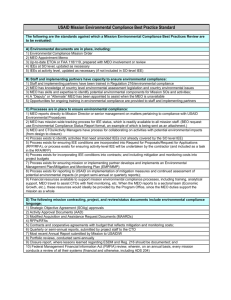
furanoid sugar amino acid and -hGly
advertisement

Synopsis SYNOPSIS The thesis entitled “Synthesis of unusual amino acids as building blocks in bioactive peptidomimetics” is divided into three chapters. CHAPTER-I: This chapter deals with Introduction to -peptides and synthesis of linear and cyclic peptides of cis--furanoid sugar amino acid and -hGly. CHAPTER-II: This chapter describes the synthesis of protected (2R,3R,4S) 4,7-diamino 2,3-dihydroxy heptanoic acid, a constituent of Callipeltins A and D. CHAPTER-III: This chapter deals with the introduction to antidepressants and the synthesis of antidepressant ‘Glaziovine-Sertraline hybrid’. CHAPTER-I: Introduction to -peptides and Synthesis of Linear and Cyclic peptides of cis--furanoid sugar amino acid and -hGly. The secondary structure of peptides, i.e. helices, turns, and sheet like conformations are determinant factors of their biological properties, both in and amino acid derivatives. -peptides are composed of amino acids with the carboxylic acid functionality at C rather than C The difference in the chiral center allows the peptides to resist hydrolysis by proteases even though they are amide-linked oligomers with the side chains similar to those in dietary proteins. Typically, -peptides make poor drugs due to low bioavailability as the body readily breaks them by proteases. Thus, biomimetic polymers hold promise for new biomaterials and therapeutics. Originally, it was proposed that -peptides would be more flexible than peptides because they contain an additional CH2 between the amine and carboxylic acid groups. Surprisingly, -peptides have exhibited greater conformational stability than peptides. -peptides can form stable helices with only four to six residues, where as a peptide of that length would be disordered. By understanding the conformational behavior of these interesting molecules, we may develop a means of controlling their structure. This would allow us to use -peptides as building blocks in new therapeutics to target almost any protein recognition event (proteolysis, protein-protein association, phosphorylation in signaling pathways, ribosomal translation, etc.). Recently, the relevant antimicrobial and hemolytic activities of amphiphilic -oligomers have also been shown I Synopsis thus prompting the research in this field. Thus, we have turned our attention towards the design and synthesis of new class of -peptides using sugar amino acids. The sugar amino acids are part structures of several natural products and very important components. Importance of such unusual aminoacids and non-availability from ‘chiral pool’, prompted synthetic organic chemists to develop new and efficient strategies for the synthesis of such amino acids enroute to the synthesis of both natural products and peptidomimetics. Synthesis of cis--furanoid sugar amino acid: The synthesis of cis-f-SAA was started from cheap and commercially available D-Glucose 1. It was protected by acetonide using H2SO4 in acetone. Inversion of configuration at C-3 position was carried out by oxidation (PDC/CH2Cl2) followed by reduction using NaBH4/MeOH. The inverted alcohol 3 was converted into its tosylate using TsCl/Py. Tosylate was subjected to reaction with NaN3 at 135 oC in DMF for 6 h to give azido derivative 4. The exocyclic hydroxylic groups of azide 4 are selectively deprotected to diol 5 using 0.8% H2SO4 in methanol (Scheme 1). Subsequently, the diol 5 is oxidatively cleaved using NaIO4, followed by NaClO2, NaH2PO4, H2O2 oxidation to afford azido acid 6. The azido acid 6 was converted to methyl ester 7 using ethereal diazomethane followed by reduction with Pd/C 10% afforded free amine ester 8, which was protected using di-ter-butyl dicarbonate to give Boc protected sugar monomer 9 in 96% yield (Scheme 2). HO OH O OH i) acetone, CuSO 4 OH HO O H2SO4 O O O HO 1 O 2 i) PDC, Ac2O, CH2Cl2 ii) NaBH4, MeOH, 0oC O O O O 3 ii) NaN3, DMF, 135 oC O O HO O MeOH O N3 OH 0.8 % H2SO4 O 5 Scheme-1 II O O N3 4 O HO D-Glucose i) TsCl, Py, CH2Cl2 O Synopsis OH HO O O O O N3 5 i) NaIO4, THF-H2O O HO ii) NaClO2, NaH2PO4 CH3CN O CH2N2 dry Ether O N3 6 O O O O MeO O O MeO EtOAc O N3 Pd-C/10%, H2 O O H2N O MeO (Boc)2O EtOAc O BocHN 9 8 Scheme-2 7 O Having successfully synthesized new class of -amino acid monomer, cis-furanoid sugar amino acid (cis-fSAA or fSAA), the attention was then focused to synthesize new class of -peptides using this monomer. Hence, homooligomers of cisfSAA were synthesized and their secondary structure pattern was studied. Synthesis of homo oligomers of cis--furanoid sugar amino acid: Homo oligomers of cis-furanoid sugar amino acid -peptides were prepared using 3-Azido–3-deoxy-1, 2-O-isopropylidene-α-D-xylo-furanoic acid 6 and free amine ester 8 by adopting segment condensation method. Dipeptides were prepared by condesation of two monomers. Tetrapeptide was prepared by coupling of two dipeptides. Hexapeptide was synthesized by condensing tetrapeptide acid with dipeptide amine, whereas octapeptide was obtained by coupling of two tetrapeptides. Coupling of azido acid 6 and free amine ester 8 in the presence of EDCI-HOBt in dry dichloromethane gave dimer azide 10 (Scheme 3). The above dimer azide on hydrolysis gave corresponding dimer azido acid 11, where as hydrogenation with PdC/10% in ethyl acetate gave the corresponding dimer amino ester 12 (Scheme 4). O O O O HO O O N3 6 O + MeO H2 N O O EDCI-HOBt N3 N H CH2Cl2 O O O 8 Scheme-3 III O O 10 O OMe O Synopsis O O O N3 11 O R THF-H2O(3:1) O 12 O O H2, EtOAc O OR' O Pd-C/10% O O N H OMe N H O O O LiOH O O 11 R = N3, R' = H 10 12 R = NH2, R' = Me Scheme-4 Coupling of these two (11 and 12) gave tetramer azido ester 13 which on reduction followed by treatment with Boc anhydride yielded tetramer 14 (Scheme 5). The same sets of reactions were carried out for hexamer 15 and octamer 16. O O R i) EDCI-HOBt + 11 12 O O N H O H N O O O OMe N H O O O CH2Cl2 O O O O O 13 R = N3 Pd-C/10% H2, (Boc)2O EtOAc 14 R = NHBoc Tetramer Scheme-5 O O BocHN O O O H N N H O O O O O O H N N H O O O O O O OMe N H O O O O O O O 15 Hexamer O O BocHN O O O H N N H O O O O O O O O O O H N N H O O O O O 16 Octamer IV O O H N N H O O O O OMe N H O O O O O O Synopsis The above synthesized homooligomers from cis-fSAA were characterized by circular dichroism (CD), NMR and molecular dynamics, which displayed well-defined helical structures characterized by series of 14-membered hydrogen bonded rings (14Helix). J. Am. Chem. Soc. 2004, 126, 13586. Synthesis of hetero (or mixed) oligomers of cis--furanoid sugar amino acid and hGly. The preceeding section described the synthesis of homo oligomers of cis-fSAA and study shown that the cis-fSAA oligomer adopts in solution a well defined righthanded 14-Helix. Our next interest was to check for the conformational rigidity of this sugar amino acid. Hence we proposed to synthesize mixed -peptides using cis-fSAA and -hGly alternatingly to provide conformational freedom to the rigid cis-fSAA peptides. -homoglycine is the only one naturally occurring -amino acid, which is known to destabilize helices because the unsubstituted -homoglycine is highly flexible. Accordingly, mixed -peptides (23, 25, 26 having cis-fSAA at the N-terminus, while 29, 32, 33 with -hGly at N-terminus) were prepared by conventional peptide coupling procedure. The commercially available -homoGlycine (-hGly) was esterfied in presence of acetyl chloride in methanol under reflux conditions to give -hGly ester 17. The synthesis of mixed -peptides having cis-fSAA at the N-terminus was started with the coupling of azido acid 6 and -hGly ester 17 under standard reaction conditions in presence of coupling agents EDCI, HOBt and DIPEA in CH2Cl2 to afford dipeptide 18 (Scheme 6). Methanol H2 N COOH CH3COCl COOMe ClH.H2 N 17 O O O O OH + ClH.H2 N N3 COOMe EDCI-HOBt DIPEA,CH2Cl2 O 6 17 Scheme-6 V O N3 O H N O 18 OMe O Synopsis The dipeptide 18 was hydrolyzed using LiOH in 1:3 THF:H2O solution at 0 oC to produce acid 19 without epimerization, whereas hydrogenolysis in the presence of Palladium on charcoal afforded amine 20, which was protected with Boc anhydride to give Boc protected dipeptide. The dipeptide 21 was hydrolyzed using LiOH in 1:3 THFwater solution at 0 oC to produce acid 22 (Scheme 7). O LiOH O O N3 H N OMe O O Pd-C10%, H2 O 20 EtOAc 18 O 19 THF-H2O O H N OR' R O 19 R=N3, (Boc)2O O R'=H 20 R=NH2, R'=Me 21 R=NBoc, R'=Me O O LiOH 21 THF-H2O O H N OH BocN O 22 Scheme-7 O Coupling of Boc protected dipeptide acid 22 and monomer amine 8 gave trimer 23. Same sets of reactions were carried out for Boc protected hetero -tetramer 25 (Scheme 8) and hetero -hexamer amine 26. O 22 + O EDCI-HOBt 8 CH2Cl2 O H N O H N OMe BocHN O O O O O 23 O 19 + 20 EDCI-HOBt O CH2Cl2 O H N R O O 24 R = N3 Pd-C/10% H2, (Boc)2O EtOAc 25 R = NHBoc Scheme-8 VI O O H N O O O N H OMe Synopsis O O O O H 2N H N O O O O H N N H O O O N H O H N OMe O O O 26 After preparation of mixed -peptides using cis-fSAA at N-terminus and -hGly at C-terminus, another set of mixed -peptides were prepared using -hGly at N-terminus and cis-fSAA at C-terminus. Accordingly, for the preparation of dipeptide, -hGly Nterminal was protected with Boc using 5% aqueous NaOH and di-ter-butyldicarbonate to afford Boc-protected -hGly acid 27, which was used directly for the coupling reaction. Boc-protected -hGly acid 27 was coupled with free amino esters 8, 20 using EDCI-HOBt to give Boc-protected hetero dipeptide ester 28 and Boc protected hetero tripeptide ester 29 respectively (Scheme 9). (Boc)2O, NaOH COOH H2 N COOH BocHN THF-H2O 27 O O O O + 27 O O OMe H2 N 8 EDCI-HOBt BocHN CH2Cl2 O O 27 + O 28 O O O OMe N H O H2 N H N O OMe O O EDCI-HOBt CH2Cl2 O 20 Scheme-9 BocHN O H N N H O OMe O 29 Boc-protected hetero dipeptide ester 28 was hydrolyzed with LiOH to give the carboxylic acid 30 almost quantitatively, where as removal of the Boc-protecting group from 28 with TFA furnished the TFA salt 31 (Scheme 10). Subsequent activation of the carboxylic acid function in 30 with EDCI/HOBt/DIPEA and reaction with the TFA salt VII Synopsis 31 produced the Boc-protected -hetero tetramer 32. Same sets of reactions were carried out for hexamer 33. O O O O BocHN LiOH THF-H2O O OMe N H O O 30 R O 31 TFA-CH2Cl2 (1:1) 0 oC 28 O OR' N H O 30 R = NHBoc, R' = H 31 R = NH2.TFA, R' = Me O 30 + EDCI-HOBt 31 O H N BocHN O CH2Cl2 O N H O O O BocHN N H N H O O N H O O O O O O OMe O O O O N H O 32 Scheme 10 O O N H O O N H OMe O 33 The above synthesized series of mixed peptides comprising of alternating cisfSAA and -hGly were characterized by circular dichroism (CD), NMR and molecular dynamics. The structural data showed that the mixed peptide heterooligomers 25, 26 and 29, 32, 33 form robust right-handed 14-helical secondary structures in solution. These results indicate that the secondary structure resulted exclusively from the cis-fSAA and the presence of the incorporated highly flexible -hGly did not affect the secondary structure. J. Am. Chem. Soc. 2005, 127, 9664. Cyclic homo and hetero oligomers of cis--furanoid sugar amino acid and -hGly: The self-assembly of peptide motifs into nanotubular objects mediated by hydrogen bonding has become an important area of research in supramolecular VIII Synopsis chemistry. These nanotubes exhibit unique structural and functional properties and have tremendous potential in biomedical and material sciences. Nanotubes based on cyclic peptides were first proposed in 1972 by Hassall et al. and predicted that cyclic tetramers of alternating - and -amino acids would assemble through backbone-backbone hydrogen bonding to yield hollow cylindrical constructs. Sugar amino acids (SAAs) have also been extensively studied as potential building blocks for the design of peptide nanotubes. Several groups have synthesized cyclic homooligomers from furanoid and pyranoid sugar amino acids but there were no reports of sugar peptide nanotubes. This motivated us to synthesize sugar peptide nanotubes Accordingly, cyclic homo oligomers of cis--furanoid sugar amino acid were synthesized by the coupling of azido acid 6 and dimer amino ester 12 in the presence of EDCI-HOBt in dry dichloromethane to give trimer azide 34 (Scheme 11). O O EDCI-HOBt 6 + 12 N3 O N H CH2Cl2 H N OMe O O O O O O O O O 34 Scheme-11 The trimer azide 34 was hydrolyzed by using LiOH in THF-H2O, followed by reduction of azide on Pd-C at 1 atm hydrogen pressure to give trimer amino acid, which was cyclized by using (8 eq) EDCI-HOBt under high dilution condition to yield 12membered (C3-symmetric) homo tricyclic compound 35 (Scheme 12). O 34 O H N O i) LiOH, THF-H2O, 0 oC ii) Pd-C/10%, EtOAc, H2 O O O HN iii) EDCI-HOBt (8eq) CH2Cl2, 48h O NH O O O 35 Scheme-12 IX O O Synopsis Similarly, homo tetra amino acid 36 was prepared from tetramer azido ester 13. The cyclization of homo tetra amino acid 36 was attempted with various coupling reagents, however it turned out that FDPP, in acetonitrile at room temperature was best to afford homo tetra cyclic 37 in 60% yield (Scheme 13). O O i) LiOH, THF-H2O, 0 O H2 N oC O O N H 13 O O ii) Pd-C/10%, H2 EtOAc O O H N O O O O O OH N H O O 36 O O O H2N O O O O H N N H O O O O O O OH O O FDPP, DIPEA O O H N O N H O O O O NH NH CH3CN O O O O O O O O N H O 36 O 37 Scheme-13 The cyclic hetero oligomers were synthesized from the trimer 23 by hydrolysis using LiOH in THF-H2O, followed by deprotection of the Boc to give trimer amino acid, which was cyclized by using (8eq) EDCI-HOBt under high dilution condition yielded 12membered homo tri-cylic peptide 38 (Scheme 14). O O O O H N O H N OMe BocHN O O O O O i) LiOH, THF-H2O, 0 oC ii) TFA:CH2Cl2 iii) EDCI-HOBt (8eq) CH2Cl2, 48h O H N O O O HN O NH O 23 38 Scheme-14 X O O Synopsis The hetero -tetramer azide 24 was hydrolyzed by using LiOH in THF-H2O to yield -tetramer azido acid 39. This azido acid 39 was transformed into the pentafluorophenyl ester followed by hydrogenation to give cyclic hetero -tetra peptide 40 in 75% yield (Scheme 15). O O LiOH, THF 24 H2O, 0 oC N3 O H N O O O N H OH O O O H N O 39 O O 39 C6F5OH, EDCI, CH2Cl2. O H N O O NH NH Pd/C-10%, H2 EtOAc O O N O H O O 40 Scheme-15 The self assembly of the above synthesized cyclic -peptides to form nanotubes was characterized by NMR spectroscopy, FT-IR, Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM) and Differential Interference Contrast (DIC) microscopy. Among the four synthesized cyclic -peptides, two cyclic peptides (35, 40) showed nanotube formation. XI Synopsis CHAPTER II: Synthesis of protected (2R,3R,4S) 4,7-diamino 2,3-dihydroxy heptanoic acid, a constituent of Callipeltins A and D. Callipeltin A, a cyclic depsidecapeptide isolated from the marine Lithisda sponges Callipelta sp. and Latruncula sp., was shown to possess antifungal, anti-HIV activity and cytotoxicity against selected human carcinoma cell lines, as well as powerful inhibition of the Na/Ca exchanger in guinea pig left atria. The structure of callipeltin A 1 was determined by Minale and co-workers; it contains a number of novel amino acids and a novel fatty acid. Comparison of the bioactivity of 1 with the related compound callipeltin B indicates that the side chain attached to the macrocycle, recently isolated as callipeltin D is essential for anti-HIV activity. The key residue of the side chain is the novel amino acid, (2R,3R,4S)-4-amido-7-guanidino-2,3-dihydroxy heptanoicacid (AGDHE, 2). In continuation of our ongoing research towards the synthesis of biologically active compounds, we planned to construct the AGDHE fragment of callipeltin A and D and in this chapter we report an efficient synthesis of AGDHE fragment starting from readily available L-ascorbic acid. 2R,3R,4S AGDHE NH H2 N N H O NH2 OH O N NH OH H O O OH O H N N H HN O O O OH MeN MeO O O O O O NH N H NH NMe N H CONH2 NH2 NH HN NH2 OH COOH NH2 OH 2 AGDHE OH Callipeltin A 1 The retrosynthetic analysis of AGDHE fragment revealed that the C2-hydroxy protected aldehyde (Scheme 1) was the key intermediate, which inturn would easily be obtained from L-ascorbic acid. XII Synopsis Retrosynthetic Analysis of AGDHE fragment of Callipeltin A: NH CALLIPELTIN A (1) H2 N N H HO NHPG3 MeOOC NH2 OH NHPG2 NHPG3 OPG1 OPG1 O OH OH NHPG2 OH OH O OH HO O O O OPG1 OH O O CHO OPG1 HO OH L-Ascorbic acid Scheme 1 Our synthetic approach to AGDHE fragment 2 involves chiron approach via cheap and commercially available L-ascorbic acid as the starting material. The 5,6-diol of L-ascorbic acid was easily protected as acetonide. The acetonide was purified through crystallization method to get 80-85% of yield. The enone moiety in acetonide was cleaved using H2O2, K2CO3 to afford potassium salt, which on treatment with ethyl bromide in acetonitrile at reflex conditions afforded (2R,3S)-hydroxy ester 3 in good yield. Inversion of configuration at C-2 in 3 by the Mitsunobu reaction provided ethyl (2S,3S)-hydroxy ester 4 in 65% yield for two steps (Scheme-2). OH O HO HO O 2 steps OH O O (a) chloro acetic acid, O O COOEt b) DIAD, TPP, THF, 0 oC Et3N, EtOH OH 3 COOEt OH 4 Scheme 2 Benzoylation of 4 with benzyl bromide, silveroxide in boiling acetonitrile in the presence of catalytic amount of Bu4NI, O-Benzyl ester 5 was isolated in 80% yield after 12 h. Since a direct reduction of these esters to the corresponding aldehyde gave XIII Synopsis mixtures, the two-step procedure was applied. The reduction of 5 with LiAlH4 in THF furnished the alcohol 6 (Scheme 3). O O COOEt OH b) Ag2O, BnBr, O O COOEt CH3CN (c) LiAlH4, CH2 OH THF, 0 oC OBn 4 O O OBn 6 5 Scheme 3 Oxidation of the primary alcohol 6 using IBX provided the corresponding aldehyde 7 in good yield. The aldehyde 7 was treated with allyl bromide and Zn powder in THF at 0 oC using saturated aqueous NH4Cl as catalyst resulted in the formation of anti-homoallylic alcohol as major isomer in 85% combined yield, as an inseperable syn:anti (1:5) diastereomeric mixture. The relative stereochemistry of the new stereogenic center in the major isomer was determined by a short deprotection-protection protocol. After confirming the stereochemistry of the major isomer in the homoallylic alcohols 8 (a/b), they were esterified with methane sulphonyl chloride to give the mesylates in 92% yield. At this stage, the two diastereomers resulting from the preceeding allyl zinc addition were easily separable by column chromatography to afford mesylate 10 as a major single diastereomer (Scheme 4). O O CH2 OH OBn 6 O d) IBX O CHO O OBn 8 (a/b) syn : anti (1:5) OMs O O + cat DMAP 0 oC OBn 8 (a/b) OH O OBn 7 e) MsCl, Et3N, OBn allylbromide Zn, THF, 0 oC THF, 0 oC OH O O O 9 (minor) OMs O OBn 10 (major) Scheme 4 The mesylate 10 was converted into its azide 11 using NaN3/DMF. Azide thus obtained was reduced to amine using LAH/THF/0 oC and protected as its tert-butyl carbamate derivative (Boc) by treatment with 1N NaOH/(Boc)2O to give 12 (Scheme 5). XIV Synopsis 10 f) NaN3, DMF, 80 O N3 oC O g) LiAlH4,THF O (Boc)2O, 0 oC OBn NHBoc O OBn 11 12 Scheme 5 Hydroboration of 12 with BH3.Me2S followed by H2O2 furnished primary alcohol 13, which was subsequently converted to azide 14, followed by reduction on Pd-CaCO3 under 1 atm hydrogen pressure and treatment with chlorobenzylformamate gave diamino protected compound 15 in 85% yield. After obtaining the protected diamine functionality, 1,2-isopropylidene was selectively cleaved with 0.8% H2SO4 in MeOH at ambient temperature to result the diol 16. The diol primary alcohol was selectively oxidized by using TEMPO/NCS and NaClO2,, NaH2PO4 to yield the acid, which was protected as its methyl ester 17 using ethereal diazomethane (Scheme 6). O NHBoc O OBn O h) BH3.DMS NHBoc O OH NaOH, H2O2 Cbz-Cl CH2Cl2 DIPEA O OBn 14 NHBoc O NHCbz k) 0.8% H2SO4 MeOH OBn OH NHBoc HO NHCbz OBn 16 OH l) TEMPO-TBACl NHBoc N3 13 15 Na2CO3, NaHCO3 CH2Cl2 m) NaClO2, NaHPO4, H2O2 O O ii) NaN3, DMF 80 oC OBn 12 j) H2, Pd-CaCO3 EtOAc i) TsCl, Et3N, 0 oC CH2Cl2, DMAP NHBoc NHCbz MeOOC OBn 17 n) CH2N2, Ether Scheme 6 In conclusion, we have developed an efficient strategy for the synthesis of protected unusual amino acid (2R,3R,4S)-4,7-diamino-2,3-dihydroxyheptanoic acid 17 from cheaply available L-ascorbic acid. This synthetic strategy could be useful in making reasonable quantities of the key residue 17, which can be used for the total synthesis of callipeltins A and D. XV Synopsis CHAPTER III: Introduction to antidepressants and the synthesis of antidepressant ‘Glaziovine-Sertraline hybrid’ Depression is a major mental disorder and one of the most frequent chronic illness that effects people of all ages. There are very few drugs available for the treatment of depression. Use of lithium salt as drugs for this disease has shown some positive effect. Drug development activities resulted in introduction of first-generation tricyclic antidepressants (imipramine, amitriptyline), which were found to induce severe side effects such as anti cholinergic and cardiovascular effects. They were replaced in 1980s by the selective serotonin reuptake inhibitors SSRIs. The synthesis of hybrid molecules made up of two different molecular units, has recently gained importance because many of them exhibit promising physical, chemical and biological properties as well as being novel architectures. In this work, we designed and synthesized a new hybrid antidepressant 1, having Sertraline 2, one of the clinical compound that acts on CNS as SSRI and the key building block of antidepressant glaziovine 3, one of the important alkaloid of lauraceae family. NH-CH3 MeO MeO MeO Cl N-Boc N NH-Me HO Cl O Cl Cl 1 2 O 3 Our synthetic strategy for the key building block of glaziovine was started from vanillin 4. Methylation and bromination of vanillin using methyl iodide and K2CO3 in acetone, molecular bromine in methanol at less than 40 oC gave the bromoaldehyde 5. Nitroaldol reaction of bromoaldehyde 5 with nitro methane gave unsaturated nitrocompound 6. Double bond was reduced in compound 6 using sodium borohydride to give saturated nitrocompound 7 (Scheme 1). Saturated nitro compound 7 was reduced to amine using LAH/THF/0oC and protected as its tert-butyl carbamate derivative (Boc) by treatment with 1N NaOH/(Boc)2O to afford 8. This amine product was subjected to heck reaction with ethyl acrylate using triethyl amine and palladium diacetate in polyethylene glycol at 80 oC to give the unsaturated ester 9 (Scheme 2). XVI Synopsis CHO MeO HO ii) Br2,MeOH <40 0C MeO acetone,reflux 4 CHO MeO i) MeI,K2CO3 MeO CHO MeO Br 5 iii) CH3COONH4, MeO AcOH,CH3NO2 100 0C MeO NO2 Br 6 iv) NaBH4, MeO MeOH 0 oC MeO Br NO2 7 Scheme-1 Azamichael reaction was carried out by using TFA:CH2Cl2 1:1, then basified with Na2CO3 to give cyclic ester 10. This ester was hydrolyzed with LiOH to achieve acid 11. The acid 11 was directly coupled with commercially available Sertraline 2 to give hybrid product 1 (Scheme 2). MeO MeO Br NO2 NaOH,0 0C, (Boc)2O MeO MeO NH-Boc NH-Boc Pd(OAc)2,Ph3P PEG-400,1000C MeO vii) TFA:CH2Cl2 (1:1) Na2CO3,(Boc)2O 9 Br 8 7 MeO vi) CH2CHCOOEt, MeO v) LiAlH4, THF LiOH, THF N-Boc MeO COOEt H2O-MeOH OEt 10 O NH-Me MeO EDCI-HOBt N-Boc + MeO CH2Cl2 OH MeO N-Boc MeO O MeN Cl 11 O Cl 2 Cl Scheme-2 1 Cl In conclusion, we synthesized a new hybrid antidepressant from key building block of glaziovine and highly potent antidepressant sertraline. This hybrid compound has shown a significant antidepressant activity in the biological tests. XVII