practices (GCP) 1. PROVISIONS AND PREREQUISITES FOR A CLINICAL TRIAL 1.1 Justification for the trial 1.2 Ethical principles 1.3 Supporting data for the investigational product 1.4 Investigator and site(s) of investigation 1.5 Regulatory requirements 2. THE PROTOCOL 3. PROTECTION OF TRIAL SUBJECTS 3.1 Declaration of Helsinki 3.2 Ethics committee 3.3 Informed consent 3.4 Confidentiality GCP 4. RESPONSIBILITIES OF THE INVESTIGATOR 4.1 Medical care of trial subjects 4.2 Qualifications 4.3 Selection of trial subjects 4.4 Compliance with the protocol 4.5 Information for subjects and informed consent 4.6The investigational product 4.7 The trial site 4.8 Notification of the trial or submission to the DRA 4.9 Review by an ethics committee 4.10 Serious adverse events or reactions 4.11 Financing 4.12 Monitoring, auditing and inspection 4.13 Record-keeping and handling of data 4.14 Handling of and accountability for pharmaceutical products for trial 4.15 Termination of trial 4.16 Final report 4.17 Trials in which the investigator is the sponsor GCP 5. RESPONSIBILITIES OF THE SPONSOR 5.1 Selection of the Investigator(s) 5.2 Delegation of responsibilities 5.3 Compliance with the protocol and procedures 5.4 Product information 5.5 Safety information 5.6 Investigational product 5.7 Trial management and handling of data 5.8 Standard operating procedures 5.9 Compensation for subjects and investigators 5.10 Monitoring 5.11 Quality assurance 5.12 Study reports 5.13 Handling of adverse events 5.14 Termination of trial GCP 6. RESPONSIBILITIES OF THE MONITOR 6.1 Qualifications 6.2 Assessment of the trial site 6.3 Staff education and compliance 6.4 Data management 6.5 Case-report forms 6.6 Investigational product 6.7 Communication 6.8 Notification of the trial or submission to the regulatory authority 6.9 Reports 7. MONITORING OF SAFETY 7.1 Handling and recording adverse events 7.2 Reporting adverse events 8. RECORD-KEEPING AND HANDLING OF DATA 8.1 Responsibilities of the investigator 8.2 Responsibilities of the sponsor and the monitor 8.3 Archiving of data GCP 9. STATISTICS AND CALCULATIONS 9.1 Experimental design 9.2 Randomization and blinding 9.3 Statistical analysis 10. HANDLING OF AND ACCOUNTABILITY FOR PHARMACEUTICAL PRODUCTS 10.1 Supply and storage 10.2 Investigational labelling and packaging 10.3 Responsibilities of the investigator 10.4 Responsibilities of the sponsor and the monitor 11. ROLE OF THE DRUG REGULATORY AUTHORITY 11.1 General responsibilities 11.2 On-site inspections 12. QUALITY ASSURANCE FOR THE CONDUCT OF A TRIAL ) CLINICAL Good Practices for Quality Control Laboratories (GPQCL) Part One. Management and infrastructure 1. Organization and management 2. Quality system 3. Control of documentation 4. Records 5. Data processing equipment 6. Personnel 7. Premises 8. Equipment, instruments and other devices Part Two. Materials and set-up of equipment, instruments and other devices 9. Specifications archive 10. Reagents 11. Reference materials 12. Calibration, validation and verification of equipment, instruments and other devices 13. Traceability GPQCL Part Three. Working procedures 14. Incoming sample 15. Analytical worksheet 16. Testing 17. Evaluation of test results 18. Retained samples Part Four. Safety in pharmaceutical control laboratories 19. General rules Good Laboratory Practices (GLP) INTRODUCTION TO GLP AND ITS APPLICATION The history of GLP What is GLP? GOOD LABORATORY PRACTICE TRAINING INTRODUCTION THE FUNDAMENTAL POINTS OF GLP Resources Rules Characterization Documentation Quality assurance RESOURCES Facilities: buildings and equipment Personnel RULES FOR THE CONDUCT OF STUDIES General aspects The study plan or protocol Standard Operating Procedures (SOPs) GLP CHARACTERIZATION6 The test item Test system DOCUMENTATION – RAW DATA AND DATA COLLECTION Carrying out procedures and recording observations Records and recording QUALITY ASSURANCE UNIT Protocol (or study plan) review SOP review Planning (Master schedule, inspection plan) Audits and inspections Quality assurance statement QAU inspections of suppliers and contractors The distribution and archiving of QAU files and reports Guidelines This presentation will focus on guidelines for CROs, then GCP and GLP What is a CRO: WHO: "any organization involved in the conduct or analysis of in vivo bioequivalence studies". Per ICH Tripartite Harmonized Guidelines: "a person or an organization (commercial, academic or other) contracted by the sponsor to perform one or more of a sponsor's trial-related duties and functions" Research Organizations Scope: Guidance to organizations involved in the conduct and analysis of in vivo bioequivalence (BE) studies Note: BE studies should be performed in compliance with: • General regulatory requirements • Good practices in the WHO bioequivalence guideline, • Good clinical practice (GCP) • Good laboratory practices (GLP) Research Organizations Guideline provides information on: - - organization and management; study protocols; clinical phase of a study; bio-analytical phase of a study; pharmacokinetic and statistical analysis; study report. Research Organizations • ORGANIZATION & MANAGEMENT • COMPUTER SYSTEMS Hardware Software Data Management • ARCHIVE FACILITIES • PREMISES • CLINICAL PHASE • CLINICAL LABORATORY • PERSONNEL • QUALITY ASSURANCE Research Organizations • ETHICS COMMITTEE Informed Consent • MONITORING • INVESTIGATORS • RECEIVING, STORAGE AND HANDLING OF INVESTIGATIONAL DRUG PRODUCTS • CASE REPORT FORMS • VOLUNTEERS, RECRUITMENT METHODS • DIETING Research Organizations • SAFETY, ADVERSE EVENTS, ADVERSE EVENT REPORTING • SAMPLE COLLECTION, STORAGE AND HANDLING OF BIOLOGICAL MATERIAL • LABORATORY PHASE (BIOANALYTICAL DATA) • DOCUMENTATION • PHARMACOKINECTIC & STATISTICAL CALCULATIONS • CLINICAL STUDY REPORT Research Organizations Organization and management • Legal requirements • Organization chart •Key positions, names, authorized • Job descriptions and responsibilities • List of signatures Research Organizations Computer systems • Hardware •Sufficient •Data entry and handling, calculations, reports •Capacity and memory •Access control • Software •Suitable program •Written procedures: program, virus tests, archiving, back-ups Research Organizations Software can manage: • Word processing, • Data entry, • Databases, • Graphics, • Pharmacokinetics and • Statistical programmes • Computer systems validated Research Organizations Data management: • • • • • Includes transfer of the data from case report forms (CRF), analytical data for pharmacokinetic and statistical analysis and reporting SOPs designed to prevent errors Double entry of the data Data validation methodology (proofreading, double data entry, electronic logical control) in writing Changes to data entered in database - authorized persons only - specified and documented Research Organizations ARCHIVE FACILITIES Sufficient and appropriately secure storage space, fire proof, archiving trial-related documentation and product samples • SOP for archiving. • Access to areas restricted and controlled • Archiving period - documentation including raw data - product samples retained - defined in the SOP Research Organizations PREMISES • Conditions to ensure (consideration) adequate safety for the subjects stage of development of the product potential risk involved • Sufficient space (personnel and activities) • Adequate facilities, including laboratories • Clinical phase: Areas well organized, activities in order Entry restricted and controlled logical Research Organizations • Laboratories with sufficient space to avoid mixups, contamination and cross-contamination, adequate, suitable storage space for samples, standards, solvents, reagents and records. • Alarm system or adequate monitoring system to control the temperature of the critical stage areas. • Automatic alarm system tested regularly • Daily monitoring and all the alarm checks should be documented. • Access to telephone, E-mail and facsimile facilities to ensure proper communication and necessary office equipment (printer, copymachine) to perform the required activities Research Organizations Clinical Phase • Sufficient space • Where appropriate, beds should be available (overnight stay/ type of trial/ investigational drug) • Facilities for: changing and storing clothes Washing and toilets - easily accessible and appropriate Research Organizations Other rooms or areas: • Volunteer screening; • "Clinic" for volunteers; • Ancillary areas; • Pharmaceutical operations (e.g. storage, repacking) • Administration of investigational drug(s) and sample collection; • Sample processing (e.g. plasma separation) and storage (freezer); • Controlled storage areas for study materials, medication and documentation including CRFs; • Preparation of standardized meals; • Emergency or first-aid equipment and appropriate rescue medication for emergencies Research organizations CLINICAL LABORATORY • A qualified clinical laboratory for analysing the screening samples. • As per protocol: Haematological tests, urine analysis and other tests • Information about analytical methods used, a dated list of laboratory normal ranges and accreditation certificate of the laboratory, if available. • Curriculum vitae of the responsible analyst • Actual original results (including raw-data) of all the tests performed should be documented and should be included in the CRFs Research organizations PERSONNEL • Sufficient number of qualified personnel Key persons with appropriate responsibilities: • Medical/Scientific director • Principal investigator • Quality assurance manager • Technical manager • Quality Control manager • • • • • Quality assurance should be independent, reporting structure Contract workers allowed Current curriculum vitae and training records Appropriate qualifications and sufficient knowledge Records for training and assessment - GCP and GLP Research organizations QUALITY ASSURANCE • Appropriate quality assurance (QA) system QA unit responsible for: • Verifying all activities; • Quality assurance systems, SOPs; • Verifying data for reliability and traceability; • Planning and performing self-inspections; • Contract facilities - including auditing of such facilities. The CRO should allow the sponsor to monitor the studies and to perform audits of the clinical and analytical study and sites Research organizations ETHICS COMMITTEE • Trials approved beforehand • Independent from the promoter, the investigator, the CRO • Discussions, recommendations and decisions in detailed minutes of the meeting • Sufficient time for reviewing protocols and ICFs Informed consent • Language and a level understandable • Both orally and in writing • Given by the subject, documented, before start • Participation is voluntary, the right to withdraw without having to give a reason • Compensation paid pro rata temporis • If reasons given, included in the study records • Subject access to information about insurance, and other procedures for compensation or treatment Research organizations MONITORING Note: Monitoring is an essential part of the clinical trial. Qualified monitor • Ensure compliance with the protocol, GCP, GLP and applicable ethical and regulatory requirements • Completion of CRFs and verification of the accuracy of data obtained • Pre- and post-study visit as well as a monitoring visit during the conduct of the trial • Written report after each site visit • CRO: SOPs concerning the visit procedures, extent of source data verification, drug accountability and adherence to the protocol. • Monitor: SOPs (with checklists) - initiation visit, routine monitoring visits and a closing visit Research organizations INVESTIGATORS • Principal investigator: overall responsibility for the clinical conduct of the study • Appropriate qualifications, trained, experience • At least one investigator practice medicine by law • Responsible for the integrity, health and welfare of the subjects during the trial, and the accurate documentation of all trial-related clinical data. • Permanent employees or external investigators contracted and adequately trained Research organizations RECEIVING, STORAGE AND HANDLING OF INVESTIGATIONAL DRUG PRODUCTS • Records: for receipt, storage, handling and accountability of investigational and comparator products – all stages of the trial. • Information about: the shipment, delivery, receipt, storage (including storage conditions), dispensing, administration, reconciliation, return and/or destruction • Product used: dosage form and strength, lot number, expiry date, and other coding that identifies the specific characteristics of the product tested. Research organizations RECEIVING, STORAGE AND HANDLING OF INVESTIGATIONAL DRUG PRODUCTS • Samples in the original container retained • Suitable location within the CRO (pharmacy) • Under appropriate storage conditions • In a securely locked area accessible only to authorized persons • Randomization and dispensing, including the labelling of drug products - SOP and records • Reconciliation verified by a second responsible person Research organizations CASE REPORT FORMS • • • • • • • Case report forms (CRFs) to record data on each subject Procedure for designing CRFs Sample CRF should be appended to the protocol. Guarantee preservation, retention and retrieval of volunteer information Reflect the actual results obtained during the study and allow easy access to verification, audit and inspection of the data. Investigator's certification of the accuracy of CRFs Errors or omissions – clarified, corrected, dated and signed and explained Research organizations VOLUNTEERS, RECRUITMENT METHODS Note: Pool of healthy volunteers - medically tested and selected. •Informed consent for any screening procedures required to determine eligibility for the study, in addition to informed consent for participation in the research portion of the study. •Subject selection criteria (inclusion and exclusion criteria) and recruitment procedures should be described in the clinical trial protocol. Research organizations DIETING Meals can significantly affect absorption of drugs • Fasting and meals should be standardized and adequately controlled • Arrange for standardized meals, snacks and drinks protocol. • Records should be maintained for timing, duration and amount of food and fluids consumed. Research organizations SAFETY, ADVERSE EVENTS, ADVERSE EVENT REPORTING • Appropriate study planning - evaluation of risk • First-aid emergency equipment and appropriate rescue medication • Adequate facilities of the proper care • Investigator(s) responsible for: medical decisions notifying the relevant health authorities, the sponsor and, when applicable, the EC, without delay in the case of serious adverse events. • Adverse event registration and reporting forms Research organizations SAMPLE COLLECTION, STORAGE AND HANDLING OF BIOLOGICAL MATERIAL • Samples (serum, plasma, or urine), sampling method, volume and number of samples - in the clinical trial protocol and the information provided to the volunteer. • SOPs for the collection, preparation, transport and storage of samples • Actual sampling times and deviations recorded. • Labelling of samples clear - identification and traceability Research organizations SAMPLE COLLECTION, STORAGE AND HANDLING OF BIOLOGICAL MATERIAL • Storage conditions of samples • All storage conditions (e.g. temperature in the freezer) protocol - controlled, monitored and recorded throughout the storage period and transportation. • System failure. •Storage and retrieval of samples •Duplicate or backup samples - stored and shipped separately. •Local requirements for the handling and destruction Research organizations BIOANALYTICAL DATA (LABORATORY PHASE) Note: Same CRO or contracted to another laboratory or CRO • • • GLP to non-clinical safety studies - general principles Laboratory with established quality assurance systems Accredited laboratories should be used when possible. Premises and equipment • Sufficient space and infrastructure • Utilities such as water, air, gas and electricity - adequate, stable and uninterrupted. • Equipment qualified and methods described validated. • SOPs for the operation, use, calibration and preventive maintenance of equipment - records maintained. • Equipment used should be identified - ensure traceability. Research organizations • Validation requirements for the analytical method with SOPs for analytical method validation. • Stability of the samples under the stated conditions and period of storage • Chemicals, reagents, solvents and solutions should be labelled to indicate identity, purity concentration (if appropriate), expiry date and specific storage instructions, information concerning source, preparation date and stability should be available. Quality assurance (QA) •QA unit - independent from the person(s) responsible for analytical work and which should ensure that the analytical method in use is validated and current Research Organizations DOCUMENTATION • All original analytical raw data (e.g. calculations, chromatograms, etc.) documented • Traceable to the sample number, equipment used, date and time of analysis and the name(s) of the technician(s). • Each data point should be traceable to a specific sample, including sample number, time of collection of the sample, time of centrifugation, if applicable, time when the sample was placed in the freezer, time of sample analysis, etc, to be able to determine whether any aberrant results might have been due to sample mishandling. • Coding techniques and methods to perform blinded analysis when relevant. Research Organizations PHARMACOKINETIC AND STATISTICAL CALCULATIONS • Calculations should be made by qualified persons • Calculation methods should be specified in the study protocol and data analysis should conform to the protocol requirements. • Computerized systems can be used Research Organizations CLINICAL STUDY REPORT • Reflect the complete study procedures and results in an accurate manner. • Well written and presented • All deviations reported • No discrepancies between the results in the report and the actual original (raw) data • Comply with regulatory requirements as applicable and be in a standard format Research Organizations CLINICAL STUDY REPORT • Cover at least the items listed in the International Conference on Harmonization (ICH) guideline (Topic E3. Structure and Content of Clinical Study Report) • Specifies the procedure for approval by the investigator and sponsor approved (signed and dated) by the responsible persons • Monitoring report and audit report available before release of the final study report Research Organizations • GCP WHO Technical Report Series, No. 850, 1995 (pp. 97-137) • GLP OECD Principles on Good Laboratory Practice (as revised in 1997). Organization for Economic Co-operation and Development. ENV/MC/CHEM(98)17. 26.Jan, 1998 International Conference on Harmonization (ICH) Guidelines. Tripartite Harmonized Guidelines on Good Clinical Practice, Step 4, May 1996. Program • Group sessions •Clinical •Bio-analytical Good Clinical Practices (GCP) • Q