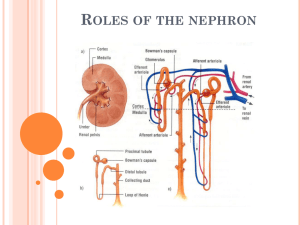
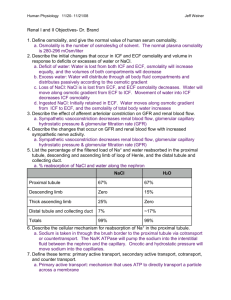
Respiratory and Renal Review
advertisement

Respiratory Physiology Mechanics of Respiration: Overview: • Events in one lung can occur in isolation from the other... • The intrapleural space is filled with liquid (2-10mL) ! two appositional pleura can move with respect to one another. • In steady state the rate of O2 consumed and CO2 produced by tissues of the body matches their respective rates of removal from or addition to alveolar gases. • Structure of the Airway: o o o • • Nose ! mouth ! pharynx ! larynx ! trachea ! two branches ! 20-23 divisions ! 5 million terminal alveoli. " Trachea is surrounded by horseshoe shaped cartilage " Bronchi have broken rings or plates of cartilage surrounding them " Bronchioles have no supportive cartilage holding them open or patent. • Bronchi and bronchioles are subject to collapse. Alveoli: tiny sacs (one cell layer thick) that provide surface for diffusive gas exchange between lungs and blood. Divisions 17-23 contain alveoli and ! is the respiratory zone (300 million alveoli make up surface of 70m2. Lungs are covered by the visceral pleura and the chest wall by the parietal pleura. The Intrapleural space is filled with a small volume of intrapleural fluid (2-10mL) ! the two appositional pleura can move with respect to one another - ! FRC (obstruction) = Musculature of the Chest Wall: o Inspiration: external intercostals (pull lower ribs toward ! C = emphysema, upper ribs) and diaphragm (moves down, increasing asthma vertical dimensions of the thorax). - # FRC (restriction)= o Expiration: internal intercostals and rectus abdominus. # C = # surfactant, " During respiration, the lung and chest wall move edema, fibrosis together because of interpleural cohesive forces (see above). Elastin-collagen latticeworks allow for expansive properties. Distending Pressure and Functional Residual Capacity (FRC): o Distending (transmural) Pressure: Pinside - Poutside = PAlveolar - PIntrapleural. This is caused by a decrease of pressure outside of the lung (PIP). " PIP is negative (subatmospheric) because the lung, which adheres to the chest wall by a thin layer of fluid, tends to recoil, pulls intrapleural space (causing PIP to fall). " The larger the lung volume, the greater the lung recoil forces, and the lower the PIP. Lung Volume " Pvolume IP drop o Equilibrium (end of quiet expiration) is called the functional residual capacity (FRC). " At FRC, the tendency of the lung to recoil is exactly balanced by the tendency of the chest wall to expand and therefore the lungs remain inflated. • Change in volume from FRC requires use of respiratory muscles. • If air/blood is introduced into the intrapleural space (pneumo-/hemothorax), PIP rises until it reaches PATM ! chest wall expands and lung recoils (until distending pressures are zero). • PIP is made less negative by any factor that decreases lung elasticity (age, emphysema, etc). Tendency of the lung to recoil (white arrow) is balanced by that of the rib cage to spring out (black arrow). PIP is subatmospheric. Pneumothorax, which occurs when the intrapleural space becomes atmospheric, allows the lung to collapse and the thorax to spring out until the distending pressure for each is zero. The pressure in the air space is greater than total venous gas pressue (! recovery) During contraction, !V! … PA 1/" Volume. During inspiration, PA ~ -2. PIP is measured via an esophageal balloon. Compliance: • A compliance curve is generated by plotting lung volume against distending pressure (Palv - PIP) (compliance is the slope of the curve). Compliance of the lung decreases at large lung volumes. • When the distending pressure for the lung is zero, it still contains air; it is not completely collapsed. • With saline, Vmax is reached at lower pressure. # surface tension is • C=!V/!P eliminated by surfactant. Lung compliance (slope) differs at different "VL pressures ! #C at mid-lung pressure/inflation. CL = • Hysteresis (path-dependent compliance): inflation and deflation curves of "( Palv ! PIP ) the air-inflated lung are not the same (due to surface tension/surfactant – at least in part). - Emphysema and age ! C - Edema and ! pulmonary BP # C • • • CL " Ease of lung inflation CL 1/" Distending pressure required at any given lung volume. o $C requires higher than normal distend pressures to expand the lungs. Lung with $ C recoils from the chest wall more forcibly at any given volume (PIP is more negative) and # inspiratory effort required. " Interstitial fibrosis, !pulmonary BP, pulmonary edema ! $C. " Emphysema and age ! !C (! compliance ! greater than normal resistance to expiration). Surface Tension: significantly effects CL. Surface tension (attractive forces between water molecules) impel air-water interfaces to have minimum surface area ! # recoil forces in alveoli ! shrinkage (resist expansion). o Pressure exerted by an alveolus 1/" Radius at a given surface tension (Law of Laplace). • The law of Laplace states that small alveoli ($radius) have a tendence to collapse into larger alveoli. This is overcome by surfactant - reduces the surface tension of the aqueous air-water interface proportional to the area it covers. ! surface tension of an alveolus decreases as the alveolus gets smaller and pressure does not increase as it shrinks (! alveolar capillary filtration forces $). • Interdependence states that the tendency of one alveolus to recoil is opposed by the recoil forces of surrounding alveoli. • Surfactant is produced by Type II alveolar cells (begins to appear in the lung at the 26th week of gestation. Deep breathing stimulates production. Comprised of dipalmitoyl phosphatidyl choline (DPPC), and other lipids and proteins. The effect of surfactant can be replicated by detergents. • Surfactant lowers surface tension and reduces tendency of alveoli to shrink ! surfactant reduces the pull on the underlying interstitial fluid (makes the interstitial fluid pressure less negative) ! reduces tendency for fluid to be drawn out of capillaries ! prevents pulmonary edema. Lack of surfactant ! Respiratory Distress Syndrome/Hyaline Membrane Disease (FRC $) – Cortisol # rate of surfactant production. o In the absence of surfactant, a greater distending pressure is needed to inflate lungs. • #P $P Compliance of the Lung and Chest Wall: o Transmural (distending) pressure across the chest wall and lungs together = (Palv - Patm) - in cm H2O o Transmural (distending) pressure across the chest = (PIP- Patm) o Transmural (distending) pressure across the lung = (Palv - PIP). " o At FRC, transmural (distending) pressure across the combined chest and lung is zero (force of the elastic recoil of the lung is balanced by that of the chest wall expanding outward). • At greater volumes, transmural pressures across the combined lung and chest wall are positive • At smaller volumes, transmural pressures are sub-atmospheric. • Transmural pressures across the lung are always positive because the lung tends to recoil inwards. Transmural pressures across the chest wall are negative because the chest tends to recoil outwards at most volumes. o At very large lung volumes (~ 80% of vital capacity), the chest wall is overstretched and instead tends to recoil inwards. Compliance in Series: 1/CT = 1/C1 + 1/C2 etc… ! Compliance of the lung and chest wall is less than that of either the lung or the chest wall alone. Respiratory Pressure Cycle (Inspiration, Expiration, and Ventilation): • Inspiration: o (1) Prior to lung inflation: PA = 0, PIP = -5cm H2O ! PDistending = PA – PIP = 0 – (-5) = +5 (2) During inspiration: volume of chest and lungs # ! PIP $ ! PA $ ! PDistending (transmural) # ! pressure gradient that o promotes flow of air into the lungs. (3) End inspiration is the state of the lung after pressure gradients between alveolar gases and the outside air are o dissipated and the flow of gases into the lungs has stopped. Transpulmonary pressure has risen to +9 cm H2O, i.e., Palv – PIP = 0 – (-9) = +9 cm H2O. " Lung volume is greatest. " During inspiration, expansion of the lungs $PA relative to PATM ! gas flows into lungs. During expiration, recoil of the lung and chest wall # PA and reverses flow of air. # lung volume during inspiration are also associated with # P Distending of the lung. During expiration lung volume and PDistending $. B and D have the same lung volume because they have the same PDistending (+6 cm H2O). During inspiration there is an # distending pressure on the airways. • Expiration: o (1) End Inspiration: Muscles of inspiration relax and lungs and chest wall passively return to FRC. Alveolar air is compressed and PA exceeds PATM !flows into atmosphere. o (2) Active Expiration (exercise or forced expiration): Internal intercostals and rectus abdominus increase intraabdominal pressure to $ chest volume. " " • PIP does not change linearly with volume despite nearly linear lung compliance during normal breathing cycles. • Airway resistance prevents instantaneous equilibration of gas pressures between alveoli and outside air. During respiratory excursions, when the PA are below or above PATM, PIP at a given lung volume will be more negative during inspiration than during expiration. This explains, the non-linear relationship between PIP and lung volumes during respiratory cycles. Ventilation: P " Patm !P Flow = alv = o Poiseuille’s Equation for Laminar Flow of Gases: R = (Viscosity x L) / r4 R R " R " Viscosity " R " Length of the Airway " R 1/" radius4 o Primary Resistance to Flow: " 25-50% of total airway resistance is in the nose, nasopharynx, and larynx (upper airways) " Greatest resistance in the tracheobronchial tree is the medium sized bronchioles (up to 7th generation) and not in the very small bronchioles (because there are so many in parallel) – total resistance $ with number of parallel airways. " Overall resistance in the respiratory tree is low enough that small pressure differences [(Palv - P atm) of less than 2 cm H2O] allows 500 ml of air to move in and out of the lungs during quiet breathing. o Radial Traction: connective tissue fibers pull out on the sides of airways, holding them Q = (PA – PATM) / R open. This force is # during inspiration. Radial Traction Force " Inspiration o Cross-sectional area of airways (! resistance) can be affected by transpulmonary pressures and factors that affect radial traction. Transpulmonary pressures (Palv - PIP) that distend the alveoli distend the airways as well and prevent the smaller non-cartilaginous airways from collapsing during quiet breathing. At large lung volumes the small airways (like alveoli) are more distended than they are at small lung volumes. • • Emphysema: the airways collapse because of loss of radial traction. Fibrosis: radial traction is excessive and increases airway caliber above normal at any given lung volume. Dynamic Airway Compression: can lead to airway collapse during forced expiration • Forced expiration can ! collapse of non-cartilaginous bronchioles (if PIP exceeds Pairway). As air flows through airways, Pairway $ as frictional forces dissipate energy as heat. • As Pairway $, PDistending $. If Pairway decreases below PIP (the equal pressure point) the airway will collapse (dynamic compression). As resistance then rises, flow drops. • ! # expiratory effort raises PA and PIP (and ! P on airway) by the same amount #! Effort Independent Flow. o Tendency for airway collapse is reduced if PIP During forced expiration, the large positive intrapleural pressure is low $ airway collapse is less likely during collapses the airway just downstream forced expiration at ! lung volumes (# PIP) of the “equal pressure” point. At than at # lung volumes (! PIP). vital capacity (expiration o When PIP # above Pairway non-cartilagenous beginning), flow is effort dependent. sections of the airway close ! low rate of flow. Flow becomes effort independent o At the highest lung volumes, when PIP remains once expiratory effort ! (terminal velocity). negative (or only slightly positive), flow rises with greater expiratory effort. At mid-low volumes flow becomes independent of effort as PIP !. Lung Volumes and Pulmonary Ventilation: • Tidal Volume (VT): Volume of air expired (or inspired – but not by definition) during a single respiratory cycle (about 500 ml). o When the rate of O2 uptake by the lung matches the rate of CO2 added to the alveoli, the inspired and expired volumes are equal. However, as mentioned above, when CO2 production is less than O2 consumption, the expired volume is slightly less than the inspired volume. Because of this discrepancy, physiologists have chosen the expired volume to represent VT. • Inspiratory Capacity (IC): The maximal volume that can be insiraed above the resting end-expiratory position. • Vital Capacity (VC): the greatest volume of gas that can be expelled after maximal inspiration (sum of the IC and ERV). • Inspiratory Reserve Volume (IRV): maximum inspiratory effort after normal inspiration that can increase lung volume by 2.5-3 L. • Expiratory Reserve Volume (ERV): maximum expiratory effort after normal expiration that can expel an additional 1.5L. • Residual Volume (RV): 1.5L remains in the lungs after a maximum expiratory effort. • Functional Residual Capacity (FRC): the volume remaining in the lungs after a passive expiration. • Forced Vital Capacity: obtained when individual exhales as rapidly, forcefully, and completely following maximal inspiration. • FEV1: amount of air expired in first second of the FVC • FEV1/FVC: Normally 80%. Lower than 80% = obstruction. o All except residual volume, total lung capacity and FRC can be measured via spirometry • Minute Ventilation: total volume of air expired during one minute of respiration (VT x frequency) • V E (ml/min) = VT (ml/breath) x f (breaths/min) IC = IRV + TV • Inspiratory Capacity FRC FRC = ERV + RV Total Lung Capacity VC = IRV + TV + ERV (or IC + ERV) Vital Capacity TLC = IRV + TV + RV + ERV (or VC + RV). Anatomic Dead Space (VD): Air remaining in the conducting airways (where no gas exchange occurs) that did not enter alveoli. o Due to anatomic dead space, the tidal volume is actually the sum of the volume expired from the dead space plus that expired from the alveoli (VA) [VT = VD + VA]. Minute Ventilation (VE) $ is the sum of dead space ventilation (VD x f) plus the alveolar ventilation [(Tidal Volume – Dead space) x f] . . . VE= f .VD + VA • (VA x f = V A ) AIR FLOW = LAST IN FIRST OUT. The Fowler Method: Single-breath nitrogen washout. One-breath inhalation of 100% O2, holds for one second, then exhales through a one-way valve into a nitrogen and volume-measuring device. Initially, only O2 (from airways), then N2 and O2 are both exhaled. Physiologic Dead Space: Dead space composed of the alveolar dead space (the alveoli that are not perfused and $ do not participate in gas exchange) plus the anatomic dead space (alveolar dead space is created when dead space volume exceeds the volume in the conducting airways). PACO2 can be estimated by ParterialCO2. VT is a mixture of gas from the anatomic dead space (VD) o o • VD PACO 2 ! PECO 2 = VT PACO 2 • and a contribution from the alveolar gas (VA). PACO2 # when it is diluted in the total expired volume that includes the dead space volume. For a given minute ventilation, increasing the depth (VT) of respiration is more effective in elevating alveolar ventilation than increasing the frequency. It is more effective to take less frequent, deep breaths than to take shallow, frequent breaths, because a fixed amount of each breath remains in the dead space. In exercise VT increases as does frequency. Diffusion and Alveolar Ventilation: Gas exchange occurs both during inspiration and expiration. • Ventilation: bulk flow of air - occurs between the atmosphere and terminal bronchioles. o gas movement results from the total pressure gradient. " linear velocity of flow # in the upper airways and $ as airways branch and combined crosssectional area increases. " between the trachea and the alveolar ducts, cross-sectional area increases ~5,000x ! if initial linear velocity of gas entering the upper airways is 200 cm/sec (ventilation), it will $ to 0.04 cm/sec (near diffusion) in the alveolar ducts. $ further in the alveoli. • " • fine particulate matter settles near the 16th generation ! velocity slows here. bulk flow does not completely disappear in the respiratory zone (it is slow). Diffusion: dominant mechanism of transport within the respiratory bronchioles and alveoli (the respiratory zone). Diffusion of gases in the gas phase is followed by diffusion of gas across the alveolar-capillary barrier (gas in liquid). This is followed by a second diffusion step (capillaries ! mitochondria in cells - gas in liquid). o each gas moves according to its own partial pressure gradient (from an area in of high partial pressure to an area of low partial pressure). o diffusion of gases is capable of effecting the subsequent transport of gas between the alveolar ducts and the alveoli. This accounts for the rapid uniformity of the gas composition that occurs within the alveoli. " Diffusion of Gas in the Gas Phase: • Movement from ! concentration ! # concentration. • Avagadro’s Law: partial pressure " concentrationgas • Diffusion is very quick over short distances (equilibration over 0.5mm will occur within 0.002 seconds). If there is enlargement of small airways (emphysema, etc) diffusion distances # and gas transport by diffusion is impaired. o " Rate of Diffusion 1/" Distance2 Diffusion of Gas in the Liquid Phase (at alveolar capillary barrier and beyond): • CO2 (more soluble) diffuses through the liquid phase about 20 times more rapidly than O2 for a given partial pressure gradient. Diffusion of gas through the liquid phase occurs through: 1) surfactant layer, 2) alveolar epithelium, 3) interstitium, 4) pulmonary capillary endothelium, 5) plasma, and 6) red cell membrane. Total thickness of the aggregate barrier (primarily water) is ~0.5-1.5 µm. o Solubility of O2 is linear with respect to pO2. Fick’s Law of Diffusion: • DSA (P1 - P2 ) V gas = T Direction of net diffusion of a gas across a barrier depends on ! partial pressure " Gas phase: partial pressuregas " concentration. " Liquid phase (Henry’s Law): Cx = Px · Sx " Fick’s Law of Diffusion: D = diffusion coefficient; S = solubility in liquid; A = surface area of the barrier; T = thickness; P = partial pressure of gas (P1 – P2 ! across the barrier) • " Limitations on Gas Transfer: Blood spends ~0.75 sec in pulmonary capillaries (normal exertion), traversing ~2-3 alveoli in this time. o • Abnormal barely reaches equilibrium (PAO2 = PcapillaryO2). Grossly abnormal does not reach equilibrium. PAO2 does not = PcapillaryO2 Graham’s Law: Diffusion coefficient is dependent on molecular weight of the gas. For a given metabolic rate, ! air-flow into alveoli ! PO2 and # PCO2. In steady state, neither the flow of gas nor the mean alveolar partial pressures of CO2 or O2 ! much over time. For O2, equilibrium is achieved within 0.3seconds. CO2 has a smaller driving pressure, but reaches equilibrium in the same period of time (due to greater solubility). o " Diffusing capacity " volume of pulmonary capillaries ($ !surface area). " Diffusing capacity " number of pulmonary capillaries recruited " Diffusing capacity " hemoglobin concentration " Diffusing capacity " surface area of functioning alveoli (! tidal volume) RULE: If O2 diffusion is sufficient, then it can be assumed that the CO2 diffusion is sufficient. Rule: two gases with the same partial pressure in the gas phase will have the same partial pressure in the liquid phase) " Diffusion to equilibrium is much faster than 0.75seconds ! in exercise, blood flow # (blood clearance ~ 0.25 seconds in capillary) but this is enough time for equilibrium to be reached. • If the alveolar-capillary barrier is thickened by disease (interstitial fibrosis or pneumonia) O2 diffusion is impeded and rate of rise of pO2 is slow and equilibrium may not be reached (not likely to occur for CO2 given ! solubility). o o Perfusion Limitation (normal O2, CO2, and N2O): " Gas equilibrates early along length of pulmonary capillary (PAO2 = PaO2) " Diffusion of gas can only be ! if blood flow ! • o Blood can equilibrate in under 0.3 seconds $ blood flow can double (transit time 0.375 seconds – enough for equilibration of O2) and twice as much O2 would have been transported. Diffusion Limitation (O2-emphysema, fibrosis, and exercise, CO): " Gas does not equilibrate by the time blood reaches the end of the pulmonary capillary (e.g. in emphysema, surface area for diffusion $, or fibrosis – thickness #). The partial pressure difference between alveolar air and pulmonary capillary air is maintained and diffusion continues as long as the gradient is maintained. " Transit time was insufficient to allow equilibration and that the limitation on gas exchange had become diffusion limited. • • Alveolar Ventilation Equation: o • The number and size of capillaries can be ! during exercise by recruitment and distension. • V A = V CO 2 PB ! 47 mm Hg PACO2 VA = alveolar ventilation; VCO2 = amount of CO2 produced/minute. PB = barometric pressure; pH2O=47mmHg (correction for water vapor pressure). Alveolar ventilation 1/" PACO2. Uses PCO2. If alveolar ventilation is halved and CO2 production is constant, PACO2 and ! PaCO2 will double. Central chemoreceptors that detect partial pressures of CO2 in blood, and reflexively adjust ventilation to keep the PACO2 regulated, normally at about 40 mm Hg. Alveolar (O2) Gas Equation: complicated because more O2 is normally removed from alveoli than is replaced by CO2 [the respiratory quotient (R) = VCO2/V O2 (rate of CO2 produced/rate of O2 consumed) is usually less than 1]. PAO2 = PIO2 – PACO2/R When R = 1; PAO2 = PIO2 – PACO2 If R= 1, hyperventilation causes the # PAO2 = $pCO2 Removal of O2 and addition of CO2 “converts” fresh inspired air reaching alveoli to alveolar gas. PIO2 drops to PAO2, and PICO2 (normally 0) increases to PACO2. If R = 1, the quantity of O2 removed from alveoli (i.e., from the alveolar ventilation) equals that of the CO2 replacing it. PIO2 will, therefore, decrease quantitatively by the same amount that PICO2 increases, i.e., from 0 to PACO2. The CO2 stores (including that in the form of bicarbonate) are greater than the O2 stores (O2 bound to hemoglobin and myoglobin). Therefore, the alveolar PCO2 takes longer to come to equilibrium, and during the non-steady state, the R-value of expired gas is high, as the CO2 stores are washed out. Obviously, the opposite changes occur if alveolar ventilation is decreased. Transport of O2 and CO2 in Blood: o o Alveolar Air Equation: PAO2 = (760-47)FiO2 –PACO2/0.8 where FiO2 = 0.21; 0.8=R, and 47 = pH20 A-a gradient can be up to 10mmHg Numbers: o PB varies with altitude. PB (Sea Level) = 760mmHg. o Atmospheric air consists of 21% O2, 79% N2, and CO2 (0.03 to 0.04%). " Rest VO2 = 250ml/min PO2 = PB x FractionO2 = 760mmHg x 0.21 = 160mmHg ; CO2 = 0.2-0.3mmHg " Rest VCO2 = 200ml/min " Exercise VO2 = 3000ml/min " R = VCO2/V O2 = 0.8 Oxygen Transport: carried in two forms – (1) dissolved in liquid (small amount) and (2) bound to hemoglobin o Concentration of oxygen dissolved in arterial blood = 0.32ml O2/100ml blood (0.3%). Gas solubility 1/" temperature o Hemoglobin is not normally fully saturated with O2. Saturation is a function of PO2. o Hemoglobin blood concentration = 15 g % (g/100 ml blood). One gram of Hb binds ~1.34mL O2. (HbO2 / O2 capacity of Hb) x 100 = % saturation of Hb or [(total O2 content – O2 dissolved) / O2 capacity of Hb] x 100 = % saturation of Hb Substance Dry Air Arriving at Alveoli (saturated w/ H2O vapor Equilibrated in Alveoli Arterial Blood Venous Blood o O2 (mmHg) 158 150 100 100 40 CO2 (mmHg) 0.3 0.29 40 40 46 Oxyhemoglobin Dissociation Curve: (1) flat portion at the upper-right provides for constant arterial O2 saturation despite wide fluctuations in PAO2 (2) steep slope in the center allows for the release of a large quantity of O2 from Hb as PO2 decreases in the tissues (! delivery) (3) arterial blood is ~98% saturated with O2 means that if we want to increase the amount of O2 on Hb we have only a 2% increment available. (4) P50 ~ 26mmHg. (5) !PO2 of pure O2 delivery (medical ~ 600mmHg) does not saturate Hb significantly, but puts more O2 into solution (dissolved O2 ! ! delivery). • • • • Right Shift: # Hb-O2 affinity at a given PO2 (! delivery). o # pH (!H+), ! temperature, ! [2,3-DPG], !pCO2 (the Bohr Effect). " The above bind more strongly to deoxygenated Hb than to oxygenated Hb. By stabilizing the deoxygenated configuration, they make O2 binding more difficult. Left Shift:! Hb-O2 affinity at a given PO2 (# delivery). Myoglobin and fetal Hb have a greater affinity for O2 than does HbA (adult). P50myoglobin = 5 and P50 - HbF = 15-20 mm Hg. Carbon Monoxide (CO) binds Hb with an affinity of 240x that of O2 ($pCO of ~ 0.16 mm Hg, CO occupies ~75% of Hb). In the steady state CO occupies a small fraction of our Hb (~1-2%). o Carbon Monoxide: In addition to binding Hb with an affinity much greater than that of O2, CO shifts the dissociation curve of the remaining HbO2 to the left. An individual with " of Hb in the form of CO-Hb will be able to unload O2 in peripheral tissues less effectively than an anemic individual with half the normal amount of Hb • Carbon Dioxide Transport: CO2 is carried in three forms: (1) dissolved CO2 (HCO3- = 90% of CO2 in arterial blood), (2) bicarbonate ions, and (3) in combination with protein (carbamino compounds). Of the total venous-arterial CO2 difference (i.e. CO2 exchanged), about 60% is attributable to HCO3-, 30% to carbamino-Hb, and 10% to dissolved CO2. catalyzed by carbonic anhydrase non-enzymatic #pCO2 drives reaction to right the globin of hemoglobin forms carbamino-hemoglobin in the RBC. o Total CO2 content is a function of PCO2. Within the usual physiologic range of PCO2, the relation is essentially linear. The locus of the CO2 dissociation curve depends on the hemoglobin saturation. The lower the saturation of Hb with O2, the larger the CO2 content for a given PCO2 (the Haldane effect). Uptake of O2 aids in unloading of CO2. Oxygenated hemoglobin is a stronger acid than non-oxygenated hemoglobin and gives up H+ ions that combine with HCO3- and drive the carbon dioxide sequence of reactions toward CO2. In the tissues, the Haldane effect (increased CO2 content) is due to deoxygenated hemoglobin moppping up H+ ions produced when carbonic acid dissociates and by the greater facility of deoxygenated hemoglobin to form carbamino-hemoglobin, favoring the diffusion of CO2 from the tissue into blood. Buffers and Respiratory Regulation of pH: - acidity is protected by three lines of defense: (l) body buffers, (2) pulmonary regulation of [CO2 ], and (3) renal regulation of [HCO3-] in extracellular fluids by minimizing ! in pH of body fluids and correcting acid-base balance by appropriate retention or excretion of HCO3- and hydrogen ions. Buffers: minimize range of pH ! by giving up or accepting protons. Acids are proton donors and bases are proton acceptors. The pulmonary system interacts with the buffer system to regulate CO2 content. 50% of the total buffering occurs inside cells by phosphate and various organic anions, including proteins. This process is slower than extracellular buffering, (hours rather than minutes). Normal pH = 7.4 and normal [H+] = 40nM/L o Buffer Pairs: " HCO3- – H2 CO3 " Protein- – H-protein " HPO4-2 – H2PO4" Hb- – H-Hb millimols of strong acid • The curve has a point of inflection at which changes in the amount of acid added (or removed) from the solution has little effect on pH (at equilibrium point, [A-] = [HA]). lo o o o pH hi For HA # ! H+ + A- K = [H+][A-]/[HA] ; pH = pK + log [A-]/[HA] ; pH = -log [H+] ; pK = -log K Isohydric Principle: There is only one H+ ion active in any given part of the body at any given time and it is this single H+ with which every buffer system interacts. The Bicarbonate-Carbonic Acid Buffer System: CO2 + H2O ! H2CO3 ! H+ + HCO3- ! pH = 6.1 + log [HCO3- ] / 0.03 pCO2 OR [H+] = 24 pCO2 / [HCO3- ] In the range pH =7.28-7.45, the relationship between pH and [H+] is almost linear with an increase of pH by 0.0l unit corresponding to a decrease in [H +] of l nanomole/L. o Pulmonary Regulation of pCO2: protection from acidity afforded by buffering alone is often inadequate in the absence of pulmonary mechanisms of pH regulation. " Metabolic Acidosis: ! acid production (or base loss) ! metabolic acidosis. # plasma [H+] detected by chemoreceptors (carotid and aortic bodies), ! reflex respiratory response (hyperventilation) ! ($pCO2) drive [CO2] # ($ [H+] and [HCO3-]) ! # pH toward normal. " Metabolic alkalosis: ! hypoventilation: loss of H+ from the body by vomiting. #pH causes reflex $ in alveolar ventilation and # pCO2 ! generation of H+ and HCO3- ! compensatory $ pH. Ventilation Perfusion Relationships (V/Q mismatch): • • The exchange of O2 and CO2 in the lungs is determined by: o the ratio between alveolar ventilation (VA) and pulmonary blood flow(perfusion) (Q) o the rate of O2 consumption and CO2 production (VO2 and VCO2). o the gas tensions in inspired air and in mixed venous blood o chemical processes in the blood. Causes of Hypoxemia ($PaO2): o Inadequate (Hypo-) Ventilation: " Reduced or deficient ventilation of the alveoli per unit time. If O2 consumption is not correspondingly reduced, hypoventilation results in a reduction in alveolar PO2 and hypoxemia. o Diffusion Impairment: " $ diffusing capacity of the lung ! lack of equilibration between PcapillaryO2 and PAO2. (blood-gas barrier is thickened ! diffusion is so slow that equilibration is incomplete – i.e. diffuse interstitial fibrosis, asbestosis or a build up of fluid in alveoli as a result of infection). o Right ! Left Shunt (venous blood bypasses the lung): " right-to-left shunts (arterial blood is diluted with venous blood). " natural shunts occur as a result of the return of the bronchial circulation to the left heart via the pulmonary veins and from the return of some coronary venous blood to the left ventricle via the thebesian veins. most common shunts are extra-pulmonary (atrial or ventricular septal defects or a patent ductus arteriosus). o the final PO2 that results from a mixing of blood volumes with different oxygen partial pressures cannot be calculated by simply weight averaging the respective PO2 values. Calculations require that the content of each of the two streams of blood (venous and oxygenated) be determined first. After averaging O2 content, the PO2 of the blood mixture can then be estimated. " Hypoxemia resulting from sizeable shunts cannot be corrected by breathing pure O2. Hemoglobin carries most of the O2 in blood and is nearly saturated under normal conditions. Breathing 100% oxygen adds little additional O2 to hemoglobin, it adds a small additional quantity of dissolved gas, and it adds nothing to the shunted venous blood $ breathing 100% oxygen can significantly raise the partial pressure of O2 in the oxygenated blood, but the additional content of O2 is quite small and may not compensate for the oxygen deficit in the venous blood with which it is mixed. • Breathing 100% O2 ! ! A-a difference with a lack of correction and is thus diagnostically indicative of a shunt. o Ventilation/Perfusion Mismatch (effects of gravity): " PaO2 is slightly less than PAO2 because of mismatching of blood flow and ventilation in various parts of the lung (and secondarily because of natural right to left shunts) " Normal blood flow to the lung is equal to the cardiac output and is about 5 L/min. Resting alveolar Regional arteriolar differences are ventilation is roughly 4L/min. accounted for in " VA/Q ratio for the lung = 0.8 arteries. That is, the " There is non-homogeneity in the ratio of ventilation to perfusion in each alveolus (PvO2 and PvCO2 are pO2 of the fairly homogenous with regard to location within the lung, but alveolar heterogeneity gives locational pulmonary artery is individuality to PaO2 and PaCO2 (due to gravity). the same at the apex • Final arterial values are the summation of all varying individual PaO2 and PaCO2. and the base. " In the normal lung, regional differences in VA/Q ratios are responsible for an alveolar – arterial (A-a) oxygen partial pressure difference (PAO2 – PaO2) of ~ 4 mm Hg. • Blocked Airway (restricted ventilation) $ V A/Q Normal VA/Q # V A/Q – significant $ perfusion can’t deplete O2 or enrich with CO2. Blocked Vessel (restricted perfusion) Bottom of lung (similar to venous pressures) • Top of lung (similar to inspired air) Regional Variation in V/Q ratio: o Gravity and Ventilation: at any given lung volume, alveoli are more distended at the top of the lungs than at the bottom. Ventilation is better at the bottom of the lung than at the top. " Because of gravitational forces, PIP is more negative at the top of the lung than at the bottom (weight of lung pulls down from the apex of the thoracic cavity – lowered PIP ; weight of the lung compresses the base – increased PIP). ! a gradient of distending pressures (PA – PIP) from the top of the lung to the bottom. • Larger alveoli at the top of the lung would be on a less compliant portion of the pressure-volume curve and would be less distensible than smaller ones at the bottom • An inspiratory effort that # PIP everywhere in the lung will cause the greatest volume change in the alveoli with the highest compliance $ for a given ! PDistending, ventilation will greater at the base of the lung than at the apex. • By gravity, the basal lung is relatively compressed in its resting state but expands more readily on inspiration than the apex. o Gravity and Perfusion – top of the lung is poorly perfused, and the bottom of the lung is well perfused. " Pulmonary circulation has low vascular pressure and low flow resistance. Mean pulmonary artery pressure is about 15 mm Hg. As blood flows through the pulmonary circulation, frictional forces cause energy to be lost as heat, and pressures continually drop along the arterial tree, the arterioles, capillaries, venules and veins. • Pressures are influenced by gravity as well. For each cm we move upward above the level of the heart, the hydrostatic pressure in the blood vessels decreases by 1 cm of H2O. • Distending pressure for vessels within the lung is the difference between the pressure within the vessel and that within alveoli (Pa - PALV). • Zone 1 (PA > Pa > Pv): At the top of the lung, arterial pressures would be above alveolar pressure (when PALV = 0 cm H2O) for only a part of the cardiac cycle. When arterial pressure fell (due to runoff) below 0 cm H2O, vessels would be compressed and flow would stop ! the pulmonary artery may be insufficient (during part of the cardiac cycle) to maintain blood flow at the apex of the lung (when PA > Pa, all downstream vessels are collapsed). Complete cessation of flow throughout the cardiac cycle does not occur in Zone 1 unless arterial pressure is reduced (hemorrhage) or if PA is ! (positive pressure ventilation). Ventilated but unperfused areas contribute to alveolar dead space. • Zone 2 (Pa > PA > Pv): Ppulmonary artery # because of # hydrostatic pressure (exceeds PA). Pressure in downstream venules falls below PA and they collapse (thus act as Starling resistors - when venules collapse and flow stops, fluid energy is no longer dissipated as heat and pressure rises. As soon as pressure in the vessel exceeds that in the alveoli, the vessel opens and flow is reestablished. However, with flow, the fluid energy (pressure) drops and the vessel collapses once again. Pressure in the venule, therefore, oscillates slightly above and below alveolar pressure, and the gradient for flow effectively becomes the difference between arterial and alveolar pressures) • Zone 3 (Pa > Pv > PA): pressures have increased by 1 cm H2 O for every 1 cm of descent. All pressures in the vasculature exceed PA, and all vessels remain open. Distending pressures for vessels is greatest in this zone. Vascular resistance is lowest and perfusion greatest in this region of the lung. There is a vertical distribution of VA/Q. The top of the lung is relatively poorly perfused (with respect to ventilation) and the bottom of the lung is relatively poorly ventilated (with respect to perfusion). Though ventilation (VA) and perfusion (Q) each alone decrease near the top of the lung, the V/Q ratio improves near the top of the lung. BUT, given the perfusion at the bottom of the lung, relatively more gas exchange occurs there. At the bottom of the lung, VA/Q approaches zero (little or no ventilation) and at the top, VA/Q approaches infinity (little or no perfusion – alveolar dead space) Point V represents the PO2 and PCO2 of mixed venous blood (~ 40 and 46 mm Hg). Point I represents the PO2 and PCO2 of inspired gas (~ 150 and 0 mmHg). Lung units with VA/Q near zero (little or no ventilation) would reach equilibrium values near V. Lung units with VA/Q approaching infinity (little or no perfusion, i.e., alveolar dead space) would reach equilibrium values near I. AATT TTO OPP:: ppCCO O222 ##,, ppH H!!,, ppO O222!! o Effect of Regional Differences of VA/Q on Mixed Arterial Blood - The small amount of oxygen added by the unit with the high VA/Q ratio (right: PO2 = 100 mm Hg) is inadequate to compensate for the deficit created by the unit with the low VA/Q ratio (left: PO2 = ~ 38 mm Hg). Overall depression of mixed arterial PO2 to about 50 mmHg (for a Hb concentration of 15 gm/100 ml). In normal individuals ventilation perfusion mismatch accounts for the small 4 mm Hg (PA - Pa) difference. In the diseased lung, nonuniform distribution of ventilation and perfusion is the main cause of arterial hypoxemia and hypercapnia. Average weighted alveolar pO2 ~ 101mmHg. This is depressed to ~97mmHg by mixing of blood (i.e. poorly ventilated and well-ventilated areas). ! A-a difference of 4mmHg due to V/Q imbalance. $V and # Q ! #Q -A contribution of PaCO2 = 36mmHg #VA and $ Q ! $Q --contribution of PaCO2 = 36mmHg Regulation of Respiration: • Peripheral Efferent Impulses: o o o o • Augmenting Behavior: at the onset of inspiration, impulses in fibers innervating inspiratory muscles # in frequenc and then rapidly $ near the end of inspiration (“ramplike”). # Depth of Respiration: (1) # frequency of impulses (steepness of ramp#) ; (2) recruitment (activation of more motor units) ; (3) longer duration of impulse burst (prolonged inspiration) – # VT but does not # minute ventilation (period of inspiration and of expiration both increase). Force of contraction is progressively summated during inspiration until sudden termination . Rate of Breathing: Determined by duration of interval between successive discharge and the duration of the burst of impulses during inspiration (expiratory time " inspiratory time ! longer, slower breaths ! slower respiratory frequency). Muscle Control: During inspiration, the expiratory muscles are inhibited. Only during forced expiration is there an # excitation of motor units innervating expiratory muscles. Central Regulation of Respiration: - rhythmic excitation of respiratory motor neurons depends on impulses arising in higher centers (bilaterally in the pons and medulla). o Transection of the brain stem below the medulla results in respiratory arrest o Transection of the brain stem above the pons limits (but does not abolish) breathing (or the majority of reflex control) " Dorsal Respiratory Group [Nucleus of Tractus Solitarius] – associated with inspiration " Ventral Respiratory Group [medulla] – associated with inspiration and expiration " Pneumotaxic Center (Pontine Respiratory Group) [pons] – provides early cut-off of inspiration • Removal of input from the vagus or from the pontine respiratory group ! ! depth of respiration and # respiratory frequency. o If both inputs are removed, apneustic breathing results (prolonged inspiratory period interrupted by brief expiratory gasps). o Self-Cycling Firing/Quiescence Cycle Regulation: primarily in the Dorsal Respiratory Group. - The medullary inspiratory neuron (A) sends descending fibers to respiratory motor neurons in the C-spine (phrenic) and sends processes that synapse and stimulate other medullary neurons (B). B sends processes to an inhibitory interneuron (C), the offswitch, which synapses on, and inhibits the inspiratory neuron (A). A ! activates inspiratory motor activity and generates an inhibitory input to itself (negative feedback loop). The inhibitory interneuron (C) also receives input from the pontine respiratory group (pneumotaxic center) to aid in controlling duration and depth of inspiration. - Hering-Breuer Reflex: Pulmonary stretch receptors (in the airway smooth muscle layer) are activated by lung inflation. Afferent APs travel in the vagi and activate B neurons (# activation of the off-switch) ! # termination of inspiration. Tidal volume must be at least 1L to invoke the Hering-Breuer reflex (so adults can take deep and ! frequency breaths). - Neurons in the VRG have expiratory phase firing patterns that reciprocally inhibit inspiratory neurons and vice versa (accounts for the lack of firing of inspiratory neurons during expiration, and vice-versa)- The Pre-Botzinger Complex (neurons in the rostral portion of the VRG) have pacemaker activity and fire at the onset of inspiratory activity (central pattern generators) • Modulation of Neural Control: #pO2, !pCO2, #pH (![H+]) all ! ! ventilation (to return values to normal – PaO2 = 100mmHg, PaCO2 =40mmHg, and pH = 7.4) o Chemical Factors: " Chemoreceptors monitoring arterial PO2, PCO2 and pH: Chemoreceptors excite inspiratory PMN and inhibit the offswitch (C) ! ! VE (minute ventilation). • Peripheral: Carotid and Aortic bodies (100% of pO2 regulation, 80% of pH regulation, and 10% of pCO2 regulation). • Central: ventral surface of the medulla near the exit of CN IX and X. Chemicals reaching central receptors are determined by the blood brain barrier. o Changes ventilation due to PO2 and pH result from stimulation of the peripheral chemoreceptors. The central chemoreceptors modulate ventilation primarily in response to CO2. Central chemoreceptors detect changes in the acidity of the CSF that baths the o neurons (CO2 crosses the blood-brain barrier and ! CSF pH is influenced by changes in arterial PCO2) Changes in CO2 are more effective in changing ventilation than are changes in O2. (Effects of gas partial pressures on ventilation are most pronounced when PO2 and PCO2 change reciprocally (when PO2 decreases as PCO2 increases and vice-versa). Until pO2 is less than 60mmHg, Hb saturation ~ 90% … Below this, ventilation # $ humans are not as sensitive to O2 as to CO2. !pCO2 ! neural toxicity (CO2 narcosis). The response to both gases is greater than the sum of individual responses (synergistic) • • • CO2: ! PCO2 is much more effective in increasing ventilation than # in PO2. (! PCO2 of as little as 3 mm Hg ! doubled ventilation). o Relationship between ventilation and PCO2 rises steeply and reaches a maximum when PCO2 is about 70-80 mmHg. o At # levels, ventilation $ because of the toxic effects of high CO2 on central neuronal function. Inactivation of carotid and aortic bodies does not abolish or even significantly reduce the total respiratory response to inhalation of CO2. o Central chemoreceptor response to CO2 is mediated through !s in [H +] of the CSF (via carbonic anhydrase and a carbonic acid intermediate) ! when [CO2] # in arterial blood, [H+] in CSF # ! activation of central chemoreceptors. " $ pH of CSF as a result of chronic # PCO2 leads over time to a compensatory # [HCO3-], returning pH of CSF toward normal and shifting the sensitivity of the receptors so that they operate over a higher range of PCO2. " When PaCO2 is chronically $ (high altitude), ! in CSF-HCO3- are in the opposite direction, and sensitivity of the chemoreceptors is shifted to lower ranges of PCO2. " ! arterial pH is ineffective in activating central chemoreceptors, because the blood-brain barrier prevents H+ from entering the CSF. O2: carotid bodies are more important than the aortic bodies as respiratory regulatory organs. o Aortic and carotid bodies are relatively insensitive to ! PO2 (changing the fraction of O2 in air from 21% to as low as 12-14% doesn’t change ventilation significantly). At fractional O2 content less than 10%, # ventilation is pronounced " Type I glomus cells contain O2 sensitive K+ channels (close when O2 levels $ ! depolarization of the cells) ! opening of voltage gated Ca2+ channels and influx of Ca2+ ! transmitter release and activation of the afferent nerves fibers " Chemoreceptor blood flow is enormous (40x that of the brain) " Normally, PaO2 levels are monitored, not the oxygen content of the blood. When flow $ substantially (hemorrhage), AB and CB respond with increased electrical activity even though PaO2 may be normal. (Large $ flow produces a local $ in PO2 that depolarizes the glomus cells). • Glomus cells contain K+ channels that are closed not only by low levels of O2, but also by $ pH of the cell (can result from # pCO2 or metabolic acidosis). Subsequent depolarization will trigger transmitter release and activation of afferent fibers. o Denervation of the CB and AB results in complete loss of the respiratory response to ! PaO2, while the response to PaCO2 remains unchanged. Respiratory response to changes in pO2 derives entirely from the peripheral chemoreceptors, while that for CO2 is regulated by central chemoreceptors. [H+]: # [H+] ! # tidal volume and frequency (not as sensitive to pH as to O2 /CO2 o AB and CB respond to $pH (major portion of respiratory response to ! arterial pH) Central chemoreceptors have a role in response to large ! arterial pH (H+ may be able to cross the blood-brain barrier through very infrequent “breaks”). o Other Inputs to the Respiratory Center: " Cerebral cortex: can initiate voluntary respiratory activity that can # minute ventilation to 160 L/min. " Hypothalamic centers and the limbic system: can be activated by emotional states (anxiety, fear or stress) ! # ventilation. • Temperature receptors (hypothalamic and skin): ! # ventilation when body temperature #. Helps the body lose heat by warming inspired air. " Receptors in muscles and joints: cause # ventilation with exercise or simply when limbs are moved passively. E. " Protective reflexes (sneezing and coughing) " Medullary areas: block respiration during swallowing and vomiting. " Activation of baroreceptors: ! $ respiration when BP #, and # respiratory activity when BP$. Lung Receptors: o Stretch Receptors (in airway smooth muscle): # activity during inspiration and cause # firing of vagal afferents to the medullary respiratory control center (Hering-Breuer reflex). The inflation reflex is well-developed in newborn babies; it $ in effectiveness during the first 5 days of life. The reflex is weak in adults (unless the lung inflation is 1L or more). o Irritant receptors (between airway epithelial cells): respond to noxious gases, ammonia, cigarette smoke, other particulate material and agents such as histamine. " Vagal afferents ! reflex bronchoconstriction and hyperpnea. o J Receptors (in conducting airways and alveoli): respond to chemical and mechanical stimulation. " Vagal afferents (slowly conducting, unmyelinated, C-fibers) ! rapid, shallow breathing, bronchoconstriction and mucus secretion. " May play a role in dyspnea associated with left heart failure. " • Hemo-Physiology Red Blood Cells: • Primary Functions of Blood: o Transport medium for: Nutrients (glucose, amino acids, fatty acids, O2, salts, etc), waste products (CO2, urea, uric acid), hormones, defense components (antibodies, leukocytes, platelets, clotting factors), carrier proteins (albumin, transferrin, lipoproteins), heat, H+, and H2O (among others). " Centrifugation of blood ! solid red layer at the bottom (RBCs), a yellowish layer at the top (plasma), and a tan interface (the “buffy” layer – platelets and leukocytes. • % hematocrit = height of whole blood / height of RBCs. o Male: 42-52% o Female: 37-47% • A unit of blood = 500ml (75ml/kg) o Male: 5.2L o Female: 3.3L o Carrier Proteins: Triglycerides (lipoproteins), fatty acids (serum albumin), iron (transferrin), bilirubin (serum albumin), thyroxine – thyroid hormone (thyroid binding globulin). o Transport of O2: Transport of O2 principally via the carrier protein hemoglobin (15g Hb/100ml of blood). This is three times the amount of all the other plasma proteins combined. • Erythrocytes: o Overview: " Produced by precursor cells in bone marrow. Has no nucleus, no ribosomes, and no mitochondria and ! is essentially a biconcave sac filled with a 30% Hb solution (plus other enzymes and smaller molecules). " Incapable of synthesizing proteins or lipids or of carrying out oxidative metabolism. " Survives for ~120 days despite repeated deformations throughout circulation and despite repeated exposure to # turbuence. • Loss of deformability upon aging leads to death of the RBC. " The overall function of erythrocytes is to protect Hb from denaturation and degradation. The lifespan of hemoglobin in the red cell (intracorpuscular hemoglobin) is the same as the lifespan of the RBC (120 days). In the absence of a protective envelope, Hb rapidly disappears from the plasma (lifespan of seconds or minutes). • In whole blood: 15g Hb/100ml blood • HCT = 50% • In RBCs: 30g Hb/100ml cytosol (30% concentration) o Borderline crystallization (a la sickled RBCs) o Structure: Flat biconcave disk (8µ m x 1 µ m x 1µ m) NOT spherical " A sphere has the worst possible surface area/volume ! deformation ! # SA ! # surface tension ! # tearing of membrane (spherocytosis) ! RBCs are flat for protection ! $intracellular time of diffusion to and from Hb (O2 loading and unloading). o Metabolism: Insulin Independent " 90% of RBCs energy is derived from anaerobic glycolysis (conversion of glucose to lactic acid). The remainder comes from the hexose monophosphate shunt (the phosphogluconate pathway). • ATP derived from glycolysis powers the Na+-K+-ATPase (maintains Na+ and K+ gradients ! keeping H2O out). o ~30% of RBCs energy resources may be thus devoted. • ATP also fuels active removal of calcium from RBCs. Buildup of calcium ions causes crosslinking of RBC membrane proteins and decreased deformability of the cell. o NADPH (from phosphogluconate pathway) drives reduction of glutathione (GSSG). Reduced glutathione (GSH) protects the cell membrane and hemoglobin against oxidants. Oxidation of sulfhydryl groups (H2O2) associated with RBC membrane proteins ! # increased stiffness and fragility of older cells. Fe2+ can also become Fe3+ (methemglobin – metHb – cannot bind O2). " ATP depletion ! RBC crenation (deformation), swelling (H2O influx), and $ Lactose Dehydrogenase deformability (due to Ca2+ sedimentation). Renewal • Principal task of the erythrocyte -- transport of oxygen and carbon dioxide -- does not require energy (relies on passive diffusion ). Energy is utilized primarily in maintenance functions in the mature erythrocyte (strong correlation between drop in ATP concentration and impaired survival in older red cells). o Erythropoiesis (production of RBCs): At time of birth, almost all RBCs are produced by bone marrow. As we age, marrow components of long bones become filled with fat. Erythropoiesis is a self-sustaining system (maintenance on stem cells puts RBCs at risk – chemotherapy/radiation). " Precursors: • (1) Pluripotential Stem Cells: give rise to erythrocytes, platelets, monocytes, granulocytes, and lymphocytes. • (2) Committed Stem Cells: develop from pluripotential cells and give rise only to one type of blood cell. o Committed stem cells give rise to normoblasts (make Hb), which undergo four differentiating divisions (~five days). Hb synthesis occurs in normoblasts (all maturational stages) until concentration is 20g/100ml at which point cell division stops and the reticulocyte is produced ! circulation (five days after differentiation). " Ribosomes, ER, and mRNA keep making Hb for ~2days in the reticulocyte form, then the cellular machinery breaks down ! biconcave shape. o Reticulocyte Index: Normally ~ 2% of all RBCs are reticulocytes. # when production of RBCs is #. o Regulation of Erythropoiesis: Regulation of [RBC] is via rate of production, not rate of hemolysis. " # Erythropoiesis due to: • (1) # size of erythroid marrow compartment (#total number of erythrocyte precursors) • (2) # rate of maturation of erythrocyte precursors o Both of the above are stimulated by erythropoietin. Erythropoietin production ! (from endocrine cells in kidney) due to #pO2. Erythropoietin stimulates erythropoiesis during day-to-day regulation of RBC output (in addition to hypoxic crisis). EPO also stimulates Hb synthesis by normoblasts. • If one kidney is failing, it can perceive #pO2 ! !erythropoiesis ! abnormally ! [RBC]. o Hemoglobin Synthesis/Iron Metabolism: " Hemoglobin: • Globin: tetrameric protein consisting of two molecules, each of two different polypeptide chains ($ and %-globin) • Heme: porphyrin ring structure containing iron which binds oxygen (in Fe2+ form). o There are four heme groups, four iron molecules, and four O2 binding sites per Hb molecule. " and %-globin are synthesized on ribosomes in the cytosol of normoblasts. The porphryin group is synthesized in the mitochondria of normoblasts. • Sources of Iron: salvaged from degraded red blood cells > mobilized from body stores > dietary iron o Iron is complex with transferrin in the blood. Fe-Transferrin enters marrow and binds on a normoblast surface receptor. Iron is incorporated into Hb and transferrin returns to plasma. Iron is stored bound to ferritin or hemosiderin. o Regulation of iron levels in the body is exerted by rate of intestinal absorption of iron at the level of the mucosal cells (enterocytes) in the GI tract. It can be retained in mucosal cells as ferritin complex and eliminated as cells slough into feces or it can bind reversibly to mobilferrin !ferroportin ! transferrin complex. " Iron absorption is influenced by the amount of apoferritin (synthesized by mucosal cells). (! when body iron stores are abundant and # when body stores are depleted. Translation of the apoferritin mRNA that is regulated by the cytosolic iron concentration. " Hepcidin is the principal regulator of the absorption and systemic distribution of iron. It binds ferroportin (basolateral membrane of enterocytes) and initiates degradation. If [hepcidin] !, little ferroportin is available to permit iron absorption. Hepcidin synthesis ! by conditions producing iron loading and # by anemia, hypoxia or ! erythropoiesis. o RBC Destruction: 10% intravascular hemolysis (Rh-incompatibility, transfusion reactions, etc) and 90% extravascular hemolysis (splenic cord filter fragmentation, etc). " Hb and other RBC proteins are degraded to amino acids (reutilized for protein synthesis). Iron may be temporarily stored (within the macrophage) and is released and bound by transferrin (returned to the marrow or other stores). Porphyrin group is degraded via the bilirubin pathway. Hb dissociates into dimers, which bind to serum protein haptoglobin. During severe intravascular hemolysis, the capacity of haptoglobin to bind Hb dimers is exceeded, the plasma haptoglobin becomes depleted, and a portion of Hb may appear in the urine (hemoglobinuria), which will turn dark. If intravascular hemolysis #, Hb-tetramers ! methemglobin ! globin or heme (! liver via hemopexin). • The conversion of porphyrin to bilirubin yields carbon monoxide ! measurement of CO in expired air provides an index of # hemolysis. Anemia: insufficient number of circulating red blood cells or insufficient amount of circulating Hb. Anemia can develop if rate of RBC production is abnormally #, if the rate of RBC destruction of red cells is abnormally ! , or both. Anemia also results from acute loss of blood o Inadequate erythropoiesis: " • " " " Iron Deficiency: Rarely occurs in males except after bleeding episodes. Fairly common in females due to loss of iron in the menstrual flow, and, during pregnancy, because of the relative iron deficiency resulting from the demands of fetal erythropoiesis. Treated by giving iron. Vitamin B12 Deficiency/Pernicious Anemia: Inability to absorb B12 (failure of intrinsic factor). Treated by parenteral B12 administration. Folic Acid Deficiency: • B12 and folic acid deficiency are necessary for polynucleotide biosynthesis $ for proliferation of erythrocyte precursors in marrow (lack of maturation of erythrocytes. " " " o Diseases of Bone Marrow: Failure of marrow to produce erythrocytes often results from unknown causes (i.e. chemotherapeutic agents and radiation). These agents destroy stem cells and deplete marrow of its primary erythrocyte precursor cells. Defective Hb Synthesis: Thalassemia (" and % globin chains are not synthesized in equal amounts) ! destruction of erythrocyte precursors in the marrow. &-globin (fetal) can be upregulated to replace %-globin. Inadequate Erythropoietin: results from renal or liver disease. Abnormal Rate of RBC Destruction: Abnormally rapid destruction of RBCs is called hemolysis (hemolytic anemia). Marrow has the capacity to expand its rate of production of RBCs by 6x. With normal marrow function, ! erythropoiesis $ partly compensates for hemolytic conditions in which the life span of circulating erythrocytes is only 20 days " Factors # lifespan of RBCs: • • • • • • Genetic abnormalities of RBC membrane ! rigid or fragile cells o Hereditary Spherocytosis: RBCs are spherical and rigid. They suffer damage while passing through the spleen. Treated by splenectomy. Genetic abnormalities of Hb o Sickle Cell Anemia: One Glu!Val in the %-chain of Hb. In response to #pO2 and #pH, Hb polymerizes and RBC assumes a rigid (sickled) shape. Sickled cells are trapped in microvasculature and the spleen. Genetic abnormalities of RBC metabolism o Glucose-6-phosphate deficiency: activity of the hexose monophosphate shunt is # ! # production of NADPH and reduced glutathione. RBCs are sensitive to oxidizing agents ! hemolytic crises. Presence of antibodies against RBC surface antigens ! lysing o Rh-incompatability Lytic agents released by bacteria Blood Grouping/Typing: AB = universal recipient O = universal donor Rh Blood Group (D antigen): - Cells which bear D antigen are termed Rh-positive; cells without D antigen are termed Rh-negative. When an Rh+ father and an Rh mother conceive an Rh+ fetus, RBCs from the baby introduce Dantigens into maternal circulation (at birth), antibodies against the D-antigen form in the mother (anti-D antibodies develop after birth) ! in subsequent pregnancies with an Rh+ fetus, maternal anti-D antibodies cause erythroblastosis fetalis (fetal hemolysis). - At time of birth of an Rh+ child, an Rh- mother can be treated with rhogam (D-antibodies). These injected antibodies remove Dantigen from maternal circuation ! prevent maternal immune system from detecting the antigen and prevent maternal antibody production (the injected antibodies degrade). Hemostasis: (1) Vasoconstriction, (2) Platelet Plug Formation, (3) Blood Coagulation, (4) Control and Limitation of Clotting • Vasoconstriction: (crushing injuries bleed less than cutting injuries) – vasoconstrictive response " damage o Myogenic/Neurogenic Vasospasm (immediate smooth muscle reaction): " Immediate local vasoconstriction (several cm on either side of the injury site) – may cause almost complete cessation of flow. " Pain receptors can trigger spinal reflexes ! # sympathetic firing and # vasoconstriction. • Myogenic and neurogenic vasospasms follow injury immediately and help prevent acute blood loss. o Lasts no more than 20-30 minutes. Humoral Response (serotonin and prothrombin released at site of injury – minutes later): " Serotonin (5-HT), thromboxane A2, and prostoglandins all cause vasoconstriction • Humorally triggered vasoconstriction rapidly follows injury (but slower than myogenic/neurogenic vasospasm) o Lasts up to several hours. o Pre-Capillary Sphincter Plugs: control blood loss at the capillary level by contraction of the muscle sphincters at the junctions of met-arterioles and the capillaries. Platelet Plug Formation: when a wall defect forms, platelets in the vessel bind subendothelial collagen (platelet adhesion) outside of the defect. Binding ! conformation ! ! granular exocytosis (release reaction – ADP, Ca2+, and serotonin released, and Phospholipase A2 activated ! Thromboxane A2. o • (1) Platelet Adhesion: Platelets bind Von Willebrand Factor (made by endothelium), which is in turn bound to collagen [VWF coats collagen and binds Factor VIII for coagulation – VWF disease is common (~10% of population) and may or may not include some combination of platelet adhesion or coagulation defecits. o (2) Release Reaction: ! shape and release of storage granules. " Diacylglycerol (DAG) and Inositol triphosphate (IP3) are platelet 2nd messengers. • DAG activates C-kinase and IP3 ! release of intracellular Ca2+. o Synergistically, DAG and IP3 # release reaction. " Release reaction ! #ADP, Ca2+, Serotonin, and activation of Phospholipase A2 (! Thromboxane A2). • Thromboxane A2 and ADP ! # platelet-platelet adhesion (platelet aggregation) and # release reaction (! positive feedback ! # Thromboxane A2 and ADP release). o Platelet aggregation does not normally extend away from the site of injury because the endothelial cells of the surrounding vessels produce an inhibitor of aggregation (prostacyclin – PGI2). o Aspirin (acetylsalicylic acid) inhibits thromboxane A2 formation ! no clotting: " acetylates and inactivates the enzyme cyclo-oxygenase (required for thromboxane A2 formation). The function of a given platelet is irreversibly depressed following exposure to aspirin. Blood Coagulation (secondary hemostasis): o Fibrinogen ! Fibrin (protease activation to fibrin monomers, which spontaneously aggregate to polymers with assistance of fibrin stabilizing factor – CF VIII) forming the strong cables of a clot). " The Fibrinogen ! Fibrin reaction is driven by thromboplastins (activated by platelets) and Ca2+. • Thromboplastins = Clotting Factors o • Clotting factor I II III IV V VII VIII IX Synonym Fibrinogen Prothrombin Thromboplastin Calcium Proaccelerin, labile factor Proconvertin, stable factor Antihemophilic factor/globulin, antihemophilic factor A Plasma thromboplastic component, Christmas factor, antihemophilic factor B X XI Stuart Prower factor Plasma thromboplastin antecedent, antihemophilic factor C. XII XIII Hageman factor, glass factor Fibrin stabilizing factor, Laki-Lorand factor o Deficiency of factor I Syndrome Afibrinogenemia (can be congenital - rare) II Hypoprothrombinemia (usually as a consequence of vitamin K deficiency) V Parahemophilia (congenital) VII Hypoconvertinemia (congenital) VIII Hemophilia A (congenital sex-linked) IX Hemophilia B (Christmas disease) (congenital) X Stuart-Prower factor deficiency (congenital) XI PTA deficiency (congenital) XII Hageman trait (congenital) VIII Deficiency is Classic Hemophilia Intrinsic Pathway: all involved factors are found in the blood " The cascade system has tremendous amplification potential (but breakdown at any step ! inhibition of the system). • The intrinsic pathway has a requirement of Ca2+ and platelets. • There are positive feedback loops within the system (fibrinogen cleaves V ! Vactive) " Contact Activated by the binding of clotting factor XII to the subendothelial connective tissue exposed by blood vessel injury (along with prekallikrein, kininogen, and factor XI). Factor XII undergoes structural ! and with kininogen (cofactor) digests prekallikrein ! kallikrein. Kallikrein cleaves factor XII ! XIIa (kininogen – cofactor). Factor XIIa and kininogen cleave factor XI ! XIactive. Factor XIa activates Factor IX ! IXa (with Ca2+ - cofactor), which is bound to phospholipids (platelet factor three – PF3) on the surface of aggregated platelets. Factors VIII and IXa are bound to PF3 along with Ca2+ and Factor X ! Xa. While bound to PF3 Prothrombin (II) is cleaved to Thrombin (IIa) in the presence of Ca2+ (Factor V – cofactor) by Xa. Thrombin cleaves fibrinogen to produce fibrin (monomers). Fibrin then freely polymerizes to form fibrin. Polymers are rapidly strengthened and stabilized by the formation of covalent bonds within fibrin strands and between different strands under the action of factor XIII (itself activated by thrombin). o Extrinsic Pathway: one extrinsic component - tissue factor/tissue thromboplastin (derived from adventitia of blood vessels – not normally exposed to blood) " following an injury in which blood extravasates, tissue factor binds to and activates the blood factor VII. Following tissue trauma with blood extravasation, tissue factor (Ca2+ and phospholipids – cofactors) cleaves Factor VII ! VIIa. Factor VIIa activates Factor X ! Xa (in the presence of Ca2+ and phospholipids). Factor Xa activates more Factor VII (positive feedback loop). Prolonged digestion by Xa ultimately inactivates factor VII (delayed negative feedback regulatory loop). Factor Xa acts as above. The extrinsic pathway (contact activation) represents the main trigger for coagulation. Once triggered, the end product of the pathway, thrombin, has a positive feedback action, activating intermediates of the intrinsic pathway, XI and VIII, which then amplify the response, producing far greater amounts of thrombin. Thrombin can also activate V and, as noted above, XIII. Once tissue thromboplastins mix with the blood, thrombin is released rapidly, and clotting by the extrinsic pathway may require only 10-15 seconds as opposed to the 1-3 minutes required by the intrinsic system. " " Role of Calcium Ions: o Critical for calcium binding by various factors (II, VII, IX, and X) is the presence of &-carboxyglutamic acid (GLA) residues in the amino acid sequences of these proteins. Vitamin K is required for carboxylation (posttranslational modification) to form these GLA residues. In the absence of vitamin K there is no calcium binding by Factors II, X, VII and IX ! there is no coagulation. " Coumadin (anticoagulant) blocks enzymatic formation of GLA residues ! factors in the circulation cannot bind Ca2+ and ! coagulation is impaired. • EDTA/Citrate block Ca2+ in blood tubes (preventing coagulation in blood being sent to labs). Control and Limitation of Clots: o Clearance of Clotting Factors by Blood Flow: " As blood vessels dilate (site of injury is normally vasoconstricted) activated clotting factors diffuse (or are transported) away and they are readily diluted and removed by normal clearance mechanisms. o Antithrombins (potentiated by heparin): " "-2-globulin antithrombin III binds to and inhibits thrombin and activated factors IX, X, XI and XII. Its activity is enhanced over 1000X by the presence of heparin. o Protein C: " Protein C limits clotting and is activated by thrombin. Thrombin only becomes active against this target once it is itself bound to the protein thrombomodulin (a thrombin receptor protein on the surface of endothelial cells). When bound to thrombomodulin, thrombin does not act on fibrinogen, but instead activates protein C. " Protein C (and protein S), inhibits clotting factors VIII and V • ! Thrombin directly activates factor VIIIa and Va but also inactivates them, indirectly, through proteins C/S. o Tissue Factor Pathway Inhibitor: " Produced by endothelial cells, TFPI binds to and inactivates VIIa (inhibits extrinsic pathway). o Plasmin (Fibrinolysin): " Plasmin is a proteolytic enzyme present in the circulation as an inactive proenzyme (plasminogen). Tissue plasminogen activator (tPA) is released from endothelial cells following injury and cleaves plasminogen ! plasmin (Factor XIIa – cofactor). " Plasmin degrades fibrin and fibrinogen (! dissolves clots). • Genetically expressed tPA is used to degrade clots in the coronary artery. " Degradation of fibrin by plasmin is also an important means of remodeling and breaking down the clot during the healing process. o Plasminogen Activator Inhibitors (PAI) and Antiplasmin: help to modulate the fibrinolytic action. o Angiostatin: derives from part of the plasminogen molecule and inhibits growth of blood vessels Renal Physiology and Homeostasis Renal Organization and Function: " " Primary Functions of the Kidneys: (1) Regulate volume and composition of ECF (concentration of inorganic ions, osmolality, and acidity) in order to maintain homeostasis, (2) Excrete metabolic waste products (urea, uric acid, and creatinine), end products of Hb degradation, and foreign chemicals (drugs, food additives, and pesticides), and (3) Produce circulating factors (erythropoietin, renin, and 1,25-dihydroxyvitamin D3). Structural Organization: o Two retroperitoneal organs (in the back of the abdominal wall). Urine flows from kidneys ! ureters ! bladder ! elimination during micturition (via the urethra) o Each kidney contains ~1 million nephrons. o Tubular fluid from nephrons ! collecting ducts ! pyramids ! papillae (tips of pyramids) ! calyces ! renal pelvis ! ureter. Pyramids contain the collecting ducts, loops of Henle, and vasa recta Blood Supply: Renal artery divides ! interlobar and arcuate arteries ! interlobular arteries ! Afferent arterioles (perpendicular to interlobular arteries – each supplies blood to a single nephron) ! glomerulus (glomerular capillary bed) ! efferent arteriole ! peritubular capillaries and vasa recta (lg. capillaries from which emerge a network of small capillaries surrounding the medullary tubular system – less flow than cortical system) ! venules ! interlobular veins ! interlobar and arcuate veins ! renal vein ! portal circulation. " Degree of constriction of both the efferent and afferent arterioles influences pressure and flow through the glomeruli. Caliber of the efferents also influences pressure and flow through the vasa recta. Nephron: o Renal Corpuscle: Contains the glomerulus and a surrounding fluid filled capsule (Bowman’s Capsule). " As blood enters the glomerulus, a fraction (20%) is filtered into Bowman’s space, forming the ultra-filtrate. The remaining blood leaves the glomerulus via the efferent arteriole. o Tubule: A narrow cylinder made up of a single layer of epithelial cells resting on a basement membrane. The ultrafiltrate enters the tubule where it is processed before exiting the kidney as urine. " Continuous with Bowman’s capsule o Type: There are two types of nephrons " (1) Cortical Nephrons (80%): corpuscle in the cortex and short tubular systems reaching varying levels in the outer medulla. " (2) Juxtamedullary Nephrons (20%): large corpuscles located at the junction of the cortex and medulla. They have long loops of Henle that enter the inner medullary zone parallel to the vasa recta o " " " Glomerular Filtration Barrier of the Nephron: o 3 Layers: " (1) single-celled capillary endothelium with fenestrations (70nm gaps) – the principal barrier to blood cells " (2) basement membrane made of negatively charged glycoproteins (collagen, prteoglycans, laminin, etc) " (3) single-celled epithelial visceral layer of Bowman’s Capsule (epithelial cells are called podocytes). • Podocytes have a large number of foot-like extensions embedded in the basement membrane surrounding the glomerular capillaries. There are filtration slits between these processes containing pores (4-14nm in size). o Molecules of size 1.8-3.6nm are allowed to pass through the barrier. Cationic species are filtered better than anionic species. " Filtration of large molecules (including proteins) are inhibited (although small amounts of albumin may filter through). Segmentation of the Renal Tubule: o Proximal Tubule: Most of the reabsorption of H2O and solutes. o Loop of Henle: Concentration and dilution of the urine. o Distal Tubule and Collecting Duct: Fine-tuning of the urine concentration (site of action of most hormones) " " " The beginning of each distal tubule passes between (and contacts) the efferent and afferent arterioles of its own nephron. At this site there is a plaque of cells in the distal tubule (macular densa). At this site of contact, juxtaglomerular (granular) cells are found in the walls of the arterioles. Juxtaglomerular cells secrete renin. • Macula densa + juxtaglomerular cells = juxtaglomerular apparatus (JGA). o The JGA regulates RBF and GFR. " ~ Ten nephrons drain into one collecting duct. Multiple collecting ducts merge into one minor calyx. Innervation of the Kidneys: Rich sympathetic innervation (particularly the arteriolar smooth muscle and the tubules) but no parasympathetic innervation Glomerular Filtration: o The kidney receive 1200ml blood/minute (25% of cardiac output) and 20% of the incoming plasma is filtered at a glomerular filtration rate (GFR) of 130ml/min (180L/day). The remaining 80% of plasma entering the glomeruli flows into the efferent arterioles. " O2 consumption per unit gram of the kidney is higher than that of most other organs because of the high expenditure of energy required to reabsorb filtered Na +; 90% of the O2 is consumed for this purpose. " Filtrate is an ultrafiltrate of plasma (content is the same except no protein). • Plasma is filtered ~30x/day (plasma volume ~3.5L) • Renal Plasma Flow is 660ml/minute (plasma volume is ~ 55% of blood volume – if hematocrit ~45%). • Filtration rate is dependent on net Starling forces, which drive fluid from the lume of the glomerular capillaries into Bowman’s space. GFR = Kf [(PGC - PBS) -("GC - "BS)] ; Filtration Fraction = GFR/RPF The relatively high hydrostatic pressure at the afferent end of the glomerulus changes only slightly at the efferent end because of the low resistance to flow in the glomerular capillaries. Because of the large Kf, the filtration of water and retention of proteins lead to a significant, progressive increase of oncotic pressure (&GC) along the glomerular capillary. If &GC equals (PGC - PBS) at any point along the capillary, filtration ceases (filtration equilibration). In humans, the forces do not equalize and filtration continues at the efferent end of the glomerulus (afferent filtration is greater than efferent filtration – net driving forces are 17 mmHg and 8mmHg respectively). Reabsorption does not occur along the glomerular capillaries. Vasodilation ! ! PGC ! ! GFR Major pressure drops occur at the afferent and efferent arterioles (sites of # resistance) ! pressure in the glomerular capillary is regulated by arteriolar resistance. Note that glomerular capillary filtration is ~ 100x that of skeletal capillaries. " Tubular Reabsorption (RX) and Secretion (SX): - reabsorption refers to the movement of substances from the tubular lumen into the peritubular capillaries, and secretion is the movement in the opposite direction. 99% of water, 99.5% of sodium, 100% of glucose, and 44% of urea are reabsorbed. EX = FX + SX – RX " FX = PX·GFR EX = U X ·V EX<FX = net reabsorption of X by the tubules. EX>FX = X was secreted by the tubules. (net effect only - it is possible that a substance X is both reabsorbed and secreted) Renal Clearance (C X): - the volume of plasma cleared of X by the kidneys per unit time. o Inulin (freely filtered, and neither reabsorbed nor secreted): " All molecules of inulin filtered will be excreted in the urine (FINU = EINU) $ clearance of inulin is a measure of GFR. If CX less than CINU (GFR) that substance is reabsorbed. If CX greater than the CINU (GFR) that substance is secreted. o PAH (freely filtered, secreted, but not reabsorbed): " Via filtration and secretion, all of the PAH in the plasma delivered to the kidneys is removed and excreted into the urine in a single pass $ clearance of PAH is a measure of renal plasma flow. CINU = (UINU x V)/PINU = GFR " CPAH = (UPAH x V)/PPAH = RPF Tubular Fluid/Plasma Ratio (TF/P): compares the concentration of a substance in tubular fluid at any point along the nephron with the concentration in plasma. o TF/P =1.0 – either no reabsorption has occurred, or reabsorption has been exactly proportional to that of water. o TF/P < 1.0 – reabsorption has been greater than that of water (concentration in tubular fluid is less than that in plasma). o TF/P > 1.0 – reabsorption has been less than that of water (concentration in tubular fluid is greater than that in plasma). o TFINU/PINU – since inulin is neither absorbed nor secreted, any # [INU] in tubular fluid that is greater than that in the plasma (or filtrate) can only be due to water reabsorption proximal to the site of sampling in the tubule ! if TFINU/PINU in a site at the tubule is 4, it indicates that inulin has been concentrated 4 times above its plasma concentration due to reabsorption of 3/4 of the filtered water in the portion of tubule proximal to the sampling site. o UINU/PINU – indicates fraction of filtered water reabsorbed throughout the tubular system in the course of urine formation (UINU / PINU = 100 indicates that 99% of the filtered water has been reabsorbed). " Fraction of Filtered H2O Reabsorbed = 1 – (1/[TF/P]INU). " Passage of Urine into the Bladder and Micturition: Periodic contractions (initiated by pacemakers in the calyces and renal pelvis) propel the urine into ureters. Peristaltic waves (initiated by pacemakers in the ureters) occur 3/minute. Ureters enter the bladder posteriorly at the trigonum, forming oblique valvular orifices (prevents reflux from the bladder even though there are no distinct sphincters). Bladder has folds (rugae) that distend as it fills. Muscular shell is three layers, an outer longitudinal layer (neck!fundus) – the detrusor muscle, a thick circular middle layer, and a thinner inner one. At the base of the bladder, the middle muscular layer is modified into the internal sphincter at the orifice of the urethra. Striated muscle forms an external urethral sphincter where the urethra traverses the urogenital diaphragm. The bladder and internal sphincter have double innervation (sacral parasympathetic pelvic nerves, which synapse with postganglionic neurons in the bladder and postganglionic sympathetic fibers emerging from the hypogastric ganglia). Sympathetic stimulation relaxes the bladder and contracts the internal sphincter, while parasympathetic activity contracts the bladder and relaxes the sphincter. The sacral pudendal nerves innervate the external sphincter. Bladder has stretch receptors that send impulses in the pelvic nerves (synapse with motor innervation in the spinal cord and also with fibers running to higher centers). Sensory impulses lead to the desire to urinate when bladder filled 400ml or more. Stretch receptors reflexly excite the parasympathetic outflow, increase the tone of the bladder and increase the desire to urinate. Emptying of the bladder is initiated by relaxation of the external sphincter. Higher centers can initiate micturition when the bladder is not very full and conversely can inhibit the strong impulses to urinate when the bladder is very full. Such control does not exist in infants. Renal Hemodynamics: - The filtration rate (GFR) is effected by the rate of plasma flow (# plasma flow – faster – leads to # GFR – all of the capillary is used). The difference between !P and ' is also # at any point (# driving force). - Plasma flow 1/" ' - Plasma flow " GFR - The cumulative effect of vasoconstriction is $GFR and the cumulative effect of vasodilation is # GFR. Pressure in the glomerular capillaries is relatively high compared with systemic capillaries, whereas the pressure in the peritubular capillaries is relatively low. The primary pressure drops are at the arterioles. GFR RPF " Autoregulation and Other Factors Controlling RBF and GFR: o Myogenic Response: Arteriolar smooth muscle contracts on stretch and relaxes with # distending pressure. " Myogenic mechanisms adjust resistance to maintain constant flow rate. o Tubuloglomerular Feedback (TGF): ! or # in tubular flow consequent to !GFR is sensed by the macula densa and results in adjustment of GFR . " TGF prevents hyper-delivery of fluid to the distal nephron but it can be overridden by direct neuronal input (sympathetic) • The macula densa senses ! in amount of fluid or NaCl delivered to the distal nephron ! afferent arteriole vasoconstrictor ! #GFR. o Adenosine causes vasoconstriction by interacting with an afferent arteriolar smooth muscle cell receptor that is different from the one that leads to vasodilation in other parts of the CVS. • # NaCl load or flow ! afferent vasodilator ! !GFR. o NO ! vasodilation. o Vasoconstriction: " $ blood volume (or blood pressure) ! sympathetic nerve activation ! released norepinephrine interacts with "-1 receptors (in afferent arterioles) ! constriction ! $RBF and GFR ! # retention of H2O and Na+ ! # blood volume. " Sympathetic activation of #-1 receptors in granular cells in afferent and efferent arterioles # renin release. $ perfusion pressure and $ stretch of afferent arterioles ! # renin production ! formation of angiotensin II (AII). " Low concentrations of AII constrict the efferent arterioles predominantly, and higher concentrations also constrict afferent arterioles ! $ RBF and GFR. • Associated # aldosterone production retains Na+ and H2O (# Na+ reabsorption). " Endothelin vasoconstricts afferent and efferent arterioles (produced by endothelial and mesangial cells). o Vasodilation: " Nitric oxide dampens sympathetic and AII activity (produced by endothelial and macula densa cells). " Prostaglandins, released in response to norepinephrine or AII, prevent excessive constrictor responses to these agents. " ATP, Bradykinin, and Histamine stimulate the release of NO. " Dopamine is another vasodilator of renal vasculature. " Atrial natriuretic peptide dilates afferent arterioles, constricts efferent arterioles and thereby increases GFR. Reabsorption of Sodium, Glucose, and Water: " Sodium: 67% absorbed in proximal tubule (PT), also reabsorbed at the thick ascending limb (TAL) the distal tubule (DT), and the collecting duct (CD). All sodium transport is transcellular. Entry into the cell is always downhill and basolateral exit is always uphill (Na+-K+ - ATPase). 90% of energy used in the kidney is used for Na+ transport (basolateral). o PT: " Early: co-transport with organic solutes (glucose, amino acids, lactate, phosphate, etc) and via a Na+ - H+ exchanger. " Late: Na+-H+ exchanger is coupled with a Cl- —OH- exchanger (net NaCl reabsorption). o D-LOH: impermeable to Na+ o TAL: Na+-K+-2Cl- co-transporter and Na+-H+ exchanger o DT: " Early: Na+-Cl- cotransport (thiazide sensitive) " Late: epithelial Na+ channel (amiloride sensitive and aldosterone regulated) o CD: epithelial Na+ channel (amiloride sensitive and aldosterone regulated) " Glucose: 100% reabsorbed in the proximal tubule via energy-free coupling with Na+ transport. SGLT2 is a low-affinity/highcapacity transporter (1:1) in the early PT. SGLT1 is a high-affinity/low capacity transporter (2Na+:1glucose) in the late PT. Glucose exits basolaterally by facilitated diffusion through two Na+-independent transporters (GLUT2- early PT; GLUT1 – late PT). When Na + binds SGLT1/2, the affinity for glucose ! drastically. The kidneys do not regulate plasma glucose, they simply absorb what is available until Transport Maximum (Tm) is reached. If plasma glucose increases to high values (normal = 1g/L) - such as diabetes mellitus, filtered load exceeds capacity of the tubules to reabsorb glucose. ! transporter saturation ! unreabsorbed glucose appears in the urine. Tm for glucose = 380 mg/min. " Water: In euvolemic conditions, 67% of filtered H2O is reabsorbed in PT. Obligatory water loss = 0.4 L/day. Water permeability is conferred by the presence of water channels (aquaporins) at cell membranes. o PT: H2O is reabsorbed isosmotically by Standing Osmotic Gradients - continuous reabsorption of Na+ and Clcreates a small increase in the osmolality of the intercellular spaces that drives the reabsorption of water across the highly water permeable cell membrane and tight junctions. o D-LOH: D-LOH permeable to water (23%). Driving force for water reabsorption at this segment is the high osmolality of the medullary interstitium (near 1200mOsm/L). o TAL: impermeable to water o Early DT: impermeable to water o Late DT and CD: Impermeable to H2O in the absence of antidiuretic hormone (ADH) # ADH # water permeability of these segments ! reabsorption ! excretion of small volume of concentrated urine. Driving force for reabsorption of water in DT is ! in osmolality between the tubular and interstitial fluids. With low or zero plasma ADH, permeability to water of late- DT and CD, and there is no reabsorption of H2O ! excretion of a large volume of dilute urine. o Aquaporins: AQP1 is present in PT and D-LOH. ADH induces the insertion of AQP2 into late-DT and CD. AQP3 and AQP4 are present at the basolateral membrane of the water permeable tubular segments. Glomerulo-Tubular Balance: # filtration of Na+ and water are compensated by # reabsorption to avoid severe Na+ and water depletion. When sodium balance is normal, Na+ and water reabsorption increase in parallel with an increase in GFR and the increased load of Na+ (a constant fraction of the filtered water and salt (about 2/3) is reabsorbed at the PT). Two mechanisms. (1) Starling forces at the peritubular capillaries !GFR at constant RBF ! ! concentration of proteins and oncotic pressure in the peritubular capillaries --! reabsorption of water and Na+. (2) Following ! GFR, ! filtered load of glucose, amino acids and other organic solutes that are cotransported with Na+ at the PT ! ! Na+ and water reabsorption $ GT-balance ensure a constant fraction of reabsorption of Na+ and water at the PT (minimizing ! in body salt and water that may result from ! GFR). Secretion: Many organic acids or bases - end products of metabolism or exogenous (drugs) - are avidly secreted by the tubules (in addition to H+ and K+ - see later). Most of secreted anions and cations share a common carrier located at the basolateral and lumenal membranes of the tubular cells. Substrate competition can lead to $ rate of excretion (prolong the biological half-life of a drug). Most of the secretion of anions and cations takes place at the late proximal tubule. Sodium Regulation: The amount of Na+ in the body determines the volume of the ECF. Volume of the ECF determines perfusion pressure of the CVS $ [Na+] is critical for proper perfusion of the body. " " Distribution and Balance: o 40% of total body Na+ is in bone (but fixed ! not available for ordinary metabolic processes) o 90% of the available Na+ is in the ECF. o Typical sodium intake ~100 mEq/day (varies from <1mEq/day - 400 mEq/day). Mechanisms that regulate intake are unclear. " 1 Eq Na+ = 23 g Na+ = 58.4 g NaCl). o Kidneys are the major excretory paths for Na+ " stool and sweat losses are negligible (< 10 mEq/day). o Renal excretion of Na+ is adjusted to match the amount ingested in the diet. " [Sodium]: ! 140mEq [Na+] = +/- 1kg body weight (1L water) o Na+ intake ! ! plasma [Na+]! ! ! Osmolality ! " (1) H2O moves from ICF to ECF ! ECF! " (2) !Thirst ! ! H2O intake ! ECF! " (3) ADH secretion ! ! H2O retention ! ECF! In steady state, Na+ ingestion and excretion are equal (balance), but during adjustment periods (several days following ! or # intake) a person can be either in positive balance (ingestion exceeds excretion) or in negative balance (excretion exceeds ingestion), and there are resulting changes in total body Na+, ECF volume and body weight. Urinary Na+ excretion (dotted line) lags behind the step changes in intake and parallels the change in ECF volume reflected in the body weight. The grey area between the intake and excretion curves represent the amount of Na+ gained (about 140mEq) after increasing the intake. This results in the gain of near 1 L of water (1 kg of body weight) o o Receptors: - volume or stretch receptor rather than direct monitor of Na+ content. " Carotid Sinus and Aortic Arch: respond to # blood pressure (secondary to expansion of effective blood volume or effective circulating volume (ECV) by reducing sympathetic nervous system activity). $BP ! $ stretch ! # sympathetic signaling. o ECV is not a measurable volume, it reflects the degree of fullness of the circulatory system (pressure perceived by baroreceptors). o Under normal conditions, ECV varies directly with ECF, but in some pathological situations (CHF, cirrhosis) ECF is expanded while ECV is depleted – i.e. excessive tissue fluid retention. " Cardiac Atria: respond to # stretch by releasing atrial natriuretic peptide (ANP) o Secretion of ANP is reduced if atrial filling is decreased by reductions in effective circulating volume. Cardiopulmonary receptors also alter sympathetic efferent activity; # with $ stretch and $ with # stretch. " Afferent Arterioles: volume/stretch receptors. o $ stretch ! renin secretion (by juxtaglomerular apparatus) o # transmural pressure (stretch) ! $ renin secretion. o Renin ! AII (a potent vasoconstrictor that stimulates secretion of aldosterone. Effector Mechanisms: - sodium excretion by the kidneys is accomplished by ! in filtered Na+ load and tubular Na+ reabsorption. " Na+ filtered: 140 mEq/L x 180 L/day (GFR) = 25,200 mEq/day o total ECF Na+ is filtered 10+ times/day. " 99+% of filtered Na+ is reabsorbed. o large ! in excretion can result from small % ! in filtered load or reabsorption " PT and TAL have large capacity to reabsorb Na+ (can be adjusted in response to Na+ load/delivery) " DT and CD have relatively limited capacity to reabsorb Na+ (5-10 % of filtered Na+) but “fine-tune” reabsorption so that the final urinary Na+ matches intake. " Regulation of Filtered Sodium: Filtered Na+ is a function of GFR (plasma [Na+] varies little). Hemodynamic mechanisms (RBF, glomerular capillary pressure) control GFR. ! body Na+ $ECF and plasma volume ! !GFR. ! Na+/ECF ! ! GFR and # Na+/ECF volume ! # GFR and $ filtered load of Na+ #sympathetic activity (due to $ Na+/ECF volume) ! vasoconstriction ! $GFR. In low concentrations, Angiotensin II principally constricts the efferent arteriole, while higher concentrations also constrict the afferent arteriole. Efferent constriction by AII tends to maintain GFR in the face of low renal perfusion pressures, while afferent constriction tends to reduce it when the fall of blood pressure is more pronounced. ANP dilates the afferent arteriole and constricts the efferent arteriole ! # GFR. Nitric oxide (produced locally by vascular endothelium) ! dilates renal vasculature. " Regulation of PT Sodium Reabsorption: - # Na+ content and ECF volume $ fraction of filtered Na+ reabsorbed proximally (normally 67%). The fraction of Na+ reabsorbed by PT can ! from 50% (ECF expansion) to 80% (ECF contraction) due to the following: o Sympathetic Nerves: Innervate the PT. # sympathetic stimulation (response to ECF volume $) stimulates PT Na+ reabsorption. o Angiotensin II: stimulates PT Na+ reabsorption (partially mediated by stimulation of the Na+-H+ exchanger) " Secreted in response to renin secretion (stimulated by # Parteriolar and ! sympathetic activity) + o Starling Forces: When Na and H2O are retained in ECF/plasma, plasma protein concentration ! plasma oncotic pressure in peritubular capillaries $ ! reduced uptake of reabsorbed Na+ and water ! slows Na+ reabsorption by the tubule. o Nitric Oxide: inhibits Na+ reabsorption at several tubular sites (including the PT) - produced by tubular cells. Sodium Reabsorption in the Loop of Henle (load dependent): D-LOH is impermeable to Na+. Na+ movement in TAL is relatively small and is important in urinary concentration and dilution. TAL responds to ! or # in delivery of tubular fluid Na+ by immediate changes in Na+ reabsorption in the corresponding direction (load dependent). o ADH: Stimulates Na+ reabsorption by TAL. Secretion of ADH stimulated by small # of plasma osmolality and by large ($10%) reductions in ECF volume. o Prostanoids: released locally by tubular cells and by medullary interstitial cells inhibit Na+ reabsorption by the TAL. o Nitric oxide (NO): inhibits Na+ reabsorption by # medullary blood flow. Prostanoids and NO are released when ECF/Na+ volume #. Sodium Reabsorption by DT and CD (Renin-Angiotensin-Aldosterone System): Na+ reabsorption in is influenced by a number of factors that fine-tune reabsorption. Under euvolemic conditions, with small changes in Na+ intake, aldosterone is the primary regulator of Na+ reabsorption by its effects on distal segments of the nephron, where reabsorption of Na+ is " " adjusted to match excretion to dietary intake. 3% of Na+ (750mEq) is absorbed in DT/CD (intake range is 1-400mEq unless a significant ! in ECF occurs). o Renin-Angiotensin-Aldosterone System: " Rate limiting factor is renin (secreted by JGA in response to $ perfusion pressure and $ stretch of afferent arterioles as well as sympathetic activation of #-1 receptors in granular cells in afferent and efferent arterioles). o Angiotensin II: o stimulates aldosterone secretion o arteriolar vasoconstrictor o stimulates Na+ reabsorption by PT o stimulates of ADH secretion and thirst o Aldosterone: acts on principal cells of DT/CD ! o # recruitment of apical Na+ channels and basolateral Na+-K+-ATPase pumps (30-60 minutes latency) o # synthesis of apical Na+ channels and basolateral Na+-K+-ATPase pumps (~4-6 hours latency) " Aldosterone secretion is also stimulated by a rise in plasma potassium concentration, and it enhances the secretion of K+ by the distal segments of the nephron. Renin Secretion from juxtaglomerular cells # due to: (1) # sympathetic activity (due to $ ECF/Na+) (2) $ perfusion pressure (afferent arterioles behave is high-pressure baroreceptors) (3)$ NaCl delivery to the macula densa $ sympathetic nerve activity, # renal perfusion pressure, or # NaCl delivery to the macula densa ! inhibition of renin secretion " Natriuretic and Local Factors: o ANP: Produced and stored by atrial myocytes ! released upon atrial stretch (! ECF) ! vasodilation and ! NaCl and water excretion by the kidneys. " # GFR and Na+. " inhibits renin secretion by juxtaglomerular cells. " inhibits aldosterone secretion. " inhibits (directly) Na+ reabsorption by CD. " inhibits ADH secretion. o Urodilatin: secreted by DT and CD (acts locally inhibiting Na+ reabsorption by these segments). o NO, Prostanoids, and Kinins: produced in kidneys and inhibit Na+ reabsorption. Day to day balance of Na+ ($ ECF volume) is primarily due to aldosterone action upon the distal tubules. When significant ! occurs, other factors and locations become involved. Acid-Base Regulation: A large amount of carbonic acid formed from the ~20,000 mmoles of CO2 created during normal metabolism (as H2CO3 is in equilibrium with CO2, it is referred to as volatile acid). Respiratory excretion of CO2 (and thus H2CO3) allows elimination of this load $ under normal conditions metabolic CO2 does not contribute to acid load of the body. Normal metabolism of meat diets produces ~60 mmoles of non-volatile (fixed) acids daily. Production of fixed acid must be compensated by kidneys. • Protection from [H+] Imbalance: o Prompt: acute respiratory regulation of CO2 (prevent H+ incompatible with life in minutes) – cannot eliminate nonvolatile (fixed) acids or restore buffer supplies. o Homeostasis: renal regulation of H+ excretion and HCO3- reabsorption (days) • Renal Regulation of Acid-Base Balance: o Preservation of acid-base balance requires the reabsorption of about 4300 mEq of filtered HCO3- each day [(24 mEq/L) x (l80 L/day)]. o The kidneys must also create ~60 mEq of HCO3- each day (replace HCO3- destroyed by reaction with fixed acids to form CO2 and H2O). " Both of these functions require secretion of H+ from the body into the urine and reabsorption of HCO3-. • H+ secretion: o (1) Ion Exchange: Na+-H+ and Na+-NH4+ (PT) depend on function of a basolateral Na+-K+ ATPase to maintain low intracellular [Na+] creating a gradient for passive apical Na+ entry (in exchange for H+ or NH4+). " Little ! in luminal pH (buffered by HCO3-) o (2) Primary H+ Transport: Apical H+ pump (DT and CD). This lowers luminal pH beyond levels that can be reached in the PT (no HCO3- buffer). • HCO3 Reabsorption: (most H+ secreted is used for HCO3- reabsorption) and every H+ secreted is associated with transport of an HCO3- into the cell. o H+ that is transported into the lumen is formed in the tubular cells along with HCO3(which is transported into blood basolaterally in a Na+ cotransporter – 3HCO3- : 1Na+). Filtered HCO3- in the lumen combines with secreted protons ! CO2 + H2O. The CO2 diffuses into tubular cells and is combined with H2O to form HCO3- + H2O ! Luminal HCO3- is salvaged. " For every H+ that moves into the lumen, a HCO3- moves into the ICF ! for every HCO3- filtered, an HCO3- is reclaimed with out ! luminal pH (this primarily occurs in the PT). This mechanism prevents loss of bicarbonate. It also permits release (excretion) of excess H+. • Even with plasma HCO3- levels as high as ~28 mM, all filtered HCO3is reabsorbed. (bicarbonate threshold) – this threshold may vary: the rate of reabsorption of HCO3- is directly related to the level of the pCO2. Alterations of K+ and Na+ balance and aldosterone production are also important determinants of the HCO3- threshold. • Replenishment of Depleted HCO3- Stores (accounting for fixed acid): - Bicarbonate buffering of fixed acid depletes the ECF HCO3- levels. Kidneys restore the [HCO3-]ECF by excreting H+ o The minimum urine pH is 4.4 (0.04mEq/L) = 1500L urine/day. This is impossible ! urinary buffers are needed " Buffers: • (l) Phosphate: present in the luminal fluid (immediate but inflexible – limited by HPO42- present – cannot account for acidemia) o At normal pH of glomerular filtrate, HPO42-/H2PO4- = 4. At maximal acidification of tubular fluid, HPO42-/H2PO4- = 0.01 " ! phosphate is the primary titratable acid of the urine. " Titratable acid accounts for 33% (20mEq) of acid excretion in the urine (! in acidosis is minimal). !pH occurs as tubular fluid moves from Bowman’s capsule (pH=7.4) ! late CD (pH=4.4). • (2) Ammonia: derived from the metabolism of glutamine and other amino acids (predominantly in PT cells) o In tubular cells of PT, NH3 forms from glutamine (and other amino acids). NH3 is protonated ! NH4+ enters the lumen of PT (Na+-NH4+ exchange). In TAL, NH4+ is reabsorbed, (substitutes for K+ on Na+K+2Cl- co-transporter). NH4+ in equilibrium with a very small concentration of NH3, accumulates in the medullary interstitium. In CD, lipid- " " " soluble uncharged NH3 diffuses down its concentration gradient across the cell and the lumenal membrane into the tubular fluid. It reacts with H+ ! NH4+, $ [free H +] in urine. Charged NH4+ ions poorly permeate renal tubule cells ! ammonium trapping at # concentration in the urine. o at pH less than 9.3, NH4+ excretion exceeds that of NH3 Accounts for about 67% (40mEq) of acid excretion in the urine. NH4+ does not contribute significantly to T.A. (titrating acidified urine back up to pH 7.4 converts little NH4+ to NH3. At this pH, over 98% of NH3 + NH4+ exists as NH4+. Highly flexible to acidemia (enzymatic adaptation ! # NH3 synthesis ! #NH4+ excretion) • In steady state, the rate of fixed acid production equals the rate of urinary acid excretion. Urinary excretion of total ammonia is influenced by both the urine pH and the systemic state of acid-base balance. In alkalotic states the kidneys can excrete HCO3 and achieve urine pHs up to about 8. • Cells of the DT/CD: o Principal (granular) Cells: carry out Na+ reabsorption - no direct role in regulation of acid-base status. o Intercalated (carbonic-anhydrase rich) Cells: regulate acid-base status " Type " Intercalated Cells: Secrete H+ into the lumen (contain lumenal proton pumps and basolateral Cl-HCO3- exchangers) • Up-regulation of " cells during acidosis ! acidification of urine. " Type % Intercalated cells: Secrete HCO3- (contain luminal Cl- - HCO3- and basolateral proton pumps) • Up-regulation of # cells during alkalosis ! alkalinization of urine. • Acid-Base Disorders: o Metabolic Acidosis (pulmonary response): )[HCO3-] is associated with )pCO2. o Metabolic Alkalosis (pulmonary response): compensatory (pCO2 in association with ([HCO3-]. o Respiratory Acidosis: (pCO2, and ([HCO3-] o Respiratory Alkalosis: )pCO2 and )[HCO3-] o Mixed metabolic and respiratory acidosis (cardiopulmonary arrest) ! tissues are inadequately perfused and resort to anaerobic metabolism (production of lactic acid) ! cessation of respiration and CO2 retention. o Mixed metabolic acidosis and respiratory alkalosis (overdose of aspirin) - pH is normal, but [HCO3 ] and pCO2 are abnormal. Metabolic acidosis is the result of the uncoupling of oxidative phosphorylation by salicylic acid (active ingredient of aspirin) and an accumulation of lactic acid and ketoacids; respiratory alkalosis is the result of hyperventilation secondary to salicylic acid directly stimulating the respiratory centers. TABLE I Changes in plasma [H+], [HCO3-] and pCO2 in acid-base disorders Primary disorder [H+] [HCO3-] pCO2 Cause of HCO3change Resp. acidosis (pCO2 + renal compensation impaired lung function Resp. alkalosis )pCO2 + renal compensation increased ventilation Metab. acidosis HCO3- loss, H+ gain reflex respiratory compensation Metab. alkalosis HCO3- gain, H+ loss reflex respiratory compensation Potassium Balance: • Cause of CO2 change Distribution of Potassium in the Body: variations in [K+] ! arrythmias and paralysis • • Most abundant cation in the body (50 mEq/Kg body weight), 98% is located inside cells (concentration ~150 mEq/L). Most intracellular K+ is in muscle cells (>2600 mEq), with ~300 mEq each found in RBC’s, liver and bones. ~65 mEq (2%) of total body K+ is in the extracellular fluid (ECF). Shifts of K+ from ICF "!ECF can cause large ! [K+]plasma $ [K+]plasma is not a good indicator of total body K+. Normal range of [K+]plasma ~3.5-5 mEq/L. Greater than 5 mEq/L = Hyperkalemia. Less than 3.5 mEq/L = Hypokalemia. • • Functions of Potassium: o Cofactor for enzymes of intermediary metabolism (particularly protein and glycogen synthesis) o Regulator of muscle blood flow during exercise. o Sets cell membrane resting potential. " ! extracellular [K+] can cause severe disturbances in excitation of nerve cells and muscle (cardiac arrhythmias, paralysis). Extra-Renal Potassium Homeostasis (fast response): Excretion of K+ by kidneys after ingestion is slow (hours) $ uptake of ingested K+ is essential to prevent life-threatening hyperkalemia in response to acute K+ load. o ! [K+]Plasma = KIngested / ECF volume = 30mEq(one meal)/15L = 2mEq/L ! ingested K+ must be quickly taken up into cells (under hormonal control) so as to not create hyperkalemic states. - Daily intake of K+ is ~100 mEq (of which 5-10 mEq are lost in the feces and less in the sweat) ! 90 mEq of K+ are absorbed daily by the GI tract. - Cells (primarily skeletal muscle and liver) take up this K+ by activation of Na+-K+ ATPase (30 minutes post-ingestion, 50% has already been moved intracellularly. 1 hour post-ingestion, 97% is intracellular). -Uptake is stimulated by epinephrine, insulin, and maybe aldosterone • Hormones Mediating K+ Cell Uptake: o Epinephrine: When treated with # adrenergic blockers, # [K+]Plasma after K+ intake is greater and more prolonged than in normal subjects (epinephrine and other catecholamines enhance cellular K+ uptake by binding to #2 receptors). Administration of epinephrine or #2 adrenergic agonists reduces [K+]Plasma $ Epinephrine ! ! K+ uptake by cells. o Insulin: (secreted by the pancreas) Insulin secretion is normally # after a meal (because of # plasma glucose levels after ingestion of saccharides) ! insulin is the most important hormone promoting K+ uptake by cells after a meal. In insulin-deficient diabetic patients, # [K +]Plasma after a K+-rich meal is greater than that in normal persons. Stimulation of K+ uptake by insulin is useful in the treatment of hyperkalemia (administering glucose to release endogenous insulin or insulin (together with glucose to prevent hypoglycemia) quickly results in $[K+]Plasma. " Epi and insulin stimulate the Na+-K+ ATPase pump to ! pump turnover rate (via receptormediated activation of cytosolic kinases - PKA, PKC and/or ERK1/2 MAP kinase). o Aldosterone: Role is unknown. Small elevations in plasma K+ (as little as 0.2-0.3 mEq/L) cause # aldosterone secretion ! enhanced colonic and renal K+ excretion. " Patients with aldosterone deficiency (Addison’s disease) are prone to developing hyperkalemia. Uncontrolled secretion of aldosterone (adrenal tumor) ! hypokalemia. • Non-Hormonal Factors Mediating K+ Cell Uptake/Release: not regulatory mechanisms $ do not contribute to K+ homeostasis o Acid-Base Balance: " Acidemia: important fraction of #[H+] is buffered within the cells. As H+ moves into the cells, K+ moves out ! #[K+]Plasma. Drop of 0.1 pH unit ! ~0.6 mEq/L# [K+]Plasma. • Mechanisms include release of protein-bound cellular K+ with a drop in pH and inhibition of the pH sensitive Na+-K + ATPase. " Alkalemia: as H+ moves out of the cells and buffers the alkali ECF, K+ moves into cells, ! hypokalemia (decrease in [K+]Plasma is less prominent than the increase seen during acidemia for the same absolute pH change). o Exercise: #[K+]Plasma (K+ is released from muscles during exercise ! # blood flow to the exercising muscle). " The ! [K+]Plasma is reversed after several minutes at rest. " In individuals taking #-adrenergic blockers, [K+]Plasma can rise by a life-threatening 2-4 mEq/L during exercise. o Osmolality (! osmolality ! hyperkalemia; # osmolality ! hypokalemia): " # osmolality of ECF ! K+ moves out of cells. The result of cell shrinkage (#[K+]ICF ! # chemical gradient for K+ efflux. $ osmolality of ECF ! K+ moves into cells. The result of cell swelling ($[K+]ICF ! # chemical gradient for K+ influx. Cell Lysis: Burns, rhabdomyolysis, etc ! K+ leakage ! hyperkalemia. " o • Intra-Renal Potassium Homeostasis (slow response to [K+]Plasma and aldosterone): to remain in K+ balance an individual must excrete an average of 90 mEq of K+ daily. This renal excretion is regulated by plasma K+ and aldosterone. Potassium is freely filtered at the glomerulus (rate of 600-900 mEq/day). • Tubular Reabsorption and Secretion: • Proximal tubule through TAL – reabsorption only. • Distal tubule and collecting ducts (IMCD = reabs. CCD = secretion) – reabsorption and secretion. • Rate of excretion of K+ largely reflects the handling of K+ by the distal nephron. o PT (80%): Reabsorption is intracellular (tight junctions) secondarily to H2O and consequent elevation of tubular [K+]. The transcellular pathway is not involved in K+ reabsorption. " o o o K+ movement from the lumen into the cell is against its electrochemical gradient or uphill, since the negative cellular electrical potential is not enough to overcome the opposing concentration gradient driving K+ out of the cell D-LOH: No K + reabsorption or secretion. TAL (10%): Na+-K+-2Cl- cotransporter (inhibited by loop diuretics) at the lumenal cell membrane. K+-Cl- cotransport and K+ channels at the basolateral membrane. Some of the K+ entering the cell leaks back into the lumen through apical channels. DT and CD: " Principal (light) Cell: Responsible for K+ secretion (and Na+ reabsorption) • Secretion takes place mostly at the late DT and cortical CD o Secretion involves the K+ uptake by the Na+-K+ ATPase across the basolateral membrane of the principal cell and diffusion of K+ through lumenal channels from the cell into the tubular fluid. Although K+ channels are also present in the basolateral membrane, the overall conductance of the apical channels is larger, and K+ preferentially diffuses across the apical membrane into the tubular fluid. " " - Intercalated Cell: Mediates K+ reabsorption. • Reabsorption occurs at the medullary CD (MCD). o Secrete H+ in exchange for K+ (lumenal H+-K + ATPase). Once inside the cell, K+ exits by diffusion across basolateral channels. If lumenal [K+] is high at the MCD, reabsorption may also take place via the highly K+permeable paracellular pathway. With normal K+ intake, secretion of K+ prevails at the DT and rate of K+ excretion amounts to 10-15% of filtration rate. During periods of high K+ intake, secretion of K+ by DT ! , and rate of excretion may exceed the rate of filtration. With K+ depletion, DT reabsorbs K+, and excretion may be reduced to 1% of the filtered load (kidneys cannot reduce K+ excretion to the same low level that they can for Na+ - 0.2% of the filtered load - $ hypokalemia can develop in individuals placed on a K+-deficient diet. • Regulation of Potassium Secretion: Three factors control rate of K+ secretion: (1) activity of the Na+-K+ ATPase, (2) electrochemical gradient for K+ movement across the apical membrane (3) permeability of the apical membrane to K+. • Plasma [K+] (independent of aldo): o Hyperkalemia (stimulates K+ secretion): " stimulates K+ secretion by stimulating Na+-K+ ATPase !# [K+]ICF ! # driving force for K+ efflux across the lumenal membrane. " #permeability of lumenal membrane " Induces aldosterone secretion. Hypokalemia (inhibits K+ secretion): " inhibits Na+-K+ ATPase !$ [K +]ICF ! $ driving force for K+ efflux across the lumenal membrane. " $ permeability of lumenal membrane " Inhibits aldosterone secretion Aldosterone (#aldo ! hypokalemia): secretion is stimulated by plasma elevation of Angiotensin II and [K+]. Aldosterone secretion is inhibited by hypokalemia and atrial natriuretic peptide (ANP). o Primary effect is to # recruitment (hours) and synthesis (days) of transport proteins (Na+-K+ ATPases, Na+ channels, K+ channels) in principal cells of the DT/CD !# Na+ reabsorption and # K+ secretion. o # Na+ entry also depolarizes the lumenal membrane, favoring K+ secretion from cell to lumen. Rate of Tubular Fluid Flow (Flow " K+ Secretion - # flow ! # secretion): Distal flow functions as an autoregulatory mechanism that protects K+ balance from the physiological responses to alterations in ECF volume and water balance. o # flow of tubular fluid (diuretic treatment or expansion of ECF) ! # K + secretion within minutes. $ tubular flow (ECF contraction produced by hemorrhage, severe vomiting or diarrhea) $ K+ secretion by the distal nephron + o As the tubular flow #, it rapidly washes away the secreted K and maintains a favorable chemical gradient for the secretion of this ion across the lumenal membrane. " # flow ! # delivery of Na+ ! more Na+ reabsorption ! # secretion of K+ (entry of Na+ stimulates the basolateral Na+-K + ATPase and depolarizes the lumenal membrane) o Diuretics: # rate of tubular flow through the distal nephron ! enhance the rate of K+ secretion (an unwanted side effect that requires the administration of K+ to maintain K+ balance). K+-sparing diuretics (spironolactone, triamterene, amiloride) avoid this unwanted effect. " Spironolactone is a competitive inhibitor of aldosterone whereas the other two inhibit the lumenal Na+ channel at the distal nephron (inhibiting K+ secretion). o Aldosterone and Flow: Aldosterone (participates in the regulation of the ECF and plasma [K+]), does not disturb K+ balance when secreted to correct ECF volume. When ECF volume $, the reninangiotensin-aldosterone system ! aldosterone secretion !# distal reabsorption of Na+ and secretion of K+ (should ! hypokalemia) but, $ ECF volume ! # AII ! proximal tubular Na+ reabsorption ! # water reabsorption in the PT ! $ distal flow ! tubular [K+] is higher and the gradient for K+ secretion is less favorable. (If aldosterone is chronically administered it will stimulate K+ secretion and cause hypokalemia). o ADH and Flow: ADH # water reabsorption (late DT/CD) ! $ distal flow. This does NOT ! $ K+ excretion because ADH directly stimulates Na+ reabsorption and K+ secretion by the principal cells (counteracts negative effect of a reduced distal flow on K+ secretion) o • • Diuretics: # solute excretion ! # Cosm above normal and $ CH20 by raising the osmolarity of the tubular fluid flowing out of the TAL. Diuretics also # K + secretion (# rate of tubular flow). • Acid-Base Status: o Acidosis (# K+ secretion): inhibition of DT basolateral Na+-K+ ATPase and lumenal K+ permeability. " In chronic (days) metabolic acidosis, acute inhibition of K+ secretion is reversed. • Inhibition of Na+-K+ ATPase at the PT cells by $ pH depresses NaCl and water reabsorption ! # tubular flow (greater Na+ delivery) to the distal nephron. • Acidosis ! #ECF volume ! renin secretion ! aldosterone secretion ! offset initial $ K + secretion. (Rate of K+ excretion in chronic acidosis is highly variable - usually it is higher than normal). o Alkalosis (! K+ secretion): upregulating Na+-K+ ATPase and # lumenal K+ permeability. Osmoregulation and Volume Regulation: o Introduction: Volume and concentration of body fluids are controlled by: o (1) Thirst - Obligatory renal and non-renal water losses would lead to negative water balance if not for thirst. " Very sensitivy to slight ! osmolality – hyperosmolarity ! stimulation of the hypothalamic thirst center ! water ingestion (returning osmolality to normal). o (2) Secretion of ADH - modulates urine volume and concentration " Very sensitivy to slight ! osmolality – hyperosmolality promptly ! secretion of ADH ! $ urine volume (and # urine concentration). • without ADH – [urine] = 50mOsm/kg • with ADH – [urine] = 1200mOsm/kg o Establishment of Osmotic Stratification (Countercurrent Multiplication of the Single Effect): o PT: 2/3 of H2O reabsorbed isosmotically (PT reabs. is isosmotic at all levels of plasma osmolality). " PT reabsorption of H2O influences urine concentration by influencing the rate of fluid delivery to the distal nephron. o D-LOH and TAL: Osmotic stratification is largely created in ascending limb of the loop of Henle. " D-LOH: H2O reabsorbed without Na+ " TAL: Na+ reabsorbed without H2O. Transport mechanisms at the TAL that result in the reabsorption of NaCl. These mechanisms can maintain an osmotic difference of 200 mOsm between the lumen of the TAL and the interstitium (‘single effect’). o o Osmotic equilibration between the interstitium and the D-LOH results in an osmolality difference (osmolality of the ISF and that in the D-LOH exceeding that of the fluid in the TAL by ~200 mOsm/kg at every level. The creation of a 200 mOsm/kg gradient between the lumen of the ascending limb and that of the adjacent descending limb at each level along its length is termed the single effect and leads to countercurrent multiplication. Multiplication depends on countercurrent flow whereby tubular fluid moving down the descending limb passes fluid moving upward in the adjacent, parallel ascending limb. o Due to osmolality of the interstitum, there is a progressive increase in osmolality as tubular fluid flows downward toward the hairpin loop and progressive decrease in osmolality as it flows upward, after the turn, through the ascending limb. " Osmolality in the lumen of the cortical portion of TAL ~100 mOsm/kg. Osmolality of the interstitium at the same level is 300 mOsm/kg. Interstitial osmolality progressively rises to ~l200 mOsm/kg from the cortical to the papillary region. Establishment of these gradients permits either dilution or concentration of the urine, depending on the level of plasma ADH. • Length of the D-LOH " maximal urine concentration (some animals – not humans – can adjust loop length to assist osmotic stratification). o Role of Urea: In the inner medulla, urea contributes to osmolality (it is ~50% of solute there). D-LOH is not permeable to urea. TAL is permeable to urea. " Antidiuresis (high plasma ADH): ~50% of interstitial osmolality due to urea (~50% due to NaCl). " Diuresis (low plasma ADH $ low urea reabsorption from the IMCD): significant fraction of filtered urea is excreted ! contribution to the interstitial osmolality is less important • Urea Transport: markedly ! by ADH; # protein intake ! #urea formation !#urine concentration; urea infusion ! prompt ! concentrating ability. As the fluid moves through the D-LOH, H2O moves into the hyperosmotic interstitium. Solute concentration in the tubular lumen (mainly salt) ! until its osmolality balances that of the interstitium (due to salt ions and urea) ! lumenal fluid with the same osmolality (but higher salt concentration) as the medullary interstitium. When fluid in the D-LOH turns at the tip of the loop and moves into the ascending limb (permeable to salt), salt moves into the inner medulla (down its concentration gradient) ! salt added to the innermost regions of the medulla (maintains high osmolarity). -Distal tubules and collecting ducts are impermeable to water. -Lumenal fluid flowing from LOH is diluted (120 mOsm/kg), water is unable to cross the tubules, and continuing salt reabsorption in the DT and CD reduces lumenal fluid osmolality even further. -The osmolality of the voided urine may be as low as 50 mOsm/kg with volume as high as 18L (10% of GFR) -Higher medullary and papillary interstitial osmolality (late DT and CD are highly permeable to H2O due to the presence of ADH) and osmotic equilibration occurs between the fluid in the CD and the adjacent increasingly hyperosmotic interstitium. -The osmolality of the voided urine may be as high as 1200 mOsm/kg with volume as low as 0.4L (obligatory water loss) -Plasma Osmolality " [ADH] Plasma -Thirst " [ADH]Plasma -ECF 1/" [ADH]Plasma - but not very sensitive (at least 10% of ECF volume must be depleted before ADH# highly potent once activated. -Mechanism of action of ADH on water permeability: ADH binds a basolateral receptor ! adenylate cyclase (guanine nucleotide binding protein) ! cAMP ! phosphorylation of lumenal membrane proteins ! incorporation of aquaporins (AQP2) into CD lumenal membranes ADH: (1) stimulates TAL Na+ transport (#interstitial osmolality ! H2O reabsorption from adjacent CD), (2) depresses vasa recta blood flow (reduces washout of interstitial solute protecting osmotic stratification) and (3)enhances IMCD urea transport (urea contributes to interstitial osmolality). o Maintenance of Osmotic Stratification (Passive Countercurrent Exchange – Vasa Recta): o Blood flow through the medulla dissipates the interstitial osmolality gradients. Counteracted by passive countercurrent exchange of solutes and water between the closely apposed descending and ascending vasa recta (high permeability to water and small solutes). Plasma in the descending vasa recta gets concentrated as it flows into the medulla and diluted in the ascending vasa recta as solutes leave and water enters the vasa recta (minimizes, but does not abolish, solute washout). Descending Limb: solutes are concentrated (! permeability to H2O in medulla) Ascending Limb: solutes leave vasa recta (! ISF) and H2O enters (solutes are diluted). Osmolality in A-VR always exceeds that in D-VR at the same level NaCl and urea diffuse out of the ascending limb of the vessel and into the descending limb, while H2O diffuses out of the descending and into the ascending limb of the VR. o o o o Plasma solute concentration at the end of the VR is greater than at the beginning Regulation of ADH Secretion: o Inhibitory: " Plasma osmolality $, the osmoreceptors' rate of firing $ ! ADH secretion $. • ! dilution of body fluids ($osmolality) " Inhibitory inputs from baroreceptors and cardiopulmonary receptors (normal state). " #ANP o o Excitatory: " Osmoreceptors are exquisitely sensitive to the plasma osmolality, # firing when osmolality # ! #ADH. " Under extreme depletion of both salt and water (or disease states in which volume receptors and baroreceptors are under-stimulated) ADH secretion # ! renal water reabsorption. (requires a volume depletion of more than 10%) Under this condition, ADH secretion is stimulated to such an extent that it overrides the osmoreceptor regulation and can lead to a lowering of osmolality well below normal. " #Angiotensin II Free Water Clearance: amount of water excreted in excess of that which must be excreted in order for urine to be isoosmotic with respect to plasma and ECF. CH2O = V- C OSM Hypoosmotic Urine (dilute): Water is removed from the urine to make it isoosmotic with plasma ! UOSM is smaller than POSM, COSM is smaller thanV, and CH2O is positive Isoosmotic Urine: UOSM = POSM, COSM = V and CH2O is zero COSM = (UOSM · V) / POSM Hyperosmotic Urine (concentrated): Water is added to make the urine isosmotic ! UOSM greater than POSM, COSM greater than V, and CH2O is negative. Hemorrhage: hemorrhagic shock is defined as the inability to properly perfuse the body. It can be external (penetrating trauma) or internal (e.g. bleeding peptic ulcer). It may be slow and mild, or rapid and severe. o Symptoms: o Skin: pale, cold, and sweaty o Pulse: tachycardic (rapid) and weak (thready) o BP: mean arterial pressure is normal or low, and pulse pressure is always low o Respirations: rapid and shallow o Kidneys: $urine output (oliguric) o Causes of Shock: o Hypovolemic shock: caused by $ blood or plasma volume due to loss of fluid through hemorrhage, diarrhea or vomiting. o Cardiogenic shock: results from an acute organic impairment of cardiac function such as myocardial infarction o Vasogenic (neurogenic) shock: vasodilation leading to faint and syncope. " Septic shock: caused by bacterial infections which release toxins that impair CVS " Anaphylactic shock: immunologically-triggered allergic reaction to antigens. o Immediate Short-Term Reflexes to Hemorrhage ($blood volume): o ) Blood Volume % ) Venous Pressure % ) Venous Return (PRA)% ) Atrial Pressure % ) Ventricular End Diastolic Volume % ) Stroke Volume % ) Arterial Pressure % ) Flow (Cardiac Output). " $PRA ! $cardiopulmonary pressure receptor firing ! #sympathetic activity ! $vagal activity. • (1) ! arteriolar vasoconstriction (skin, muscle, splanchnic, renal, etc) ! #TPR ! $run-off ! maintained MAP ! # ejection fraction • (2) ! #HR and #contractility ! #venomotor tone ! autotransfusion ! $ venous compliance ! $ESV and drives venous blood into arterioles. o Cardiac output begins to fall when blood loss exceeds 10%. MAP is maintained until loss of greater than 20%. There is an immediate and progressive drop in pulse pressure. No ! MAP in 1st 10% of blood loss (but venous return and CO # $ PP#). -At 20% loss, MAP falls (but still better maintained than CO). -At 30% loss, MAP# becomes significant (50% of normal). The plateau is due to the cerebral ischemic response (maximal sympathetic output). Left: If blood pressure does not drop below 45 – 50 mm Hg, patient compensated (non-progressive shock) although recovery time " blood If pressure drops below 45 mm Hg, ! progressive state of irreversible shock ! died, decompensation (progressive shock) Right: as MAP$, perfusion of baroreceptors $ ! hypoxia ! #lactic acid production (! acidosis ! # respiration and maybe tidal volume via carotid and aortic bodies – where PaO2 is normal) OR #chemoreceptor firing (!acidosis ! # respiration and maybe tidal volume) o Responses to a Mild Hemorrhage: In spite of loss of 500 ml of blood (10%) and immediate $ venous return, there is no drop in MAP due to rapid compensatory mechanisms. o Blood loss is detected by cardiopulmonary receptors that respond to the decreased cardiac filling ! $ cardiopulmonary discharge ! removed activation of the cardio-inhibitor center and the depressor area ! $ vagal tone and # sympathetic activity ! #HR, # contractility (!ejection fraction), areteriolar constriction (#TPR), and venoconstriction to $ venous compliance to move venous blood into the heart and the arteries) – this all maintains MAP (CO is NOT restored). Metabolic acidosis and stagnant hypoxia ! ! ventilation ! # pCO2. o Carotid sinus and aortic arch baroreceptors also contribute to reflex responses Their discharge rate is reduced because of $ pulse pressure (due to $ stroke volume). Baroreceptors ! relay information to the cardiovascular control center consistent with the information from low-pressure receptors. " Cardiopulmonary and baroreceptors work together and trigger, qualitatively, the same responses. o Hormones: # sympathetic activity stimulates epinephrine secretion epinephrine and norepinephrine (epinephrine does not cause vasodilation of arterioles if they are simultaneously activated by noradrenergic firing.) " Sympathetic activation of kidney and $ renal perfusion pressure ! renin release ! angiotensin II ! aldosterone. " $ nerve activity from both cardiopulmonary receptors and baroreceptors removes inhibition of secretion of ADH (potent vasoconstrictor at high concentrations). ADH also causes NO-mediated dilation of cerebral and coronary vessels Transcapillary and Transcellular Fluid Flow: -Arterioles constrict (#TPR) ! capillary hydrostatic pressure$! PFiltration $! #reabsorption (ISF moves to plasma – $ 10% volume loss hemorrhage is corrected in 30minutes -500-600ml) ! $hematocrit ! $O2 carrying capacity of blood (although $hematocrit allows for easier flow through #resistance vessels). A Time Limited Response. -$POncotic and $ PISF resist this recovery (but ICF=2xECF). Bleeding does not " ECF osmotic pressure so the ICF is naturally used as a compensatory mechanism. ECF osmotic pressure is " by sympathetic stimulated synthesis and release of glucose by the liver ! # ECF osmolarity ~20mOsm/kg (moves ICF ! ECF ! # PISF(ECF)) o Long-Term Adaptations: o Restoration of Water and Electrolytes: Body H2O can generally be replaced in 12-72 hours. The liver replaces plasma protein concentration within days. Erythropoietin activity ! due to renal hypoxia and #[RBC] restores [RBC] within four weeks. " Sympathetics: vasoconstriction ! #GFR and ! PT solute reabsorption • (# delivery and ! reabsorption! ! blood volume) " AII: ! ! ADH, ! thirst, ! aldosterone " ADH: ! vasoconstriction, !H2O reabsorption (DT/CD), ! Na+reabsorption (TAL – in turn ! more H2O reabsorption) • # blood flow to the vasa recta (prevents washout) provides for maximal H2O reabosorption o secreted in response to AII and to reduction of firing rates of cardiopulmonary receptors and baroreceptors " Aldosterone: ! ! Na+ reabsorption in the DT/CD. Maintains ECF osmolarity. " ANP: released in response to atrial stretch (hypervolemia) ! vasodilation, !GFR, excretion of Na+, inhibition of aldosterone and ADH. Inhibited by hemorrhage (# atrial distension). o Decompensation (Progressive Shock): If blood loss in a patient exceeds 30% and has lasted for a period of 3 to 4 hours before fluid replacement begins, patient enters decompensated (progressive) phase of shock. Even if the volume lost is completely replaced, patient may progressively decline. o Gray pallor: due to vasoconstriction (!sympathetic activity) and ! reduced hemoglobin in capillaries and subpapillary venous plexus (sluggish circulation in the skin). o Cold sweat and goose pimples: ! sympathetic activity. o Dryness of the mouth: vasoconstriction of arterioles in salivary glands and stress-induced inhibition of salivary gland secretion. o Rapid breathing: reflex manifestation of carotid and aortic body stimulation by the anoxia resulting from sluggish circulation in the carotid body. o Thready pulse: # stroke volume associated with # venous return and ! heart rate. o $ urine output: (1) # GFR (both with # renal perfusion pressure and afferent arteriolar constriction – due to ! sympathetic tone and ! AII) (2) ! Na+ and (isosmotic) water reabsorption in PT (stimulated by AII and ! sympathetic nerve activity) (3) ! ADH (! water retention) Decreased cardiac output ! # systemic flow (including coronary and cerebral vessels) ! # tissue perfusion ! hypoxia ! lactic acidosis ! cellular tissue damage ! release of toxins Decreased cerebral flow ! hypoxia and acidemia ! !pCO2 (toxic) and # cerebral activity (including the cardiovascular control center) ! # sympathetic activity ! vasodilation ! # MAP and !capillary permeability ! ! capillary pressure ! ! filtration ! ! tissue H2O (even blood transfusion adds to tissue H2O – doesn’t remain in vasculature) ! hypovolemia. Decreased coronary artery flow ! hypoxia and acidemia of cardiac tissues ! # cardiac muscle function (along with effects of toxins) ! Starling curve shifts Down and Right within one hour (irreversible shock). With prolonged hemorrhagic hypotension the arteriolar vasoconstriction that occurred during compensatory stages reverses (except in skin) ! vasodilation ! ! rate of run-off from the arteries and # BP ! compromises delivery of blood to the tissues. Stagnation of the circulation occurs and fluid leaks rapidly into the interstitial compartment. (arteriolar vasodilation partially restores capillary hydrostatic pressure and ! capillary permeability follows tissue ischemia). Various tissues release cardiotoxic shock factors that have a negative inotropic effect on the heart. Blood transfusion is ineffective in reversing the course of the illness. Maintenance of BP by adaptive responses may give a false sense of security to the doctor in an emergency room or to a surgeon or anesthetist in the operating room. BP cannot be used as the sole criterion for assessing the state of the patient. Other clinical signs and symptoms must dictate whether interventions, such as a blood transfusion, are warranted. Altitude: Above sea level, total barometric pressure is less than 1 atmosphere. But percent of O2 [fractional content] remains the same (20.9 $ PO2 of inspired air decreases as one ascends. At high altitudes (i.e., 30,000 ft), PAO2 decreases more PATMO2. Up to 10,000 ft, even when breathing air, the arterial saturation remains around 90%. A person ordinarily can remain conscious until the arterial O2 saturation falls to 40-50% (47,000 ft with pure O2). o o Immediate Adjustments: o # Tidal Volume: o # Respiratory Frequency (! #pH): carotid and aortic bodies sense # pO2 and trigger ! ventilation. o # Alveolar Ventilation: o $ TPR: An autoregulatory response of the arterioles. Vasodilation with hypoxemia is mainly caused by increased conductance of KATP channels that open and hyperpolarize the arteriolar smooth muscle when ATP levels in the cell fall. With a shortage of O2, anaerobic metabolism would increase; lactic acid production would rise and contribute to local vasodilation.) o # Heart Rate: Heart rate ! because of ! activation of lung stretch receptors (associated with ! ventilation). ! cardiac output results from a drop in TPR o # Cardiac Output o # 2,3-DPG Concentration: With hyperventilation pH ! ! more 2,3 DPG is synthesized. o # pH Long-Term Adjustments (weeks): o # Minute Ventilation: Gradual ! ventilation further ! PAO 2. Maximal ventilation takes time to develop, because in the face of chronic low pCO2, it takes time for removal of HCO3- from the CSF to return pH to normal. " After return to sea level, hyperpnea persists for several days (due to ! sensitivity of the central respiratory center to CO2 following compensatory adjustments). After levels of HCO3- return to normal and CSF pH returns to normal, ventilation drops to normal as well. o # RBC Count: Additional erythropoietin (EPO) is secreted by the kidneys and other tissues such as liver ! ! [Hb] and helps compensate for the # pO2 and HbO2 saturation. Hematocrit ! from a normal value of 40 - 45 to ~50-65, with ! [Hb] from 15 g/dl to ~22 g/dl. ! blood volume (~20-30%), ! ! circulating hemoglobin of ~50-90%. If polycythemia progresses too far ! ! blood viscosity ! block of fine blood vessels with RBC masses or clots. o # Number of Capillaries (recruitment):# distance that O2 must diffuse between capillary and tissue cell and ! cross sectional area to ! diffusion through the capillary wall. o #Diffusing Capacity of Lungs: diffusing capacity ! ~ three-fold during exercise and at high altitude (results from ! pulmonary capillary blood volume ! capillary expansion and ! surface for oxygen diffusion. ! lung volume ! expanded surface area of the alveolar membrane. o # O2 Utilization of Cells: In those native to altitudes of 13,000-17,000 ft, certain cellular oxidative enzyme systems are more plentiful than in sea level inhabitants (! more effective O2 utilization). A similar ! in oxidative enzymes does NOT occur in those exposed to high altitudes for shorter periods of time (several years). Hypoxemia leads to hyperventilation and $arterial and tissue pCO2 ! # bicarbonate removal (respiratory alkalosis) (lessen ! blood pH to restore CSF pH toward normal). Hypoxemia ! # lactic acid production (metabolic acidosis) (restores pH balance) o Native Inhabitants: o $ Alveolar pO2: o $ Alveolar pCO2: o # Respiratory Rate: o # Hemoglobin Concentration: o # Blood Volume: o $ O2 Saturation: o # O2 Capacity: o # Plasma Bilirubin Concentration: o $ Plasma Bicarbonate (HCO3-) Concentration: Osmoregulation Upon Introduction/Loss of Substances: o o o Excessive Water Loss (dehydration): total osmolality ! and volumes of intracellular and extracellular compartments are # in proportion to original volume. Oral Ingestion of Water: water distributes uniformly between extracellular and intracellular compartments (! volume and # osmolarity). Infusion of Isotonic Saline: ECF expands without producing ! intracellular volume or in intra- or extracellular concentration o o Infusion of Hypertonic Saline: volume of ECF! and ICF# . Excessive Loss of Solute (excessive sweating followed by replacement of H2O and not salt): water moves into cells (!ICF and #ECF). Type Examples ECF Volume ICF Volume ECF Osmolarity Hct and Serum [Na+] Isosmotic Volume Expansion Isosmotic Volume Contraction Hyperosmotic Volume Expansion Hyperosmotic Volume Contraction Hyposmotic Volume Expansion Hyposmotic Volume Contraction Isotonic NaCl infusion Diarrhea # Unchanged unchanged $ unchanged unchanged High NaCl intake # $ # Sweating, Fever, Diabetes insipidus $ $ # $ Hct, $ [protein], unchanged [Na+] # Hct, $[protein], unchanged [Na+] $ Hct, $[protein], # [Na+] Unchanged Hct*, #[protein], # [Na+] Syndrome of Inappropriate ADH Adrenal Insufficiency # # $ $ # $ Unchanged Hct**, $[protein], $ [Na+] # Hct, #[protein], $ [Na+] * hematocrit remains unchanged because water shifts out of the RBCs (# volume and offsetting concentrating effect of # ECF volume) ** hematocrit remains unchanged because water shifts into RBCs (! volume and offsetting the diluting effect of the gain of ECF volume) Diarrhea ! # plasma [Cl-], $ ANP secretion, hyperventilation, # hematocrit. Potassium Secretion: • Principal cells (late DT and cortical CD) - stimulated by # plasma potassium (hyperkalemia) [# permeability of lumenal membrane and excites Na+-K+ ATPase] aldosterone (# uptake by cells and # recruitment/synthesis of Na+-K+ ATPases, Na+ channels, and K+ channels ! increased uptake of Na+ and increased secretion of K+) - # tubular flow (# K + gradient for secretion) - Diuretics (# DT flow) - Alkalemia - upregulating Na+-K+ ATPase and # lumenal K+ permeability. - Potassium Reabsorption: • "-intercalated cells (medullary CD) - stimulated by hypokalemia [$ permeability of lumenal membrane and inhibits Na+-K+ ATPase] - acute acidosis (! hyperkalemia); $ secretion by inhibiting basolateral Na+-K+ ATPase pumps and $ lumenal permeability. Normal K+ intake ! secretion of K+ at DT (excretion amounts to 10-15% of filtration rate). High K+ intake ! secretion of K+ by DT ! (excretion may exceed filtration). Low K+ ! DT reabsorbs K+ (excretion reduced to ~1% of the filtered load) Hyperkalemia (lack of uptake or release): • • • • stimulates aldosterone acidosis ! potassium release from cells into interstitum exercise (release of K+) # osmolality of ECF (K+ moves out of cells) Hypokalemia (uptake or lack of reabsorption): • alkalemia ! potassium moves into cells from interstitium • $ osmolality of ECF (K+ moves into cells) • loop diuretics (no uptake by Na-K-2Cl transporters) Aldosterone: • Secretion # by: o # plasma K+ o # AII ! # renin o acidosis o atrial stretch Functions: • stimulates Na+ uptake (and associated H2O retention) [maintains ECF osmolarity] + + + o # recruitment of apical Na channels and basolateral Na -K -ATPase pumps (30-60 minutes latency). # synthesis of apical Na+ channels and basolateral Na+-K+-ATPase pumps (~4-6 hours latency) • • stimulates potassium uptake by cells (preventing hyperkalemia) stimulates potassium secretion by principal cells • Secretion # by: o # renin (due to $ ECF) AII: Functions: • arteriolar vasoconstrictor o Low concentrations of AII constrict the efferent arterioles predominantly, and higher concentrations also constrict afferent arterioles ! $ RBF and GFR. • stimulates secretion of aldosterone • stimulates increased thirst • stimulates secretion of ADH • stimulates PT Na+ reabsorption (partially mediated by stimulation of Na+-H + exchanger) ADH: o Secretion # by: small # plasma osmolality and large (> 10% reductions in ECF volume) and by AII. Inhibitory inputs from baroreceptors, cardiopulmonary receptors (normal state), and Atrial Natrieuretic Peptide (ANP). Functions: modulates urine volume and concentration o induces the insertion of aquaporins into late-DT and CD. o stimulates Na+ reabsorption by TAL (#interstitial osmolality ! H2O reabsorption from adjacent CD) o without ADH – [urine] = 50mOsm/kg o with ADH – [urine] = 1200mOsm/kg o o depresses vasa recta blood flow (reduces washout of interstitial solute protecting osmotic stratification) and enhances IMCD urea transport (urea contributes to interstitial osmolality). " Urea Transport: markedly # by ADH; $ protein intake ! $urea formation !$urine concentration; urea infusion ! prompt # concentrating ability. Insulin: • stimulates potassium uptake by cells (stimulates Na+-K+ ATPase) Epinephrine: • Secretion # by $ blood volume (BP) Functions: • stimulates potassium uptake by cells " afferent arteriolar vasoconstriction (via "-1 receptors) ! $RBF and GFR " stimulation of %-1 receptors in granular cells in afferent and efferent arterioles ! ! renin release. o Stimulation can be central or in response to # arteriolar perfusion pressure ($ # stretch of afferent arterioles) Endothelin: vasoconstricts afferent and efferent arterioles (produced by endothelial and mesangial cells). Nitric oxide: dampens sympathetic and AII activity (produced by endothelial and macula densa cells). Prostaglandins: released in response to norepinephrine or AII, prevents excessive constrictor responses to these agents. ATP, Bradykinin, and Histamine: stimulates release of NO. Dopamine: vasodilator of renal vasculature. Atrial natriuretic peptide: released upon atrial stretch (! ECF) Functions: o dilates afferent arterioles, constricts efferent arterioles and thereby increases GFR. o # NaCl and water excretion by the kidneys. " # GFR and Na+. " inhibits renin secretion by juxtaglomerular cells. " inhibits aldosterone secretion. " inhibits (directly) Na+ reabsorption by CD. " inhibits ADH secretion. Urodilatin: secreted by DT and CD (acts locally inhibiting Na+ reabsorption by these segments). NO, Prostanoids, and Kinins: produced in kidneys and inhibit Na+ reabsorption.