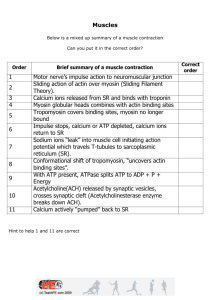
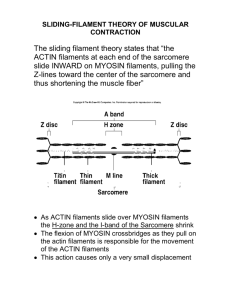
F-actin dynamics in dendritic spines
advertisement

F-­‐actin Dynamics in Dendritic Spines Mikko Koskinen Dissertationes Scholae Doctoralis Ad Sanitatem Investigandam Universitatis Helsinkiensis Neuroscience Center And Division of Physiology and Neuroscience Department of Biosciences Faculty of Biological and Environmental Sciences University of Helsinki And Doctoral Program In Brain and Mind Academic Dissertation To be presented for public examination with the permission of the Faculty of Biological and Environmental Sciences, University of Helsinki, in the lecture hall 2402 of Biocenter 3, Viikinkaari 5, Helsinki, on the 12th December, 2014 at 12 o’clock noon Helsinki 2014 Supervised by Docent Pirta Hotulainen Neuroscience Center University of Helsinki, Finland Reviewed by Professor emeritus Carl G. Gahmberg Faculty of Biological and Environmental Sciences University of Helsinki, Finland and Docent Eleanor Coffey Center of Biotechnology University of Turku, Finland Opponent Associate Professor Karen Zito Center for Neuroscience University of California Davis, USA Cover illustration: Sara Fagerstedt ISSN 2342-­‐3161 (print) ISSN 2342-­‐317X (online) ISBN 978-­‐951-­‐51-­‐0470-­‐0 (print) ISBN 978-­‐951-­‐51-­‐0471-­‐7 (online) Hansaprint ii Contents Contents .............................................................................................................................. iii List of original publications .......................................................................................... iv Abbreviations ...................................................................................................................... v 1 Summary ........................................................................................................................ 1 2 Background ................................................................................................................... 2 2.1 Neurons ............................................................................................................................... 2 2.2 Dendritic spines ............................................................................................................... 2 2.2.1 Dendritic spine development ............................................................................................. 3 2.2.2 Dendritic spine stability and maintenance ................................................................... 4 2.2.3 Dendritic spine plasticity ..................................................................................................... 5 2.3 The actin cytoskeleton ................................................................................................... 6 2.3.1 Actin organization ................................................................................................................... 7 2.3.2 Actin dynamics .......................................................................................................................... 7 2.3.3 Actin binding proteins ........................................................................................................... 8 2.4 Actin filaments in dendritic spines ......................................................................... 10 2.4.1 Organization of actin filaments in dendritic spines ................................................ 11 2.4.2 Actin dynamics in dendritic spines ................................................................................ 11 2.4.3 Actin binding proteins in dendritic spines ................................................................. 13 2.4.4 Myosin IIb in dendritic spines .......................................................................................... 15 2.5 Methods to measure actin filament dynamics and organization in dendritic spines ........................................................................................................................ 16 2.5.1 Fluorescently labeled actin ................................................................................................ 17 2.5.2 Fluorescence recovery after photobleaching (FRAP) ............................................ 17 2.5.3 Photo-­‐activatable green fluorescent protein fluorescence decay .................... 19 2.5.4 Förster resonance energy transfer ................................................................................ 20 3 Aims of the study ...................................................................................................... 23 4 Experimental procedures ..................................................................................... 24 5 Results ......................................................................................................................... 25 5.1 Development and testing of methods (I, II) ........................................................ 25 5.1.1 Fluorescence recovery after photobleaching (II, III) ............................................. 25 5.1.2 Photo-­‐activatable GFP fluorescence decay (I, II, III) .............................................. 27 5.1.3 Fluorescence anisotropy can be used to study the bundling of actin filaments in dendritic spines (II) .................................................................................................... 28 5.2 Changes in actin dynamics during the maturation of dendritic spines (II, III)….. ............................................................................................................................................ 29 5.3 Myosin IIb has a dual role in regulating actin dynamics in dendritic spines (II, III) ........................................................................................................................................... 30 6 Discussion .................................................................................................................. 32 7 Conclusions ................................................................................................................ 37 Acknowledgements ........................................................................................................ 39 References ......................................................................................................................... 41 iii List of original publications I. II. III. 1 Koskinen, M., Bertling, E., and Hotulainen, P. (2012). Methods to measure actin treadmilling rate in dendritic spines. Methods Enzymol 505, 47-­‐58. 2 Koskinen, M., and Hotulainen, P. (2014) Measuring F-­‐actin properties in dendritic spines. Front. Neuroanat. 8:74. 3 Koskinen, M., Bertling E., Hotulainen, R., Tanhuanpää, K., and Hotulainen, P. (2014) Myosin IIb controls actin dynamics underlying the dendritic spine maturation. Mol Cell Neurosci 61:56-­‐64. 1 The author contributed to the design of the research, performed all PAGFP-­‐ actin experiments, analyzed the results and wrote the manuscript with P.H. 2 The author designed the research, performed all the experiments and wrote the manuscript with P.H. 3 The author designed the research, performed the PAGFP-­‐actin, FRAP, Motility analysis, morphological analysis and immunocytochemistry experiments and wrote the manuscript with P.H. iv Abbreviations ABP ADF/cofilin ADP Arp2/3 ATP BBB CFP CNS DIV14 E17 EM Eps8 FLIM FRAP FRET GABA GFP LTD LTP MHC MLC17 MLC20/MRLC MLCK MNI-­‐glutamate NMDA PAGFP PALM PP1/PP2 P1 PSD R709C ROI siRNA STORM T203H YFP Actin binding protein Actin depolymerizing factor/cofilin Adenosine diphosphate Actin related protein 2/3 Adenosine triphosphate Blood-­‐brain barrier Cyan fluorescent protein Central nervous system 14 days in vitro Embryonal day 17 Electron microscopy Epidermal growth factor receptor kinase substrate 8 Fluorescence lifetime imaging Fluorescence recovery after photobleaching Förster resonance energy transfer γ-­‐aminobutyric acid Green fluorescent protein Long term depression Long term potentiation Myosin heavy chain Myosin essential light chain Myosin regulatory light chain Myosin light chain kinase 4-­‐Methoxy-­‐7-­‐nitroindolinyl-­‐L-­‐glutamate, N-­‐methyl-­‐D-­‐aspartate photo-­‐activatable green fluorescent protein Photo activated localization microscopy Protein phosphatase 1/protein phosphatase 2 Post-­‐natal day 1 Postsynaptic density Arginine 709 to cystein Region of interest Small interfering ribonucleic acid Stochastic optical reconstruction microscopy Threonine 203 to histidine Yellow fluorescent protein v 1 Summary Dendritic spines are small bulbous protrusions extending from dendritic shafts of neurons. These small compartments house most of the postsynaptic terminals of excitatory synapses in the mammalian central nervous system. Dendritic spines are formed during early development and their density and morphology undergoes significant changes during maturation. Even after maturation dendritic spines are not static structures but display constant changes in their morphology and stability. The shape and size of dendritic spines have been linked to synaptic transmission, coupling the form of spines to neuron function. Several neurological diseases and disabilities are characterized by abnormal spine density and morphology. The main structural component of the dendritic spines is the actin filament, F-­‐actin. Actin filaments are dynamic polymers of the monomeric protein actin. The filaments are constantly turning over and reorganizing. Both processes are regulated by actin binding proteins. All structural changes and the maintenance of dendritic spines are dependent on actin dynamics. Current research indicates that the dynamics of actin filaments do not change following spine maturation. Maturation does lead to a decrease in the movement of spines and an increase in stability, all indicating changes in F-­‐ actin dynamics. In this study I have shown that the dynamics of F-­‐actin do change during maturation. The stable pool of F-­‐actin increases in size and the turnover of the dynamic pool increases. One of the actin binding proteins with a potential to regulate actin stabilization is myosin IIb, a motor protein with capabilities to bind F-­‐actin and to introduce contractility into the filament network. Myosin IIb has been shown to regulate dendritic spine development, size and shape and play a role in memory consolidation. The molecular mechanisms of F-­‐actin regulation by Myosin IIb in dendritic spines are not known. In this study I have shown that myosin IIb regulates dendritic spine F-­‐ actin via two distinct mechanisms. Myosin IIb can bind F-­‐actin and stabilize it without affecting the turnover of the dynamic filaments. Myosin IIb-­‐mediated contractility on the other hand can facilitate the turnover of the dynamic filaments. These findings help us to understand the molecular mechanism behind dendritic spine structure regulation and possibly in the future how it is related to synaptic transmission and different pathological states. Due to their small size, dendritic spines pose unique challenges for the study of actin dynamics. Most of the available methods are based on advanced fluorescence microscopy. In this study I have made a critical evaluation of the methods used to measure F-­‐actin turnover in dendritic spines and the analysis of the data. I have also developed a novel approach to use fluorescence anisotropy to measure the level of actin bundling. The method has been previously applied to measure 1 actin polymerization. My findings have led to the conclusion that in actin-­‐dense compartments, such as the dendritic spines, fluorescence anisotropy reflects actin bundling rather than polymerization and that conclusions based on earlier research using similar techniques should be re-­‐evaluated. 2 Background 2.1 Neurons The mammalian nervous system consists of two types of cells: neurons and glial cells. Glial cells are responsible for myelinating the axons of neurons, immunological functions in the central nervous system (CNS) and formation of the blood-­‐brain barrier (BBB). Neurons fire electrical pulses and transmit information from one cell to another via synapses. In the nervous system large, complicated, networks are built between neurons. It is in these networks that the brain performs it’s computing, which controls, for example, our decisions, thoughts and movements and stores our memories. There are many different types of neurons found in the CNS. They can be labeled according to various parameters such as morphology, localization etc. In the most basic labeling scheme they can be divided into two classes based on the neurotransmitter they release: inhibitory neurons releasing γ-­‐aminobutyric acid (GABA) and excitatory neurons that release glutamate. The correct balance of excitation and inhibition is fundamental for CNS function. 2.2 Dendritic spines Dendritic spines are small bulbous protrusions from dendritic shafts of neurons first described by Santiago Ramón Y Cajal (1888). Dendritic spines are found in most of the principal neurons in the mammalian CNS (Nimchinsky et al., 2002). The size of dendritic spines varies between cell types. The dendritic spines found on a single neuron or even a single dendrite form a heterogeneous population of morphologies and sizes. The volume of spine heads is in the range of ~0.001-­‐ 1μm3 (Nimchinsky et al., 2002). A hippocampal pyramidal neuron can have 30000 spines (Megias et al., 2001). Spine density varies according to the cell type as well as the position along the dendrite (Harris et al., 1992; Megias et al., 2001). It is known that most of the post-­‐synaptic terminals of excitatory synapses are confined to dendritic spines (Bourne and Harris, 2008), although, the same dendritic spine can contain both excitatory and inhibitory synapses (Chen et al., 2012a). The correct morphology and density of spines is crucial for normal CNS function. Several diseases including Alzheimer’s disease, schizophrenia, 2 intellectual disabilities and autism spectrum disorders are linked to disturbances in the morphology and density of dendritic spines (Sala and Segal, 2014). Dendritic spines are also considered to make up the physiological sites for learning and memory (Kasai et al., 2010). The correct development and plasticity of dendritic spines are essential for the proper functioning of the CNS and therefore for the individual’s development and health. 2.2.1 Dendritic spine development 2.2.1.1 Spinogenesis Dendritic spines form during early development. During the process of spine emergence, spinogenesis, thin filopodia, i.e. thin protrusions lacking synaptic components, are constantly grown and retracted from the dendritic shaft (Dailey and Smith, 1996; Ziv and Smith, 1996; Dunaevsky et al., 1999; Garcia-­‐Lopez et al., 2010). These filopodia are sometimes observed to mature into dendritic spines (Ziv and Smith, 1996; Fiala et al., 1998). In the developmental timeline spinogenesis coincides, at least partly, with the formation of synaptic contacts between nerve cells, named synaptogenesis (Vaughn, 1989). There is evidence indicating a role of spinogenesis in synaptogenesis and therefore in the formation of brain wiring (Ziv and Smith, 1996; Yuste and Bonhoeffer, 2004). Several models have been proposed to explain the exact mechanisms of spine formations in synaptogenesis (Yuste and Bonhoeffer, 2004; Garcia-­‐Lopez et al., 2010). In the filopodial model a dendritic filopodium grows towards the axon, makes contact with the axon and matures into a spine with a functional synaptic connection (Yuste and Bonhoeffer, 2004). 2.2.1.2 Dendritic spine maturation After recruitment of the synaptic machinery into a newly formed spine, the spine acquires a mature spine shape (Dailey and Smith, 1996; Dunaevsky et al., 1999). Mature spines do not have a common shape but instead can be divided into classes based on their morphology. The different spine morphologies are denoted as thin, stubby and mushroom shaped (Figure 1). The most distinct and most studied spine shape, also considered the most mature of spine shapes, is the mushroom with a thin neck and a bulbous head. The morphology and size of the spine head and neck influence the electrical properties of the synaptic compartment (Noguchi et al., 2005) as well as diffusion between the spine head and the dendritic shaft (Tonnesen et al., 2014). 3 2.2.2 Dendritic spine stability and maintenance Even after maturation dendritic spines are not rigid structures but exhibit a change in Figure 1. Morphological classes of Dendritic spines. Filopodia constant are thin protrusions that do not contain the postsynaptic morphology and size (Fischer machinery. Thin spines have a small head that contains the postsynaptic density and a long thin neck. Stubby spines have a et al., 1998; Zuo et al., 2005). wide almost indistinguishable neck. Mushroom spines are A correlation between spine characterized by the large bulbous heads and thin necks size and morphology and spine stability has been observed with larger mushroom-­‐shaped spines being more stable than smaller thin or stubby spines (Holtmaat et al., 2005; Yasumatsu et al., 2008). The motility of dendritic spines is also dependent on their developmental state, with mature spines displaying decreased motility compared to their more immature counterparts (Dunaevsky et al., 1999; Korkotian and Segal, 2001; Takahashi et al., 2003; Oray et al., 2006). Whether this is just a result of the synaptic activity patterns experienced during development or a cellular mechanism directly influencing the motility of spines is not known. Certain spines can last a lifetime of an individual but spines keep on emerging and disappearing throughout life. The reports on the proportions of the stable vs. dynamic spines are highly variable (Bhatt et al., 2009). 2.2.2.1 Short term motility The short-­‐term motility of dendritic spines is characterized as rapid changes in size and a switching between different morphological classes (Fischer et al., 1998). Rapid morphological changes in response to synaptic activity can also be categorized as short-­‐term motility (Matsuzaki et al., 2004). The movement of spines is seen in the timescale of minutes (Fischer et al., 1998; Bertling et al., 2012). Spines fluctuate between different morphological states and show a fluctuation in their size. On average, mushroom shaped dendritic spines of rat hippocampal neurons prepared from 17 day old embryos (E17) and cultured in vitro for 14 days (DIV14) fluctuate at an amplitude of ~15% of their diameter at 1 min intervals (Bertling et al., 2012). The purpose of this constant “morphing” is not well understood. 4 Another type of short-­‐term dynamics are the rapid changes in size and morphology following synaptic activity (See 2.1.3.1). 2.2.2.2 Long term stability and dendritic spine turnover The long-­‐term spine stability is regulated by two mechanisms: (1) an activity independent intrinsic fluctuation observed at the timescale of days (Yasumatsu et al., 2008) and (2) activity-­‐dependent persistent changes (Matsuzaki et al., 2004). Intrinsic fluctuations are the activity independent changes in spine size. These changes are thought to be related to the earlier activity patterns of the synapse and to predict the stability of spines (Yasumatsu et al., 2008). A turnover of spines consisting of the disappearance of old spines and the formation and stabilization of new ones has been studied extensively in vivo and shown to be present even in adulthood (Grutzendler et al., 2002; Trachtenberg et al., 2002; Holtmaat et al., 2005). The rate of turnover has been shown to be dependent on the developmental state of the individual (Zuo et al., 2005). Turnover rate has been shown to change at forelimb projection areas during learning of new skills or exploration of novel environment leading to the hypothesis that dendritic spines are responsible for adaptation in the neural network (Yang et al., 2009; Roberts et al., 2010). Repeated bouts of practicing a task or fear have been shown to induce formation of new spines (Fu et al., 2012; Lai et al., 2012). New spines have been reported to emerge in close proximity of each other leading to a theory of spines responding to the same stimuli localizing to a spatially constrained area (Fu et al., 2012; Lai et al., 2012). However, a study of functional mapping of single synapses in dendritic spines of auditory cortex neurons in vivo did not report clustering of spines (Chen et al., 2011). 2.2.3 Dendritic spine plasticity Excitatory post-­‐synaptic terminals are the sites of functional and morphological adaptation during activity-­‐dependent mechanisms regulating synaptic conductance, long-­‐term potentiation (LTP) and long-­‐term depression (LTD) (Sala and Segal, 2014). During LTP induction a synapse receives stimulation from connected neurons that leads to a potentiation of the synapse’s response. Following LTP induction, the activation of the synapse leads to a greater effect on the post-­‐synaptic neuron’s membrane potential (Hughes, 1958). LTD, on the other hand, reduces the response of the post-­‐synaptic neuron following synaptic activation. These adaptations regulate the probability of the post-­‐synaptic neuron itself to fire an action potential and therefore alter the function of the neural network. LTP has been generally seen as a cellular mechanism connected 5 to the formation of memories and learning, and recently, experimental evidence of a causal relationship between LTP and fear learning has been presented (Nabavi et al., 2014). Therefore, dendritic spines are often seen as the site of memory storage and the functional and morphological changes of the dendritic spine and the resident synapse as their cellular and physiological counterparts (Kasai et al., 2010). 2.2.3.1 Synaptic activity-­‐dependent synaptic and morphological plasticity Synapses exhibit plasticity in their response following synaptic activation (Hughes, 1958; Malenka and Bear, 2004). The dendritic spines affected by activation patterns exhibit simultaneous morphological plasticity. Specifically, induction of LTP has been shown to result in dendritic spine enlargement (Matsuzaki et al., 2004), and LTD has been linked to spine shrinkage (Zhou et al., 2004). Of these mechanisms, LTP is spine-­‐specific and contained in the spine housing the stimulated synapse (Matsuzaki et al., 2004; Lee et al., 2009). LTD on the other hand can spread from one spine to the neighboring ones (Wang et al., 2000; Hayama et al., 2013). Changes in the synaptic conductance have been linked to changes in dendritic spine size (Matsuzaki et al., 2004). At the molecular level little is known of a direct causal link between synaptic plasticity and dendritic spine morphological changes, but evidence shows that the stabilization of LTP is blocked as a result of blocking the spine enlargement (Krucker et al., 2000). How the dendritic spine stabilization during spine maturation results from the activity patterns of the neural network, which themselves are influenced by the changes in the synapses is not known. 2.3 The actin cytoskeleton Actin is the most abundant protein in most eukaryotic cells. Three mammalian isoforms of actin are found: α-­‐actin that forms the thin filaments found in muscle, β-­‐actin that forms the main cytoskeleton of non-­‐muscle cells and γ-­‐actin that is found in the filaments of stress fibers, tightly bundled polymers spanning the cell body of many motile cell types (Dominguez and Holmes, 2011). Monomeric actin is an ATPase. In its structure an “ATPase cleft” is positioned between two lobes. adenosine triphosphate (ATP) associated with the Mg2+ is bound into the cleft and hydrolyzed to adenosine diphosphate (ADP) + phosphate (Dominguez and Holmes, 2011). Actin is responsible for many structural as well as functional properties of cells. Structures such as filopodia, lamellipodia, dendritic spines and contractile rings contain actin. Actin is responsible for maintaining and changing the cell shape of 6 all eukaryotic cells (Pollard and Cooper, 2009). It plays a fundamental role in many dynamic cellular processes as a component of a contractile element in non-­‐ muscle cells. Actin-­‐related contractility is a key component in cell motility and migration, cell division and endo-­‐ and exocytosis. Actin filaments are also used as tracks for intracellular transport (Pollard and Cooper, 2009). 2.3.1 Actin organization In cells actin is found in two forms: as monomers (globular-­‐, G-­‐actin) and as polymers (filamentous-­‐, F-­‐actin). Actin monomers have a molecular mass of 42 kDa (Dominguez and Holmes, 2011). Actin monomers polymerize to form F-­‐ actin. Actin filaments, also called microfilaments, are double stranded helices (Holmes et al., 1990; Dominguez and Holmes, 2011). The radius of the microfilaments found in the cytoskeleton is 7 nm and the symmetry of the helix is 2,17 subunits per turn (Holmes et al., 1990; Dominguez and Holmes, 2011). The actin cytoskeleton can be organized into various structures inside the cells. F-­‐actin at the cell cortex leading edge usually consists of a meshwork of branched actin filaments (Small, 1981). Filopodia contain long tightly bundled parallel filaments (Mattila and Lappalainen, 2008). Stress fibers are tightly packed bundles of filaments spanning long distances in cell bodies (Tojkander et al., 2012). The organization of the actin filaments determines the cytoskeletal structures biophysical properties (Blanchoin et al., 2014). The different types of actin structures have specific functional roles depending on the cell type and the intracellular location of the structures. 2.3.2 Actin dynamics F-­‐actin consists of polar filaments in the sense that both strands of the helix have the same orientation in their subunits (Dominguez and Holmes, 2011). Actin monomers undergo conformational changes when forming filaments resulting in different affinity at each end of the filament. ATP-­‐actin has a higher affinity towards the so-­‐called barbed-­‐ or (plus) end of the filament than the pointed-­‐ (minus-­‐) end (Pollard, 1986). ADP-­‐actin has a weaker affinity towards either end. The difference in binding affinities results in different polymerization and depolymerization rates at either end with the barbed end elongating 10 times faster than the pointed end. With the right concentration of ATP-­‐ and ADP-­‐actin this can lead to ATP-­‐bound monomer binding to the barbed end, followed by ATP-­‐hydrolysis and subsequent release of the phosphate followed by the ADP-­‐ bound monomer detaching from the barbed end (Bugyi and Carlier, 2010) (Figure 2). This constant flow of monomers through the filament is called actin 7 treadmilling. When the actin filament is anchored to the rest of the cellular cytoskeleton, polymerization can produce protrusive force against membrane or other cellular structures (Blanchoin et al., 2014). This protrusive force is how actin cytoskeleton changes cell shape (Pollard and Cooper, 2009). The rate of polymerization and depolymerization at the two ends are strictly regulated in cells and are controlled by actin binding proteins (see 2.2.3). In a simplified case, the turnover rate of the whole filament depends on the polymerization and depolymerization rates at the two ends, the filament length and the concentration of actin (Halavatyi et al., 2010). In steady state, the net rate of polymerization at the barbed end equals the net rate of depolymerization at the pointed end. 2.3.3 Actin binding proteins The actin cytoskeleton organization and dynamics are strictly regulated by a group of actin binding proteins (ABPs). ABPs can be divided into different classes depending on their function (dos Remedios et al., 2003; Pollard and Cooper, 2009). In table 1 the most common classes are presented and examples of proteins from each class are given. Selected ABPs are presented in figure 2. Due to the abundance of actin and its involvement in many fundamental cellular processes, ABPs are among the most important regulatory proteins in the cell. Disturbances in ABP functions have been linked to several diseases such as muscular dystrophy (Ozawa, 2010), Alzheimer’s disease (Penzes and Vanleeuwen, 2011) and mental retardation (Chelly et al., 2006). Table 1. Actin binding proteins Protein Function Refs. Class examples Monomer Profilin Nucleotide (Carlsson et al., binding proteins exchange 1977) Thymosinβ4 Filament depolymerizing proteins Inhibits filament elongation via sequestering of ATP-­‐actin Depolymerize F-­‐ actin ADF/cofilins Gelsolin (Paunola et al., 2002) (Lappalainen and Drubin, 1997) Cleaves F-­‐actin, (Sun et al., 1999) caps barbed ends. 8 Filament branching proteins Arp2/3 Nucleates branched actin filaments (Mullins et al., 1998) Filament cross-­‐ linking and bundling proteins Palladin Filamin Bundles F-­‐actin Crosslinks F-­‐actin into a network (Dixon et al., 2008) (Shizuta et al., 1976) Caps barbed ends (Wear et al., 2003) Capping proteins Capping protein Motor proteins Tropomodulin Caps pointed ends (Rao et al., 2014) Myosin II Actomyosin contraction Myosin V Vesicle traffic (Sellers, 2000) 2.3.3.1 Myosin IIb Myosins form a family of actin binding proteins. Myosins are motor proteins and are well known for their role in muscle contraction (Sellers, 2000). Myosins also play critical roles in many non-­‐muscle cells where they are involved in cell migration, adhesion and cytokinesis (Vicente-­‐Manzanares et al., 2009). In the human genome there are at least 40 genes coding for myosin superfamily proteins (Sellers, 2000). Myosin IIb, (encoded by the gene MYH10 in humans), belongs to the non-­‐muscle myosin II group together with myosin IIa and IIc (Vicente-­‐Manzanares et al., 2009). All of them are expressed in neurons, with IIb being the most predominant in the developed brain (Kawamoto and Adelstein, 1991; Golomb et al., 2004). Myosin IIb is an oligomer consisting of two myosin IIb heavy chains (MHC IIb), two 20 kDa myosin regulatory light chains (MLC20) (sometimes abbreviated as MRLC) and two 17 kDa myosin essential light chains (MLC17) (Vicente-­‐Manzanares et al., 2009). Myosin IIb binds actin and can introduce contractility into the filament network after activation via phosphorylation of residues threonine 18 and serine 19 of the MLC20 by the myosin light chain kinase (MLCK) (Vicente-­‐Manzanares et al., 2009). The contractile activity of myosin IIb is ATP-­‐dependent. 9 Figure 2. Actin treadmilling and actin binding proteins. A) In actin treadmilling ATP-­‐bound monomers are added to the barbed end of the filament. ATP is hydrolyzed to ADP and the phosphate released. The ADP-­‐ bound monomers are then depolymerized at the pointed end. B) Profilin is a nucleotide exchanger that catalyzes the change from ADP-­‐actin to ATP actin. Thymosinβ4 inhibits elongation by sequestering free G-­‐ actin. Capping protein caps the filament inhibiting polymerization at the barbed end. C) Arp2/3 nucleates branched filaments. Cofilin cleaves and depolymerizes filaments. D) Myosin IIb bundles filaments and introduces contractility into the system. 2.4 Actin filaments in dendritic spines Actin filaments form the cytoskeletal support structure of dendritic spines (Landis and Reese, 1983). Actin is highly concentrated in spines (Matus et al., 1982), with spines containing approximately 6 times the amount of actin found in the dendritic shaft (Honkura et al., 2008). Especially the filamentous form of actin, F-­‐actin, is found concentrated in dendritic spines (Drenckhahn et al., 1984; Honkura et al., 2008). Actin filaments play critical roles in the formation, 10 maturation, stability and plasticity of spines (Dillon and Goda, 2005; Cingolani and Goda, 2008). Dendritic spine shape, size and motility are all controlled by the actin cytoskeleton, whereas the organization and dynamics of the actin cytoskeleton in dendritic spines are regulated by various ABPs (Hotulainen and Hoogenraad, 2010; Bellot et al., 2014). Actin filaments in dendritic spines have been found to be highly dynamic displaying constant and rapid turnover (Star et al., 2002; Honkura et al., 2008; Frost et al., 2010). 2.4.1 Organization of actin filaments in dendritic spines Most of the actin in dendritic spines is F-­‐actin with only about 12% of the total actin being G-­‐actin (Star et al., 2002; Honkura et al., 2008). This is a significant shift from the dendritic shaft where approximately 70% of the actin consists of G-­‐actin (Honkura et al., 2008). Electron microscopy studies have revealed that F-­‐ actin immediately behind the postsynaptic density (PSD) in dendritic spines is organized in a meshwork of short filaments. Dendritic spines do sometimes also contain longer linear filaments (Korobova and Svitkina, 2010). Based on recent evidence produced with single molecule tracking methods, a model of F-­‐actin organization in dendritic spines was proposed (Chazeau et al., 2014). In this model the F-­‐actin meshwork is nucleated at the PSD and the changes in spine morphology coincide with a reorganization of the regulatory machinery. Dendritic spine necks have been shown to consist of long and short, linear and branched filaments with a mixture of polarizations (Korobova and Svitkina, 2010). 2.4.2 Actin dynamics in dendritic spines G-­‐actin movement in dendritic spines is rapid. The dendritic spine as a compartment does not actively limit the diffusion of G-­‐actin molecules in and out of the spine (Star et al., 2002; Honkura et al., 2008). In general, the diffusion of molecules through the spine neck is dependent on the size of the molecules (Tonnesen et al., 2014). The G-­‐actin component of total actin displays a very short (<1 s) turnover time constant in dendritic spines (Star et al., 2002; Honkura et al., 2008). Based on measured G-­‐actin diffusion rates, it has been estimated that the entire spine G-­‐actin content can be exchanged in 5-­‐670 ms (Honkura et al., 2008). This value does not take into consideration the possible rebinding of the monomers into existing filaments. F-­‐actin in dendritic spines is highly dynamic with most of the F-­‐actin having relatively short turnover times (Star et al., 2002; Honkura et al., 2008). The total F-­‐actin in dendritic spines can be divided into two pools based on their turnover 11 rate (Honkura et al., 2008). The two pools are: 1) the dynamic pool, with a fast turnover (time constant <1 min) and 2) the stable pool with a slower turnover (time constant ~17 min). The size of the stable pool correlates with the spine head size (Honkura et al., 2008). Single molecule tracking experiments have revealed a heterogeneous treadmilling rate of actin filaments in dendritic spines, with faster turnover found in areas behind the post-­‐synaptic density and at the spine edge, coinciding with the locations of the synapse and exocytosis, respectively (Frost et al., 2010). A retrograde flow of F-­‐actin from the tip towards the base of the spine has been observed in kinetic experiments (Honkura et al., 2008; Frost et al., 2010), and a general trend of inward directionality of monomer movements has been reported from single molecule tracking experiments (Frost et al., 2010). This view has recently been challenged by Chazeau et al. (2014), who have reported the directionality of monomer movement to point, not specifically inward, but away from the PSD. 2.4.2.1 Actin dynamics during maturation of dendritic spines It is known from dendritic spine motility studies that the short-­‐term spine motility decreases following spine maturation (Korkotian and Segal, 2001). Despite the fact that all the movement of dendritic spines is mediated by the actin cytoskeleton, no change was observed in the F-­‐actin turnover rate between DIV14 and DIV21 old dissociated rat hippocampal neurons (Star et al., 2002). The stability of the synapses housed in dendritic spines is dependent on the actin cytoskeleton in a developmentally regulated manner (Zhang and Benson, 2001). In younger neurons the stability of the synapse is dependent on the polymerization of actin but this dependency is reduced in older neurons. 2.4.2.2 Actin dynamics following synaptic activity Synaptic activity leading to induction of LTP has been shown to cause an increase in spine size that can be blocked by blocking actin polymerization (Matsuzaki et al., 2004). Blocking actin dynamics has also been shown to inhibit LTP stabilization (Krucker et al., 2000), linking actin dynamics to synaptic function. Actin dynamics has been shown to decrease following either application of the glutamate receptor agonist N-­‐methyl-­‐D-­‐aspartate (NMDA) or LTD inducing electric stimuli in cultured neurons, with a significant increase in the size of the stable pool of F-­‐actin (Star et al., 2002). 4-­‐Methoxy-­‐7-­‐ nitroindolinyl (MNI)-­‐glutamate uncaging accompanied with photo-­‐activatable green fluorescent protein (PAGFP)-­‐actin fluorescence decay measurements have 12 revealed an additional third pool, called the enlargement pool, of actin being transported into the dendritic spine following LTP induction (Honkura et al., 2008). In a little less than half of the experiments the enlargement pool was reported to be released from the spine as a bulk of actin through a widening of the spine neck. In the cases where the enlargement pool of actin was released from spines the LTP was not stabilized. The results of actin dynamics following spine maturation and synaptic activity are somewhat contradictory. Both maturation as well as LTP induction lead to spine enlargement and stabilization. All spine dynamics are controlled by the actin cytoskeleton but changes in actin dynamics have only been observed in conjunction with activity induced spine stabilization and not during spine maturation (Star et al., 2002). 2.4.3 Actin binding proteins in dendritic spines Just as in all other cells and compartments, actin organization and dynamics in dendritic spines are strictly regulated by ABPs. The role of several ABPs in the regulation of the dendritic spine actin cytoskeleton has been studied (Hotulainen and Hoogenraad, 2010). Some of the ABPs studied in dendritic spines and their physiological roles are found in table 2. The proteins in the table were selected based on the existence of direct measurements of the protein effects on actin dynamics. Table 2. Actin binding proteins role in dendritic spines. Protein Effect on actin Type of Physiological Refs. study function Cofilin Severs actin Loss of Facilitates actin (Hotulainen filaments, function turnover in spines et al., 2009) depolymerizes Involved in LTD (Hayama et filaments associated spine al., 2013) shrinkage and competitive selection of spines Loss of Involved in (Rust et al., function associative learning 2010) (Knock out and control of mice) extra-­‐synaptic AMPA receptor trafficking 13 Arp2/3 Eps8 Myosin IIb Gain/Loss Regulates AMPA of function receptor trafficking during synaptic plasticity Nucleates Loss of Facilitates actin branched actin function dynamics in spines filaments (Knock out Loss of function mice) leads to progressive cognitive and psychomotor disturbances in aging mice Caps actin Loss of Decreases actin barbed ends function turnover in spines Loss of function disrupts LTP induced structural and functional plasticity of spines Introduces Gain/loss Essential for contractility of function normal spine into morphology and actomyosin development structures Loss of function 14 Facilitates actin turnover in spines Essential for LTP stabilization and memory consolidation (Gu et al., 2010) (Kim et al., 2013) (Stamatakou et al., 2013) (Zhang et al., 2005; Ryu et al., 2006; Hodges et al., 2011) (Rex et al., 2010) In addition, numerous descriptive studies on the role of ABPs in regulating dendritic spine density, morphology and development have been published without investigating the direct effect on the actin cytoskeleton (Hotulainen and Hoogenraad, 2010). The roles of a number of the signaling molecules regulating the ABPs in dendritic spines have been studied extensively. For a review of the ABP signaling proteins in dendritic spines, see Dillon and Goda (2005). 2.4.4 Myosin IIb in dendritic spines Previously published findings of the role of myosin IIb in regulating dendritic spine morphology and synaptic function makes it an attractive candidate as the protein regulating dendritic spine actin during spine maturation (Zhang et al., 2005; Ryu et al., 2006; Rex et al., 2010; Hodges et al., 2011; Fromer et al., 2014). The regulatory subunit of myosin IIb, MLC20, which controls the contractile activity of the complex, is phosphorylated at residue serine 19 following synaptic activity (Rex et al., 2010). Myosin IIb has been shown to regulate spine development and morphology (Zhang et al., 2005; Hodges et al., 2011). Activation of myosin contractile activity through phosphorylation of two amino acids: threonine 18 and serine 19 is needed for spine maturation from filopodia to mushroom shape and for the enlargement of the PSD (Hodges et al., 2011). The loss of myosin IIb has been shown to lead to an abnormal, elongated, structure of the PSD that is not part of the spine head. Interestingly, results acquired through the use of the myosin IIb activity blocking drug blebbistatin do not completely match the results of siRNA knockdown experiments (Zhang et al., 2005; Ryu et al., 2006). Reasons for this discrepancy have not been investigated. In addition, knockdown of myosin IIb in the hippocampus of rats has been shown to lead to inability to stabilize LTP in brain slice field recordings as well as problems during memory consolidation (Rex et al., 2010). Based on these findings myosin IIb has been proposed to function upstream of actin polymerization (Rex et al., 2010). A single amino acid mutation in the motor domain of myosin IIb, glutamate 709 to cystein (R709C), has been shown to impair neuronal migration (Ma et al., 2004). This mutation has been shown to result in non-­‐contractile myosin IIb with a higher affinity towards actin as compared to wild type (Kim et al., 2005; Vicente-­‐Manzanares et al., 2007). Recently two different mutations in the myosin IIb motor domain have been linked to schizophrenia and autism spectrum disorders, respectively (Fromer et al., 2014). Despite the data previously presented by Rex et al (2010), produced by using myosin IIb inhibition by blebbistatin, the information on the effect of myosin IIb expression and activity on the spine cytoskeleton has been incomplete. 15 2.5 Methods to measure actin filament dynamics and organization in dendritic spines Actin cytoskeleton dynamics and organization has been studied extensively in motile cell types (Pollard and Cooper, 2009). These model systems lend themselves well towards microscopy techniques where actin structures can be visualized with the help of fluorescently labeled actin. Dendritic spines pose some additional challenges to the use of these techniques: the small volume and the high concentration of actin make the visualization of individual structures impossible with current light microscopy. At present, the only way to study the dynamics and organization of actin cytoskeleton in dendritic spines of living cells and tissues is to use advanced light microscopy techniques developed for this purpose. Techniques to study the dynamics of the actin cytoskeleton can be divided into two types: single molecule tracking techniques such as photo activated localization microscopy (PALM) and Stochastic optical reconstruction microscopy (STORM) (Frost et al., 2010) and bulk kinetic methods such as fluorescence recovery after photobleaching (FRAP) and photo-­‐activatable green fluorescenct protein (PAGFP) fluorescence decay (Star et al., 2002; Honkura et al., 2008; Hotulainen et al., 2009; Kim et al., 2013; Stamatakou et al., 2013). The single molecule tracking techniques measure the distance travelled by single molecules between two time points (Betzig et al., 2006; Hess et al., 2006; Manley et al., 2008). Velocities of a large number of tracked molecules can be acquired and the distribution of the velocities studied. Bulk kinetic methods on the other hand measure a large portion of the filaments at the same time as one group and the kinetics are derived from observations of the whole group (Reits and Neefjes, 2001). To study the organization of the actin cytoskeleton in such a small and actin rich compartment as the dendritic spine the non-­‐radiation energy transfer between fluorophores, Förster resonance energy transfer (FRET) has been used (Okamoto et al., 2004; Vishwasrao et al., 2012). FRET between different fluorophores (yellow fluorescent protein (YFP), and cyan fluorescent protein (CFP)) has been used to study the G-­‐/F-­‐actin ratio in dendritic spines following synaptic activity (Okamoto et al., 2004). FRET between two green fluorescent protein (GFP)-­‐molecules (HomoFRET) has also been used to study the polymerization state of the actin cytoskeleton (Vishwasrao et al., 2012). 16 2.5.1 Fluorescently labeled actin Actin can be visualized in cells in many ways. After the invention of fluorescence microscopy, actin filaments could be visualized with the help of fluorescently labeled phalloidin, an Amanita phalloides derived toxin that binds to F-­‐actin (Lynen and Wieland, 1938). The finding of the GFP opened up the possibilities to study the actin cytoskeleton in living cells (Shimomura et al., 1962). The fluorescent protein could be expressed as a fusion protein with actin from a plasmid DNA transfected into cells (Choidas et al., 1998). After expression, a percentage of the actin monomers in the cell are labeled with the fluorescent protein. These monomers incorporate into the actin filaments just as the endogenous ones (Choidas et al., 1998). With the known exception being a certain yeast strain where actin polymerizing ABP formin discriminates against labeled actin (Chen et al., 2012b). There are some reported adverse effects of the GFP-­‐fusion to actin in mammalian cells (Deibler et al., 2011). Problems usually increase with higher levels of exogeneous protein expression, which is why only cells with low to moderate levels of expression are selected for experiments. There are a number of different fluorescent proteins available at present but the most commonly used is still GFP (Shaner et al., 2005). Recently, a new genetically encoded peptide probe has been developed for the visualization of F-­‐actin called LifeAct (Riedl et al., 2008). This 17 amino acid peptide is fused to a fluorescent protein for use with light microscopy. While useful in visualizing the filaments this probe cannot be used to study filament dynamics. This is due to the fact that, rather than imaging F-­‐actin filaments directly, imaging LifeAct detects the dynamics of the attached probe. Furthermore, possible side effects of the probe binding due to obstruction of ABP binding sites are a concern. 2.5.2 Fluorescence recovery after photobleaching (FRAP) FRAP is based on the property of chromophore structures in fluorescent proteins to be destroyed and become non-­‐fluorescent (known as bleached) when illuminated with a high-­‐power laser at their absorption wavelength (Axelrod et al., 1976). This photon-­‐induced destruction of the chromophore is common to all fluorescent molecules, including organic chromophores and fluorescent proteins. The basic principle of using FRAP to study molecular dynamics is to rapidly bleach a specific area of the cell (known as a region of interest, (ROI)) with a high-­‐power laser and follow the recovery of the fluorescence at the ROI in question (Reits and Neefjes, 2001). The recovery of the fluorescence is a result of the bleached proteins leaving the ROI, either via diffusion or active transport, and being replaced with non-­‐bleached proteins from outside the ROI (Axelrod et 17 al., 1976). The kinetics of this process are used to study the dynamics of the fluorescently labeled protein. Using FRAP to study the dynamics of actin is based on constant treadmilling of the actin filaments. (see chapter 2.2.2). To use FRAP to study actin dynamics, an actin fusion construct containing a fluorescent protein is used (see chapter 2.4.1). When fluorescent actin is bleached from a ROI the recovery that follows contains the recovery of the G-­‐actin found in the area as well as the recovery of the F-­‐actin fluorescence (Watanabe et al., 2013). The recovery of the G-­‐actin is based on diffusion (Smith et al., 2013). The recovery of the F-­‐actin follows as the bleached monomers have moved through the filaments, depolymerized at the pointed end and diffused out of the ROI, at the same time being replaced in the filament barbed ends by non-­‐bleached monomers from outside of the ROI. FRAP has been a popular way to study the dynamics of actin in dendritic spines (Star et al., 2002; Hotulainen et al., 2009; Kim et al., 2013; Stamatakou et al., 2013). Usually the whole spine is used as the ROI. The G-­‐actin diffusion has been shown to be very rapid with time constants in <1 s range (Star et al., 2002; Honkura et al., 2008). On the other hand, the recovery of the F-­‐actin component is orders of magnitude slower with time constants in the tens of seconds range. This large difference between the recovery rates makes it possible to use FRAP to study the dynamics of the F-­‐actin component in dendritic spines (Star et al., 2002; Honkura et al., 2008). FRAP has been used to detect two differentially regulated pools of F-­‐actin in dendritic spines (Star et al., 2002; Honkura et al., 2008) as well as to study the role of ABPs in regulating actin filament dynamics in dendritic spines (Hotulainen et al., 2009; Kim et al., 2013; Stamatakou et al., 2013). 2.5.2.1 Analysis of FRAP data The analysis of FRAP data is most often influenced by previous knowledge of the protein function. As with actin, it is known that actin inside the cell exists as G-­‐ and F-­‐actin. The relative amounts of G-­‐and F-­‐actin in dendritic spines are also known from previous studies (Star et al., 2002; Honkura et al., 2008). The large difference in the time constants of the G-­‐and F-­‐actin components makes it possible to exclude the G-­‐actin component from the analysis. This is achieved by using a slow enough timescale in image acquisition while performing the experiments to reach full recovery of the G-­‐actin components before the start of the measurements (Star et al., 2002; Honkura et al., 2008). By excluding the G-­‐ actin component and having prior knowledge of the treadmilling of the filaments, the rate of filament turnover can be estimated from the FRAP recovery data (Star et al., 2002). The curves of the F-­‐actin recovery have a distinct 18 exponential shape (Star et al., 2002). The first step in the analysis is to normalize the data to the fluorescence intensity of the region outside the ROI to account for imaging-­‐induced bleaching and detector error. In the second step the post-­‐ bleach measurements are normalized to the pre-­‐bleaching fluorescence intensity values. Two methods have been used to extract values for the turnover rate. 1) Fitting the individual experiment data to an exponential function (Star et al., 2002; Kim et al., 2013) and 2) averaging the data from several spines and manually making a linear approximation of the plateau level of recovery and the recovery half time and calculating the first order rate constant (Hotulainen et al., 2009; Stamatakou et al., 2013). 2.5.3 Photo-­‐activatable green fluorescent protein fluorescence decay The photoactivatable green fluorescent protein is a green fluorescent protein with a point mutation of threonine to histidine at residue 203 (T203H) that diminishes the brightness of the fluorophore when exicited with a 488 nm laser in resting state but increases the brightness 100 times after being illuminated briefly with a ~400 nm laser or ultraviolet light (Patterson and Lippincott-­‐ Schwartz, 2002). The fusion protein of PAGFP-­‐actin is similar in behavior to GFP-­‐ actin when expressed exogeneously in cells (Honkura et al., 2008). The PAGFP fluorescence decay experiments are also based on the treadmilling of the filaments as the FRAP experiments are, except that with PAGFP, post-­‐activation fluorescence decay is measured (Honkura et al., 2008; Frost et al., 2010). Contrary to FRAP, where the recovery represents the polymerization rate of the filaments, PAGFP fluorescence decay reflects the depolymerization rate of the filaments. Therefore, the results cannot be directly compared in biological context where prolonged steady state treamilling is rare. Unlike FRAP, the PAGFP fluorescence decay assay cannot be performed by using only the PAGFP-­‐ actin fusion protein. Another fluorophore is needed to delineate the cell morphology (Honkura et al., 2008). It is common practice to use a co-­‐ transfection of a “filler fluorophore” for this purpose (Honkura et al., 2008; Frost et al., 2010). 2.5.3.1 Analysis of PAGFP fluorescence decay data The analysis of the PAGFP fluorescence decay is somewhat similar to FRAP analysis. First the fluorescence intensity of the ROI before activation is deducted from the post-­‐activation values to account for background and channel crosstalk from filler fluorophore (Honkura et al., 2008). After that all the post-­‐activation 19 values are normalized to the first post-­‐activation. Following the normalization the resulting data are commonly fitted to an exponential equation and the time constant for the decay is extracted (Honkura et al., 2008). 2.5.4 Förster resonance energy transfer The gold standard in molecular organization studies is electron microscopy (EM), which enables the visualization of single proteins. EM has provided important insight into the structure of the actin cytoskeleton in dendritic spines, but it is an “end point” –method that requires extensive manipulation of the fixed sample and is therefore not suitable for studies using live cells or tissue (Korobova and Svitkina, 2010). Advanced light microscopy techniques based on the non-­‐radiation energy transfer between fluorophores, FRET (Förster, 1948), have been developed to study actin organization in dendritic spines (Okamoto et al., 2004; Vishwasrao et al., 2012). In FRET, the energy absorbed by the first fluorophore (a donor) in the form of the excitation laser photon is transferred to another fluorophore (an acceptor) in close proximity through dipole-­‐dipole coupling (Piston and Kremers, 2007). A requirement for this energy transfer is that the emission spectra of the donor and the absorption spectra of the acceptor must overlap. The efficiency of the energy transfer is a function of the distance between the fluorophores and follows the equation: 1 𝐸= 𝑟 ! 1+ 𝑅! Equation 1. FRET efficiency, r denotes the distance between the fluorophores and the R0 the Förster distance, the distance where FRET efficiency is 50% (Förster, 1948). From equation 1 it is seen that the efficiency decreases steeply (to the power of 6) when the distance between the fluorophores deviates from the förster-­‐ distance. The distance between molecules that allows for efficient energy transfer is so small that an observed energy transfer can be interpreted as a direct interaction between the molecules (Piston and Kremers, 2007). 2.5.4.1 Methods to measure FRET Several methods have been developed to detect energy transfer between fluorophores in biological samples. Fluorescence intensity method is based on 20 measuring the fluorescence intensity ratio between the donor and acceptor emission wavelengths when only the donor fluorophore is excited (Miyawaki et al., 1997). This is based on the finding that when FRET efficiency is high the donor fluorescence decreases and the acceptor increases. This method requires the use of two different fluorophores as donor and acceptor. Another method to measure energy transfer between different fluorophores is fluorescence lifetime imaging (FLIM) (Oida et al., 1993; Ng et al., 1999). When fluorophores are excited the excitation-­‐state decays through irradiative (photon emission) and non-­‐irradiative (FRET) mechanisms with probabilistic decay rates. In fluorescence lifetime imaging the lifetime of the excited fluorophore is measured from excitation to emission (Oida et al., 1993). Increased FRET efficiency provides higher probability of decay (higher number of potential pathways), which can be seen as a decrease in the fluorescence lifetime. In this method the measurements are only made based on the donor fluorescence and as such are seen as independent of the concentration ratio between the donor and the acceptor (Sun et al., 2013) as well as excitation and emission light intensity. A third method to measure FRET is using fluorescence anisotropy (Krishnan et al., 2001; Tramier et al., 2003; Yan and Marriott, 2003; Bader et al., 2011). Unlike fluorescence intensity based method and FLIM it can be used to measure energy transfer between two similar fluorophores e.g. homoFRET (Krishnan et al., 2001; Bader et al., 2011). In terms of mechanism, homoFRET is similar to FRET between two different fluorophores and is only possible if the fluorophore excitation and emission spectra overlap. The anisotropy is a measure of the ratio of emission light polarizations. The polarization relaxation time of a fluorophore such as GFP is orders of magnitude longer than the fluorescence lifetime (Bader et al., 2011). This means that when a fluorophore is excited with a 100% polarized light, such as a laser, and in response it emits a photon the photon will have retained the polarization of the excitation light. If the energy has been transferred to another molecule via FRET the polarization is lost. In practice the emission light coming from the sample is directed into a polarization splitter and divided into channels parallel and perpendicular to the excitation light (Krishnan et al., 2001). The value of anisotropy can be calculated for any ROI from the intensity images using the following equation: 𝐼∥ − 𝐼! 𝑟!"# = 𝐼∥ + 2𝐼! Equation 2. The 𝐼∥ and 𝐼! denote the fluorescence intensities on the channels parallel (∥) and perpendicular (⊥) to the excitation light (Bader et al., 2011). 21 The measurement of anisotropy differs from fluorescence intensity based-­‐ and FLIM –methods by the use of a single fluorophore. This makes the method unsuitable for studying interactions between two different types of proteins. The polymerization of actin is an interaction between two similar proteins (G-­‐actin monomers). A mathematical model has been created for the study of actin polymerization state with fluorescence anisotropy (Vishwasrao et al., 2012). This method has been tested experimentally in cell cultures and cultured primary neurons (Vishwasrao et al., 2012). 2.5.4.2 Mathematical model for determining actin polymerization state based on fluorescence anisotropy FRET has been used to study the polymerization state of actin in dendritic spines (Okamoto et al., 2004; Vishwasrao et al., 2012). Two different FRET based methods have been used: fluorescence intensity based (Okamoto et al., 2004) as well as fluorescence anisotropy based (Vishwasrao et al., 2012). Both methods have interpreted an increase in FRET efficiency as a shift in the G-­‐/F-­‐actin ratio towards F-­‐actin. This is based on the assumption that energy transfer between labeled actin monomers is possible only if they are a part of the same filament. In FRET, the energy transfer efficiency changes rapidly according to the distance between the fluorophores. In the model for intrafilament FRET the energy transfer has its highest efficiency between positions i and i+2 (Vishwasrao et al., 2012). This corresponds to adjacent monomers in a single strand of a double stranded filament. Efficiency between all other positions is minimal with the FRET efficiency dropping to less than 1% at ranges over 10 nm. In other words, for any observable energy transfer to occur between monomers of the same filament more than 4 out of every 20 monomers would have to be labeled with a fluorophore. In the case of the fluorescence intensity based method the concentrations must be even higher because the energy transfer is only observed between specific donor and acceptor pairs (in fluorescence anisotropy this is not the case since only one type of fluorophore is used). The estimated percentage of the actin monomers labeled with fluorophores in literature is 6% with the maximum at 18% (Westphal et al., 1997). Overall, low expression levels of exogeneous proteins are usually preferred to minimize possible side effects to cell function by the fusion protein. This means that the energy transfer between labeled actin monomers has a low probability to originate from intrafilamentary interaction but instead from interfilamentary interaction, i.e. filament bundling. The authors behind the energy transfer model estimate that in actin-­‐dense compartments, interfilamentary interaction could be the dominant form of energy transfer (Vishwasrao et al., 2012). 22 3 Aims of the study 1. Dendritic spines provide a challenging environment for the study of actin dynamics. The small size of the compartments and the high concentration of actin pose difficulties in using techniques developed in other model systems. New techniques are needed to provide reliable data. In addition, the analysis of the existing ones must be evaluated to guarantee that the data is interpreted in the correct manner. The first aim of this study is to develop and test methods to measure actin filament turnover and organization in dendritic spines (I, II) 2. A decrease in the turnover of dendritic spines results from either spine maturation or following potentiation of a synapse in a mature spine. It has been previously reported that actin dynamics in dendritic spines are not affected by the maturation state of the neurons (Star et al., 2002). Nevertheless, the dynamics are slowed down by synapse potentiation (Star et al., 2002). It is also known that the short-­‐term motility of the dendritic spines decreases during spine maturation (Korkotian and Segal, 2001). All changes to the dendritic spine shape are achieved through rearrangement of the actin cytoskeleton (Dillon and Goda, 2005). Together, these observations suggest that the regulation of the actin cytoskeleton should change during spine maturation and this change should be identifiable. The second aim of this study is to use several complementary methods to identify possible changes in F-­‐actin dynamics during neuron maturation. (II, III) 3. Myosin IIb has been shown to concentrate into dendritic spines during spine maturation (Ryu et al., 2006). Evidence has also pointed to myosin IIb playing a key role in synaptic plasticity (Rex et al., 2010). Myosin IIb regulates the actin cytoskeleton through complicated mechanisms. Evidence from studies of myosin IIb in dendritic spines have provided mixed results from knockdown vs. chemical inhibition studies (Zhang et al., 2005; Ryu et al., 2006; Hodges et al., 2011). The mechanisms of actin regulation by myosin IIb in dendritic spines are not fully known. The third aim of this study is to elucidate the role of myosin IIb in the regulation of the dendritic spine actin cytoskeleton (II, III) 23 4 Experimental procedures For all experiments in these studies I used dissociated cultures of primary hippocampal neurons as a model system. Cells were prepared from E17 Wistar rat fetuses. Cells were transfected using Lipofectamine 2000. The plasmids and siRNA constructs used and their origins are described in the original publications. Detailed descriptions of the methods are found in the original publications. The methods used are summarized in table 3. Table 3. Summary of experimental methods Method Publication Neuronal cell culture I II III Live cell confocal imaging I II III Fixed cell confocal imaging II III Fluorescence recovery after photobleaching II III PAGFP-­‐fluorescence decay I II III Immunocytochemistry II III Fluorescence anisotropy II Motility analysis III Morphological analysis and classification III 24 5 Results 5.1 Development and testing of methods (I, II) The first aim of my study was to develop and test methods to measure actin dynamics and organization in dendritic spines. Here I have made a critical evaluation of the existing techniques FRAP and PAGFP-­‐actin fluorescence decay and proposed ways to improve these techniques. I have also developed a novel application of the fluorescence anisotropy microscopy for measuring actin bundling. 5.1.1 Fluorescence recovery after photobleaching (II, III) FRAP is an easy and fast method to measure F-­‐actin structure turnover in dendritic spines. As the technique is gaining popularity I decided to test the technique and find ways to improve it. The stability of the signal affects the sensitivity of the method directly. When measuring the signal stability I found that there is a rapid fluctuation in the amount of F-­‐actin found in dendritic spines in cultured cells. The GFP-­‐actin fluorescence intensity fluctuates on average 8±1.7% (mean±SD) from the mean value of the spine at 3 min timescale. This result gives a rough guideline to the magnitude of the changes in the stable pool size that can reliably be determined with FRAP. All fluorescence imaging has phototoxic effects on live cells (Magidson and Khodjakov, 2013). In the case of FRAP, a high laser power is needed to rapidly achieve sufficient bleaching. The question is, does this have adverse effects on the cells? The first effect I identified was the increasing fluctuation of fluorescence intensity after the rapid first stage recovery of the fluorescence. The standard deviation of the measurements during the first 100 s of recovery is 19% and 31% between 100-­‐315 s. After 100 s of recovery the spine fluorescence fluctuates at an average of 17% (compared to 8% in non-­‐bleached spine). This indicates that after the first 100 s the fluctuation of the fluorescence becomes too large for reliable measurements and that the FRAP recovery should be followed only the first 100 s. The second effect I observed was the enlargement of spines following photobleaching. Of 12 dendritic spines bleached, nine were enlarged after 300 s 25 recovery (mean enlargement 27% of pre-­‐bleach value). The enlargement of spines was significant and affected only bleached spines. Neighboring spines were not affected. The enlargement of spines can interfere with accurate measurement of the fluorescence intensity inside a ROI covering the whole spine. This should be taken into consideration when performing the analysis. Next I evaluated the FRAP data analysis. Two methods have been used to analyze FRAP data. The normalized data (normalized to non-­‐bleached cell region and to pre-­‐bleach values) from single spines can be fitted to a single exponential component equation with a constant term to correct for stable pool. The resulting values for the stable fraction size and dynamic fraction time constant can subsequently be averaged. The high fluctuation of fluorescence levels degrades the fitting accuracy and leads to a large variation in time constants and possibly negative stable fractions. The regular residuals from the fits range from 40% to -­‐40% with a mean value of absolute residuals of 5.5%. The high variation leads to a high n requirement to detect small differences in time constants and negates the benefits of fast and easy experimentation. The other analysis method used in the literature is to use normalized mean recovery data from several experiments. First a linear approximation of the stable component starting at t > 5 times the estimated time constant of the dynamic component is done manually (at t > 5 τ the dynamic pool has reached over 99% recovery). Then, by using the curve between the first measurement and the stable component, the recovery half time is determined and the rate constant is calculated according to equation 3. ln (2) 𝑘!"# = 𝑡! ! Equation 3. Rate constant of the fluorescence recovery. This method uses averaged data, which relieves the analysis from the problems posed by high fluctuation but does not provide the possibility of statistical analysis. Analyzing the same FRAP data set with both methods led to the following results: individual curves fitted to equation gave a time constant of 17±10 s and a stable pool of -­‐7.8±28% (mean±SD). Manual determination of stable component and halftime led to a time constant of 14 s and a stable pool of 8.5%. By using the manual method the overshooting of the fluorescence can be accounted for in the analysis. This way a more reliable estimation of the stable pool size can be achieved compared to curve fitting. 26 The possibilities to use other methods such as Kullback-­‐Leibler divergence test (Kullback and Leibler, 1951) to probe the similarity of two datasets should be investigated as a possible analysis method. 5.1.2 Photo-­‐activatable GFP fluorescence decay (I, II, III) One way to avoid the use of potentially harmful high laser power needed for photobleaching is to use photo-­‐activatable molecules. Photo-­‐activatable GPF-­‐ fluorescence decay shares many properties with FRAP. I Tested PAGFPs suitability for studying actin dynamics and its differences and similarities compared to FRAP. The main functional difference is that in PAGFP fluorescence decay the turnover rate is estimated from the release of the activated monomers from the filaments and diffusion outside the ROI. In a steady state situation the polymerization at the barbed end equals the depolymerization rate at the pointed end. Results between FRAP and PAGFP fluorescence decay from similar preparations are rarely similar. This is due to the constant changes in polymerization rates of F-­‐actin in dendritic spines. This can be observed as the fluctuating baseline fluorescence of GFP-­‐actin in spines. Therefore the absolute values for turnover rate between FRAP and PAGFP-­‐actin fluorescence decay experiments are not directly comparable. A key difference in the imaging protocols between FRAP and PAGFP fluorescence decay is the energy of the laser needed for PAGFP activation: only a brief (<1 s scan) single pulse with a moderately powered 405 nm laser is needed to activate the PAGFP molecule and increase the fluorescence 100 times. The resulting dose of radiation to the sample is significantly smaller and the adverse effects of the illumination are reduced. No spine enlargement was observed in PAGFP-­‐actin activated spines. The decay of the activated PAGFP fluorescence is very stable with only 2.2% standard deviation of the values from similarly sized dendritic spines measured for ~20 min. Furthermore the recovering fluorescence signal does not have to compete with the fluctuating baseline since the rapid activation leads to a “freeze frame” of the F-­‐actin, after which depolymerization/disassembly is followed. The signal of PAGFP-­‐actin fluorescence can be separated from background at a reasonable signal to noise ratio for a considerably longer time compared to FRAP. The PAGFP-­‐actin fluorescence decay can therefore be used to measure the properties of the stable F-­‐actin pool in dendritic spines. For analysis purposes, the raw PAGFP-­‐actin fluorescence decay data is first background-­‐subtracted and then normalized to the first post-­‐activation fluorescence intensity value. The longer recordings can be used to probe the data set to identify the number of differentially regulated pools of actin. This can be done by fitting the mean data to a number of equations having a different 27 number of exponential decay components. The fits can be evaluated based on the distribution of the residuals. A good fit has normally distributed residuals that are randomly distributed along the time axis. Also the physiological significance should be taken into account. More components will almost always lead to a “better fit” but this can easily lead to overfitting and an over-­‐complication of the model. Usually the least number of components that gives a satisfactory fit should be used. Following these guidelines led me to conclude that, in the PAGFP-­‐actin fluorescence decay experiments in the present study, the most accurate and relevant number of parameters for the PAGFP-­‐actin signal decay is two. This is in accord with previously published data (Honkura et al., 2008). The stable fluorescence and long recordings make individual measurements of PAGFP fluorescence decay data suitable for fitting into an equation. The regular residuals for the fits range from -­‐4% to 4% with a mean absolute value of 1.1%, a five-­‐fold difference to FRAP. The suitability of the individual experiment data for fitting to an equation gives rise to the possibility of studying the dendritic F-­‐actin individually in single spines. This method of analysis should be preferred especially in light of the reported finding that the size of the stable pool of F-­‐actin is correlated to spine size (Honkura et al., 2008). By treating spines as individuals, more parameters and correlations with F-­‐actin properties can be tested. These results indicate that PAGFP-­‐actin fluorescence decay is well suited to studying actin dynamics in dendritic spines, is suitable for long recordings and allows for analysis of single spine data. Compared to FRAP, the PAGFP-­‐actin has better signal to noise ratio and poses less risk of phototoxic effects but is also slower to perform and optimize. 5.1.3 Fluorescence anisotropy can be used to study the bundling of actin filaments in dendritic spines (II) Based on the mathematical model of Vishwasrao et al (2012) I hypothesized that the fluorescence anisotropy can be used to measure actin bundling instead of actin polymerization. The suitability of the fluorescence anisotropy assay to measure actin bundling was tested with U2OS cells transfected with free GFP (no interaction), GFP-­‐GFP tandem (optimized interaction) (Dopie et al., 2012), GFP-­‐ actin and GFP-­‐actin + Palladin, an ABP known to increase bundling (Rachlin and Otey, 2006; Dixon et al., 2008). The whole cell anisotropy values were calculated according to equation 2. The results are considered as border values: 0.27 for free GFP and 0.16 for optimized interaction. The GFP-­‐actin transfection led to similar whole cell anisotropy levels as free GFP (0.25 vs. 0.27). The GFP-­‐actin + Palladin on the other hand exhibited significantly lower anisotropy levels (0.22) 28 compared to GFP-­‐actin alone. In the fluorescence anisotropy images, only the most tightly bundled actin structures, the stress fibers, were visible. To test the method further, GFP-­‐actin fluorescence anisotropy images were first acquired from living cells. The cells were subsequently fixed and the F-­‐actin labeled with Alexa-­‐594-­‐tagged phalloidin and imaged. From the images it can be seen that the cell cortex, which contains a dense mesh of actin filaments that are not bundled, does not exhibit lower anisotropy values. The tightly bundled filaments containing stress fibers, on the other hand, showed lower values of anisotropy. This indicates that the energy transfer originating from intrafilament interactions is below the detection limit in cells of low to moderate expression levels of GFP-­‐actin and the observed energy transfer is mainly a result of interfilamentery interactions i.e. filament bundling. The method was further used to investigate the levels of F-­‐actin bundling in dendritic spines of DIV14 and DIV21 rat hippocampal neurons as well as DIV14 rat hippocampal neurons transfected with various myosin IIb constructs. Results of these experiments are presented in 5.3 and 5.4. 5.2 Changes in actin dynamics during the maturation of dendritic spines (II, III) Previous studies have found no changes in actin dynamics during spine maturation despite stabilization of spines (Star et al., 2002). The second aim of my study was to shed light on this contradiction. To investigate the dynamics of the actin cytoskeleton during spine maturation, FRAP and PAGFP-­‐actin fluorescence decay experiments were performed on DIV14 and DIV21 hippocampal neurons prepared from E17 rats. Only mature mushroom shaped spines were included in the experiments. With FRAP, I found that the stable fraction was significantly larger in DIV21 neuron spines compared to DIV14 (22±2.4% vs. 9.5±2.2%) (mean±SEM). Furthermore, the rate constant of the dynamic pool turnover rate was significantly larger in DIV21 compared to DIV14 spines (0.10±0.01 s-­‐1 vs. 0.057±0.004 s-­‐1) (mean±SEM). With PAGFP-­‐actin fluorescence decay, the enlarged stable fraction was observed (30±2.5% vs. 18±3.5%) (mean±SEM) but no significant difference was found in the turnover rates of neither the dynamic nor the stable pool. The inability to detect the difference in the turnover rate of the dynamic pool with the PAGFP-­‐actin fluorescence decay could be due to the lower sampling frequency used in the PAGFP-­‐actin experiments to avoid unnecessary bleaching during the longer recording. It has been previously reported that the size of the stable fraction is correlated with the size of the spine (Honkura et al., 2008). To make sure that my results were not merely artifacts of larger spine size in older neurons, I tested for correlation between spine size and stable pool size in individual spines form the 29 PAGFP-­‐actin experiments as well as between populations from the FRAP experiments. Correlation was found between head diameter and stable fraction size in individual spines, corroborating previously published results (Honkura et al., 2008). On the other hand, no correlation was found between populations indicating that the results are not caused by changes is spine size but actual changes in the regulation of the actin cytoskeleton during spine maturation. Fluorescence anisotropy measurements showed that the F-­‐actin in dendritic spines was more tightly bundled in DIV21 neurons compared to DIV14 (0.21 vs 0.25). 5.3 Myosin IIb has a dual role in regulating actin dynamics in dendritic spines (II, III) The third aim of my study was to elucidate the molecular mechanism of actin regulation by myosin IIb in dendritic spines. The effect of myosin IIb expression and contractility was studied using GFP-­‐actin FRAP following co-­‐transfection with several myosin IIb wild-­‐type-­‐ as well as mutant-­‐overexpression constructs. Myosin IIb overexpression was found to increase the stable pool of F-­‐actin in DIV14 hippocampal neurons without affecting the dynamic pool turnover rate (from 9.5±2.2% to 30±4.1%) (mean±SEM). The siRNA-­‐mediated knockdown of myosin IIb on the other hand decreased the size of the stable F-­‐actin pool (from 9.5±2.2% to 1.7±1.1%) (mean±SEM). Rescue of the siRNA-­‐mediated knockdown with overexpression of the wild type MHC IIb recovered the stable pool to control level. The increase of F-­‐actin stable pool is not dependent on myosin IIb contractility: the overexpression of a MHC IIb R709C mutant, which binds actin but cannot contract, resulted in a similar increase in the stable pool as the overexpression of wild-­‐type MHC IIb. Inhibiting the myosin II contraction with the drug blebbistatin (Straight et al., 2003) did not affect the stable pool size but did slow down the turnover rate of the dynamic F-­‐actin pool (from 0.076±0.01 s-­‐1 to 0,048±0.007 s-­‐1) (mean±SEM). Activation of myosin II contractility with the phosphatase inhibitor calyculin A (Sassa et al., 1989) increased the dynamic F-­‐ actin pool turnover without affecting the stable pool size (from 0.057±0.004 s-­‐1 to 0.10±0.02 s-­‐1) (mean±SME). As a protein phosphatase 1 and 2 (PP1 and PP2) inhibitor, calyculin A can also lead to higher levels of phosphorylated cofilin. The effects of increased cofilin phosphorylation on dendritic spine F-­‐actin would have the opposite effect on actin turnover rate (Hotulainen et al., 2009), which supports the view that the effects are mediated solely through myosin II activation. Overexpression of the constitutively active MLC20 mutant, MLC20-­‐DD, 30 also led to an increase in the turnover rate of the dynamic pool of F-­‐actin (from 0.057±0.004 s-­‐1 to 0.10±0.02 s-­‐1) (mean±SEM) without affecting the stable pool size. Overexpression of the constitutively inactive mutant MLC20-­‐AA did not have a significant effect on either stable pool size or dynamic pool turnover. Fluorescence anisotropy measurements showed that overexpression of wild-­‐ type MHC IIb as well as the non-­‐contractile mutant R709C led to an increase in F-­‐ actin bundling in dendritic spines (from 0.25 to 0.20 and 0.21 respectively). 31 6 Discussion The best available methods to measure actin dynamics and organization in live cell cultures or tissue samples are based on advanced fluorescence microscopy. As these methods are gaining popularity it is vital to continually evaluate the methods and data analysis to guarantee the reliability of the data. The bulk kinetic methods estimate the F-­‐actin structure turnover rate using the rate of either the incorporation of new monomers from outside the ROI into the filaments inside the ROI (FRAP) or the depolymerization of the activated monomers and diffusion outside of the ROI (PAGFP-­‐actin) (FIG 3). Figure 3 FRAP and PAGFP fluorescence decay. A) FRAP experiment. Before bleaching all fluorescent molecules emit light when excited. The fluorescent molecules are bleached with a high-­‐power laser. Immediately after the bleaching all of the fluorescent proteins incorporated into the network are incapable of emitting light when excited. During the recovery, the bleached monomers in the filaments are replaced with new fluorescent monomers diffusing into the spine from the outside while the depolymerized bleached ones diffuse out of the spine. Eventually only the most stable filaments still contain bleached monomers which will also depolymerize and diffuse out of the spine in time. B) Before activation the PAGFP molecules fused to actin do not emit light when excited. After a brief pulse of 405 nm light the molecules are activated and emit light when excited. During the decay the activated monomers depolymerize and diffuse out of the spine. After all the filaments belonging to the dynamic pool of actin have turned over so that all of the activated monomers have been exchanged with non-­‐activated ones only the most stable filaments still contain light-­‐emitting monomers. This decay can be followed until the fluorescence intensity cannot be separated from the background fluorescence. G-­‐actin has been omitted from the spines for clarity. 32 When using the whole spine as the ROI there is no need to consider diffusion of the filaments. Nevertheless, the view of actin turnover as solely originating from filament treadmilling is based on the assumption that the F-­‐actin structures inside the ROI are persistent within the timescale of the measurements and that there is no rebinding of the depolymerized monomers into existing filaments before diffusing outside of the ROI. Evidence from studies of cofilin (Hotulainen et al., 2009) and Arp2/3 (Kim et al., 2013) both of which do not directly regulate the polymerization/depolymerization of the F-­‐actin but rather the stability of the F-­‐actin structures suggests that the assembly and disassembly of the filaments probably plays key roles in the rate of actin structure turnover. Studies from motile cell types have suggested that filament polymerization and depolymerization alone cannot explain the turnover of the actin structures at the lamellipodia (Smith et al., 2013). A third type of actin structure (besides F-­‐ and G-­‐actin), a slow-­‐diffusing oligomer that is cleaved from the filaments and rebinds existing filaments has been suggested as a solution to the discrepancies between single molecule tracking and bulk kinetic measurements (Smith et al., 2013). It is clear that FRAP experiments suffer from phototoxic effects more than methods that employ milder illumination. The enlargement of the spines can often be seen in the FRAP data as the fluorescence intensity shooting over the pre-­‐bleach values. High-­‐power laser illumination has been found to cleave F-­‐ actin resulting in a rapid increase in new barbed ends and resulting in an increase in net polymerization (Jacobson et al., 2008; Reymann et al., 2011). The increase in polymerization would explain the enlargement of the spines. The high fluctuation of the fluorescence could be seen as overshooting while trying to regain homeostasis. The enlargement of the spines and the fluctuating baseline complicate the analysis and reduce the sensitivity of the method. Only the most robust effects in the size of the stable pool and dynamic pool turnover rate can be reliably detected using FRAP experiments. A better method is to use the PAGFP-­‐actin fluorescence decay measurements. The use of PAGFP has certain key advantages. First, the laser power needed is much smaller as compared to FRAP and the activation time is shorter. Also there is practically no baseline fluctuation to deal with, as only the disassembly of filaments present during very brief activation is visualized. This allows for long, stable, recordings to be performed. This also leads to the possibility to analyze spines individually, maybe the biggest advantage of the PAGFP compared to FRAP. More and more evidence is accumulating that spines are in fact individual and their function and stability are determined by many parameters. The use of fluorescence anisotropy to study actin bundling is a novel application of an established technique. The results and conclusions presented here should also be used to evaluate the previously published data. Okamoto et al. (2004) 33 used FRET to measure actin polymerization in dendritic spines. They observed an increase in energy transfer following LTP induction. This increase was interpreted as an increase in actin polymerization and, further, as a shift in the G-­‐ /F-­‐actin ratio towards F-­‐actin. The important question is, does this FRET signal truly originate solely from intrafilament interactions? The authors of the model for intrafilament energy transfer in F-­‐actin conclude that, for intrafilament FRET to occur with a high probability, the concentration of the labeled actin has to be high and that, in compartments of high actin concentration, interfilament FRET could be a dominant source of energy transfer (Vishwasrao et al., 2012). The percentages of exogenous actin found in transiently transfected cells are considered low (Westphal et al., 1997). Dendritic spines have a very high concentration of F-­‐actin (Drenckhahn et al., 1984; Honkura et al., 2008). Furthermore, it has been reported that the concentration of actin in dendritic spines following LTP induction rises sharply but is quickly (in seconds) restored to pre-­‐induction levels in proportion to spine size (Bosch et al., 2014). Additionally, it has been shown that: A) there is a fast diffusion of G-­‐actin between the dendritic shaft and the spine that keeps the concentration of G-­‐actin in spines and the dendritic shaft similar (Star et al., 2002; Honkura et al., 2008). B) The total actin concentration in the spine does not permanently change following LTP induction. C) The increase in energy transfer observed by Okamoto et al., (2004) happens at a timescale of tens of minutes. Thus, it is plausible that at least some of the observed increase in energy transfer is a result of interfilament interactions i.e. actin filament bundling. More research is still needed but the possibility of interfilament interactions should be taken into consideration when interpreting FRET data. It has been previously reported that no change in actin dynamics was observed during spine maturation (Star et al., 2002). This is in contradiction with spine stabilization following synapse potentiation (Star et al., 2002; Honkura et al., 2008). I have used several complementary methods to study the dynamics and organization of F-­‐actin in dendritic spines during spine maturation and the results indicate that the stable F-­‐actin pool in fact becomes larger, the turnover of the dynamic pool becomes faster and the F-­‐actin bundling increases. Why then do these results differ form those previously published? The only obvious difference between my studies and those of Star et al., (2002) is the model system used. I used dissociated cultures prepared from E17 rats, whereas the cultures used by Star et al., (2002) were prepared from postnatal day 1-­‐2 rats (P1-­‐P2). It is possible that the cells in the cultures used by Star et al., (2002) had already passed some developmental stage after which the differences in maturation state were not observable anymore. It has also recently been shown that the size of the stable pool of F-­‐actin is correlated to spine size. More mature cultures usually have a higher proportion of larger spines. This does not explain 34 the difference in results by Star et al., (2002). If the spines measured by them have followed the distribution of the spine sizes in different aged cultures, the enlargement of the stable pool size following maturation should have been even greater than when using similarly sized spines. In my work I selected only mature mushroom shaped spines of similar dimensions for the analysis, in order to rule out spine size as a source of the difference in the stable pool size. The results showed that even with no difference in spine size the more mature spines contained a larger stable F-­‐actin pool. This indicates that the process of spine maturation leads to changes in F-­‐ actin that cannot be solely explained by activity-­‐dependent plasticity following synaptic potentiation and the accompanying spine enlargement. What the exact molecular differences between an immature and mature spine are and to what extent spine maturation is dependent on synaptic activity is not currently known. The enlargement of the stable F-­‐actin fraction supports the findings of increasing spine stability. The increase in the turnover rate of the dynamic fraction can also be seen as means to control spine shape more accurately and still provide possibilities for a rapid change in morphology following synaptic activity. The logical next stages for this study would be to first repeat the experiments in a model system more closely resembling the physiological environment i.e. tissue slices and/or in vivo. Furthermore, the differences in synaptic transmission of single synapses from neurons of different maturation states should be measured to identify possible developmental changes in the form-­‐ function relationship of the dendritic spines. Myosin IIb has been shown to play a crucial role in LTP stabilization as well as memory consolidation (Rex et al., 2010). In the same study it was shown that the activation of myosin IIb contractility was increased following LTP induction. Rex et al (2010) proposed a model where myosin IIb is an upstream regulator of actin polymerization. The role of myosin IIb as an actin-­‐polymerizing agent has not been reported elsewhere. Myosin IIb-­‐induced filament depolymerization has been shown to be important for cell motility (a process dependent on F-­‐actin polymerization) (Wilson et al., 2010). The myosin IIb-­‐induced depolymerization could theoretically facilitate actin polymerization by replenishing the G-­‐actin pool in motile cells. Nevertheless, in the case of dendritic spines the diffusion of G-­‐actin in and out of spines is vey fast and the increase in actin following LTP (Honkura et al., 2008; Bosch et al., 2014) induction speaks in favor of a recruitment of new G-­‐actin into the filaments from outside the spine, rather than monomer recycling inside the spine. 35 Myosin IIb has been shown to regulate actin architecture via actin binding (stabilization) and actomyosin contractility and filament buckling (disassembly) in vitro (Reymann et al., 2012). The existence of two distinct regulatory mechanisms could explain the differences in results obtained by knockdown and chemical inhibition studies (Zhang et al., 2005; Ryu et al., 2006; Rex et al., 2010; Hodges et al., 2011). I have used wild-­‐type and mutant constructs of myosin IIb, as well as chemical inhibitors and activators to study the role of myosin IIb in actin regulation in dendritic spines. Indeed, the results point to two distinct modes of action in actin regulation by myosin IIb. Myosin IIb stabilizes actin by binding to it. The stabilization could be a result of blocking access of depolymerizing/filament cleaving proteins. This stabilization leads to an enlargement of the stable pool of F-­‐actin without affecting the turnover rate of the dynamic pool. The stabilizing effect is not dependent on myosin IIb contractility. On the contrary, myosin IIb contractility does not affect the stable pool size of F-­‐actin in spines but it instead facilitates the turnover of the dynamic pool. I therefore propose a different model for myosin IIbs role in the regulation of dendritic spine actin. Instead of functioning as a polymerizing agent in spines, myosin IIb regulates F-­‐actin via two mechanisms. First, it stabilizes the stable F-­‐ actin pool of correctly aligned filaments via actin binding. At the same time, it buckles and depolymerizes filaments at the spine periphery controlling the spine shape. Following LTP induction the contractility is increased by MLC20 phosphorylation. This leads to the reorganization of the F-­‐actin structures inside the spine so as to accommodate the new spine size and shape. The new stable F-­‐ actin pool is subsequently stabilized by myosin IIb binding. This “dual approach” mechanism of myosin IIb is supported by in vitro experiments on myosin IIb function by Reyman et al. (2012). To corroborate this model of myosin IIb’s role in actin regulation, myosin IIbs role should be studied in conjunction with synaptic activation. Using methods for LTP induction at single synapses (e.g. MNI-­‐glutamate uncaging) together with genetic and chemical manipulation of myosin IIb expression and function, the possible recruitment, activation and deactivation of myosin IIb could be investigated in real-­‐time. Results of these experiments could prove valuable in determining the accuracy of the model. 36 7 Conclusions Advanced imaging techniques have helped shed light on the complex dynamics and interactions of the actin cytoskeleton in dendritic spines. The more we learn about actin dynamics in dendritic spines, the more important the correct regulation of actin seems to become. Therefore, the methods used to measure the dynamics need to be constantly evaluated and refined to reliably detect changes. Several complementary methods should be used whenever possible to support interpretation of the data. The imaging techniques taking advantage of the energy transfer between fluorophores (FRET) offer many interesting possibilities to study molecular interaction. Proper controls and sufficient optimization of the techniques is crucial for these types of experiments. The use of fluorescence anisotropy to measure actin bundling is a novel method that, although theoretically sound, still needs more experimental characterization. These experiments should include in vitro bundling assays as well as further in vivo evaluation to determine the proportions of intra-­‐ and interfilament energy transfer along the labeling density continuum. At the time being, the results of fluorescence anisotropy should be considered in the context of other data concerning actin dynamics and organization. The results presented here shed light on yet another small piece of the machinery regulating the actin cytoskeleton in dendritic spines. The results indicate that, contrary to previous reports, the dynamics of F-­‐actin in dendritic spines change following spine maturation. The size of the stable pool increases, actin bundling increases and the turnover of the dynamic pool increases. Furthermore, these changes are independent of the increased average spine size of more mature cultures. Whether some intrinsic maturation process of dendritic spines exists or if the maturation is solely a result of synaptic activity patterns is not known. Recent results studying Arp2/3 knockout mice suggest that disturbed actin dynamics in dendritic spines in early life can lead to complications in brain function considerably later during aging (Kim et al., 2013). These results taken together with the data presented here raise interesting questions about the possible long-­‐lasting effects of the actin cytoskeleton dynamics on brain function. If actin dynamics are developmentally regulated, are the effects of disturbed regulation different at different developmental stages? If (for example) myosin IIb activity is compromised during development, how much can the other isoforms of myosin II compensate? How much can the regulation in general be compromised before pathological complications occur? How much can be 37 rescued by plastic adaptation? What are the physiological effects of disturbed actin regulation in a timespan many times the turnover time of the post-­‐synaptic proteome, possibly even the lifetime of an individual? These are all relevant and interesting questions for the future studies that need to be answered for us to better understand the molecular workings of neurons. Answers to these questions will also help us to better identify the mechanisms behind neurological diseases and mental health problems and therefore help create better means for prevention and therapy. 38 Acknowledgements As I sit here, on the last night before the printing deadline, I realize that I never started to write this chapter in time, like I had planned to. I wanted to be careful, after all, it’s not often one gets to express gratitude in writing. It’s late and I’m tired but I’m happy to realize I’ve had no problems to remember all of you. On the contrary I seem to be running out of space…. First I would like to acknowledge the people contributing to this book: Carl Gahmberg and Eleanor Coffey for reviewing the thesis and my follow up group: Heikki Rauvala and Urmas Arumäe. The ones who paid the bills deserve also to be acknowledged: Helsinki biomedical graduate school, Neuroscience Center and the University of Helsinki for financing this study. My supervisor, Pirta, I have you to thank for a lot more than helping me to make this book a reality. You believed in me and gave me a change when many might not have done it. I admire your patience, which you showed by gently guiding me while I navigated out of the dark hole I had dug myself in. It must not have been easy at times. In the end I think we did ok. The next ones to thank are of course the members of our group. Enni, in the beginning there were just Pirta, you and me, and a lot has happened since those days. We have shared countless cups of coffee together, and discussed more topics that I care to remember. Outside of work we share the passion towards the vertical world, and at times I have been privileged to tie into a rope with you. Still, I like the fact that our friendship has not been just swapping climbing stories. I have grown to value your quick wit and relaxed attitude. You have grown quite dear to me during the years. Now the group is bigger with Amr, Rimante, Iryna, Jaakko and Jonas. I feel lucky to be a part of this wonderful bunch and a bit sorry for the times when you had to listen to my rants… Moving further back, I want to thank all the wonderful people from my previous lab. Minna, Noora, Ansku, Sandra, Kirsi and everyone else, we went through a lot, but I hope you agree that we made some good friends too. I would also want to acknowledge professors Frej Fyhrquist and Jim Schröder for their help during a difficult time. Even further back is my time at the Stockholm University. I met a lot of nice people, made friends and had good times there. I’d especially want to thank my friend and lab partner Charlotte whose career in sports I have followed with interest and admiration. Another person receiving my utmost gratitude is Samir, 39 who took me under his wing in Stockholm and was the first to show me that it can be cool to be a scientist! I would also like to thank my family. My parents: Liisa and Pekka, without their unconditional and continues support I would not have been able to do science, and my siblings Matti and Minna. I would also like to extend my gratitude to the Fagerstedt family: Mikael, Nergis and Lotta, for all their help along the way. The publication of this thesis marks an end of an era, kind of a culmination of all the interactions I have had in the scientific community. But not all of life revolves around the lab, and I would never have completed this without that balance. I would like to thank the people that I share the world of physical unpleasantness with: Antti, we’ve shared countless meters together, both training in the horizontal and putting it to use in the vertical. I hope that we have many more bouts of “big game hunting” before us! All the other wonderful people I get to climb, bike, run, ski, whatever with: Markus, Nicolas, Tuomas, Iso Antti, Pirre, Kimpa, Per, Fredrik, Robban and everybody else! Suffer now, summit later! All my dear friends: Jussi, I miss our talks and all the good times, let’s go do something stupid! Simo, for being there when it counts, no questions asked. Mikko “Captain America”, for slightly intelligent conversations and being the most irritating person on the Internet. The whole of the almighty Sontalaatikko: Mikko, Henri, Teemu, Matti, Antti and everybody else. Uosu and the BMC, Sami, and everyone from the Finnish hardcore/punk scene I know. My brothers in arms: Mika, (We’re both in science now, still sometimes it feels like not much has changed), Tuomas, Tero, Juha, Juha, Juuso, Kimmo, Elmo and everyone. If I ever find myself in harms way again, I hope you’ll be by my side. Last but definitely not the least: Sara, you have taught me a lot. We have experienced so much together and I hope we get to do a lot more. I don’t care if it is mountain summit or just a cup of coffee in the morning that I get to share with you because that sharing is what makes it so special for me. I love you. I’ve met so many wonderful people in my life that unfortunately it’s impossible for me to list all here. So if you think your name should be here, you’re probably right. Here is some empty space so you can add yourself in: Cheers, /Mikko 40 References Axelrod, D., Koppel, D.E., Schlessinger, J., Elson, E., and Webb, W.W. (1976). Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 16, 1055-­‐1069. doi: 10.1016/S0006-­‐ 3495(76)85755-­‐4. Bader, A.N., Hoetzl, S., Hofman, E.G., Voortman, J., Van Bergen En Henegouwen, P.M., Van Meer, G., and Gerritsen, H.C. (2011). Homo-­‐FRET imaging as a tool to quantify protein and lipid clustering. Chemphyschem 12, 475-­‐483. doi: 10.1002/cphc.201000801. Bellot, A., Guivernau, B., Tajes, M., Bosch-­‐Morató, M., Valls-­‐Comamala, V., and Muñoz, F.J. (2014). The structure and function of actin cytoskeleton in mature glutamatergic dendritic spines. Brain Research 1573, 1-­‐16. doi: http://dx.doi.org/10.1016/j.brainres.2014.05.024. Bertling, E., Ludwig, A., Koskinen, M., and Hotulainen, P. (2012). Methods for three-­‐dimensional analysis of dendritic spine dynamics. Methods Enzymol 506, 391-­‐406. doi: 10.1016/B978-­‐0-­‐12-­‐391856-­‐7.00043-­‐3. Betzig, E., Patterson, G.H., Sougrat, R., Lindwasser, O.W., Olenych, S., Bonifacino, J.S., Davidson, M.W., Lippincott-­‐Schwartz, J., and Hess, H.F. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642-­‐1645. doi: 10.1126/science.1127344. Bhatt, D.H., Zhang, S., and Gan, W.B. (2009). Dendritic spine dynamics. Annu Rev Physiol 71, 261-­‐282. doi: 10.1146/annurev.physiol.010908.163140. Blanchoin, L., Boujemaa-­‐Paterski, R., Sykes, C., and Plastino, J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94, 235-­‐263. doi: 10.1152/physrev.00018.2013. Bosch, M., Castro, J., Saneyoshi, T., Matsuno, H., Sur, M., and Hayashi, Y. (2014). Structural and molecular remodeling of dendritic spine substructures during long-­‐term potentiation. Neuron 82, 444-­‐459. doi: 10.1016/j.neuron.2014.03.021. Bourne, J.N., and Harris, K.M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31, 47-­‐67. doi: 10.1146/annurev.neuro.31.060407.125646. Bugyi, B., and Carlier, M.F. (2010). Control of actin filament treadmilling in cell motility. Annu Rev Biophys 39, 449-­‐470. doi: 10.1146/annurev-­‐biophys-­‐ 051309-­‐103849. 41 Carlsson, L., Nyström, L.E., Sundkvist, I., Markey, F., and Lindberg, U. (1977). Actin polymerizability is influenced by profilin, a low molecular weight protein in non-­‐muscle cells. Journal of Molecular Biology 115, 465-­‐483. doi: http://dx.doi.org/10.1016/0022-­‐2836(77)90166-­‐8. Chazeau, A., Mehidi, A., Nair, D., Gautier, J., Leduc, C., Chamma, I., Kage, F., Kechkar, A., Thoumine, O., Rottner, K., Choquet, D., Gautreau, A., Sibarita, J-­‐ F., and Giannone, G. (2014). Nanoscale segregation of actin nucleation and elongation factors determines dendritic spine protrusion. EMBO J. In Press. Chelly, J., Khelfaoui, M., Francis, F., Cherif, B., and Bienvenu, T. (2006). Genetics and pathophysiology of mental retardation. Eur J Hum Genet 14, 701-­‐713. doi: 10.1038/sj.ejhg.5201595. Chen, J.L., Villa, K.L., Cha, J.W., So, P.T., Kubota, Y., and Nedivi, E. (2012a). Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 74, 361-­‐373. doi: 10.1016/j.neuron.2012.02.030. Chen, Q., Nag, S., and Pollard, T.D. (2012b). Formins filter modified actin subunits during processive elongation. J Struct Biol 177, 32-­‐39. doi: 10.1016/j.jsb.2011.10.005. Chen, X., Leischner, U., Rochefort, N.L., Nelken, I., and Konnerth, A. (2011). Functional mapping of single spines in cortical neurons in vivo. Nature 475, 501-­‐505. doi: 10.1038/nature10193. Choidas, A., Jungbluth, A., Sechi, A., Murphy, J., Ullrich, A., and Marriott, G. (1998). The suitability and application of a GFP-­‐actin fusion protein for long-­‐term imaging of the organization and dynamics of the cytoskeleton in mammalian cells. Eur J Cell Biol 77, 81-­‐90. doi: 10.1016/S0171-­‐ 9335(98)80075-­‐7. Cingolani, L.A., and Goda, Y. (2008). Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9, 344-­‐356. doi: 10.1038/nrn2373. Dailey, M.E., and Smith, S.J. (1996). The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16, 2983-­‐2994. Deibler, M., Spatz, J.P., and Kemkemer, R. (2011). Actin fusion proteins alter the dynamics of mechanically induced cytoskeleton rearrangement. PLoS One 6, e22941. doi: 10.1371/journal.pone.0022941. Dillon, C., and Goda, Y. (2005). The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28, 25-­‐55. doi: 10.1146/annurev.neuro.28.061604.135757. 42 Dixon, R.D., Arneman, D.K., Rachlin, A.S., Sundaresan, N.R., Costello, M.J., Campbell, S.L., and Otey, C.A. (2008). Palladin is an actin cross-­‐linking protein that uses immunoglobulin-­‐like domains to bind filamentous actin. J Biol Chem 283, 6222-­‐6231. doi: 10.1074/jbc.M707694200. Dominguez, R., and Holmes, K.C. (2011). Actin structure and function. Annu Rev Biophys 40, 169-­‐186. doi: 10.1146/annurev-­‐biophys-­‐042910-­‐155359. Dopie, J., Skarp, K.P., Rajakyla, E.K., Tanhuanpaa, K., and Vartiainen, M.K. (2012). Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci U S A 109, E544-­‐552. doi: 10.1073/pnas.1118880109. Dos Remedios, C.G., Chhabra, D., Kekic, M., Dedova, I.V., Tsubakihara, M., Berry, D.A., and Nosworthy, N.J. (2003). Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83, 433-­‐473. doi: 10.1152/physrev.00026.2002. Drenckhahn, D., Frotscher, M., and Kaiser, H.W. (1984). Concentration of F-­‐actin in synaptic formations of the hippocampus as visualized by staining with fluorescent phalloidin. Brain Research 300, 381-­‐384. doi: http://dx.doi.org/10.1016/0006-­‐8993(84)90851-­‐5. Dunaevsky, A., Tashiro, A., Majewska, A., Mason, C., and Yuste, R. (1999). Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci U S A 96, 13438-­‐13443. Fiala, J.C., Feinberg, M., Popov, V., and Harris, K.M. (1998). Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci 18, 8900-­‐8911. Fischer, M., Kaech, S., Knutti, D., and Matus, A. (1998). Rapid actin-­‐based plasticity in dendritic spines. Neuron 20, 847-­‐854. Förster, T. (1948). Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen der Physik 437, 55-­‐75. doi: 10.1002/andp.19484370105. Fromer, M., Pocklington, A.J., Kavanagh, D.H., Williams, H.J., Dwyer, S., Gormley, P., Georgieva, L., Rees, E., Palta, P., Ruderfer, D.M., Carrera, N., Humphreys, I., Johnson, J.S., Roussos, P., Barker, D.D., Banks, E., Milanova, V., Grant, S.G., Hannon, E., Rose, S.A., Chambert, K., Mahajan, M., Scolnick, E.M., Moran, J.L., Kirov, G., Palotie, A., Mccarroll, S.A., Holmans, P., Sklar, P., Owen, M.J., Purcell, S.M., and O'donovan, M.C. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179-­‐184. doi: 10.1038/nature12929. Frost, N.A., Shroff, H., Kong, H., Betzig, E., and Blanpied, T.A. (2010). Single-­‐ molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron 67, 86-­‐99. doi: 10.1016/j.neuron.2010.05.026. 43 Fu, M., Yu, X., Lu, J., and Zuo, Y. (2012). Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature 483, 92-­‐95. doi: 10.1038/nature10844. Garcia-­‐Lopez, P., Garcia-­‐Marin, V., and Freire, M. (2010). Dendritic spines and development: towards a unifying model of spinogenesis-­‐-­‐a present day review of Cajal's histological slides and drawings. Neural Plast 2010, 769207. doi: 10.1155/2010/769207. Golomb, E., Ma, X., Jana, S.S., Preston, Y.A., Kawamoto, S., Shoham, N.G., Goldin, E., Conti, M.A., Sellers, J.R., and Adelstein, R.S. (2004). Identification and characterization of nonmuscle myosin II-­‐C, a new member of the myosin II family. J Biol Chem 279, 2800-­‐2808. doi: 10.1074/jbc.M309981200. Grutzendler, J., Kasthuri, N., and Gan, W.B. (2002). Long-­‐term dendritic spine stability in the adult cortex. Nature 420, 812-­‐816. doi: 10.1038/nature01276. Gu, J., Lee, C.W., Fan, Y., Komlos, D., Tang, X., Sun, C., Yu, K., Hartzell, H.C., Chen, G., Bamburg, J.R., and Zheng, J.Q. (2010). ADF/cofilin-­‐mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci 13, 1208-­‐1215. doi: 10.1038/nn.2634. Halavatyi, A.A., Nazarov, P.V., Al Tanoury, Z., Apanasovich, V.V., Yatskou, M., and Friederich, E. (2010). A mathematical model of actin filament turnover for fitting FRAP data. Eur Biophys J 39, 669-­‐677. doi: 10.1007/s00249-­‐009-­‐ 0558-­‐2. Harris, K.M., Jensen, F.E., and Tsao, B. (1992). Three-­‐dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-­‐term potentiation. J Neurosci 12, 2685-­‐2705. Hayama, T., Noguchi, J., Watanabe, S., Takahashi, N., Hayashi-­‐Takagi, A., Ellis-­‐ Davies, G.C., Matsuzaki, M., and Kasai, H. (2013). GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat Neurosci 16, 1409-­‐1416. doi: 10.1038/nn.3496. Hess, S.T., Girirajan, T.P., and Mason, M.D. (2006). Ultra-­‐high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 91, 4258-­‐4272. doi: 10.1529/biophysj.106.091116. Hodges, J.L., Newell-­‐Litwa, K., Asmussen, H., Vicente-­‐Manzanares, M., and Horwitz, A.R. (2011). Myosin IIb activity and phosphorylation status determines dendritic spine and post-­‐synaptic density morphology. PLoS One 6, e24149. doi: 10.1371/journal.pone.0024149. 44 Holmes, K.C., Popp, D., Gebhard, W., and Kabsch, W. (1990). Atomic model of the actin filament. Nature 347, 44-­‐49. Holtmaat, A.J., Trachtenberg, J.T., Wilbrecht, L., Shepherd, G.M., Zhang, X., Knott, G.W., and Svoboda, K. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279-­‐291. doi: 10.1016/j.neuron.2005.01.003. Honkura, N., Matsuzaki, M., Noguchi, J., Ellis-­‐Davies, G.C., and Kasai, H. (2008). The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57, 719-­‐729. doi: 10.1016/j.neuron.2008.01.013. Hotulainen, P., and Hoogenraad, C.C. (2010). Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189, 619-­‐629. doi: 10.1083/jcb.201003008. Hotulainen, P., Llano, O., Smirnov, S., Tanhuanpaa, K., Faix, J., Rivera, C., and Lappalainen, P. (2009). Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol 185, 323-­‐339. doi: 10.1083/jcb.200809046. Hughes, J.R. (1958). Post-­‐tetanic potentiation. Physiol Rev 38, 91-­‐113. Jacobson, K., Rajfur, Z., Vitriol, E., and Hahn, K. (2008). Chromophore-­‐assisted laser inactivation in cell biology. Trends Cell Biol 18, 443-­‐450. doi: 10.1016/j.tcb.2008.07.001. Kasai, H., Fukuda, M., Watanabe, S., Hayashi-­‐Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 33, 121-­‐129. doi: 10.1016/j.tins.2010.01.001. Kawamoto, S., and Adelstein, R.S. (1991). Chicken nonmuscle myosin heavy chains: differential expression of two mRNAs and evidence for two different polypeptides. J Cell Biol 112, 915-­‐924. Kim, I.H., Racz, B., Wang, H., Burianek, L., Weinberg, R., Yasuda, R., Wetsel, W.C., and Soderling, S.H. (2013). Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci 33, 6081-­‐6092. doi: 10.1523/JNEUROSCI.0035-­‐13.2013. Kim, K.Y., Kovacs, M., Kawamoto, S., Sellers, J.R., and Adelstein, R.S. (2005). Disease-­‐associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-­‐B and II-­‐C. J Biol Chem 280, 22769-­‐22775. doi: 10.1074/jbc.M503488200. Korkotian, E., and Segal, M. (2001). Regulation of dendritic spine motility in cultured hippocampal neurons. J Neurosci 21, 6115-­‐6124. 45 Korobova, F., and Svitkina, T. (2010). Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell 21, 165-­‐176. doi: 10.1091/mbc.E09-­‐ 07-­‐0596. Krishnan, R.V., Varma, R., and Mayor, S. (2001). Fluorescence Methods to Probe Nanometer-­‐Scale Organization of Molecules in Living Cell Membranes. Journal of Fluorescence 11, 211-­‐226. doi: 10.1023/A:1012201201651. Krucker, T., Siggins, G.R., and Halpain, S. (2000). Dynamic actin filaments are required for stable long-­‐term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A 97, 6856-­‐6861. doi: 10.1073/pnas.100139797. Kullback, S., and Leibler, R.A. (1951). On Information and Sufficiency. 79-­‐86. doi: 10.1214/aoms/1177729694. Lai, C.S., Franke, T.F., and Gan, W.B. (2012). Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 483, 87-­‐91. doi: 10.1038/nature10792. Landis, D.M., and Reese, T.S. (1983). Cytoplasmic organization in cerebellar dendritic spines. J Cell Biol 97, 1169-­‐1178. Lappalainen, P., and Drubin, D.G. (1997). Cofilin promotes rapid actin filament turnover in vivo. Nature 388, 78-­‐82. doi: 10.1038/40418. Lee, S.J., Escobedo-­‐Lozoya, Y., Szatmari, E.M., and Yasuda, R. (2009). Activation of CaMKII in single dendritic spines during long-­‐term potentiation. Nature 458, 299-­‐304. doi: 10.1038/nature07842. Lynen, F., and Wieland, U. (1938). Über die Giftstoffe des Knollenblätterpilzes. IV. Justus Liebigs Annalen der Chemie 533, 93-­‐117. doi: 10.1002/jlac.19385330105. Ma, X., Kawamoto, S., Hara, Y., and Adelstein, R.S. (2004). A point mutation in the motor domain of nonmuscle myosin II-­‐B impairs migration of distinct groups of neurons. Mol Biol Cell 15, 2568-­‐2579. doi: 10.1091/mbc.E03-­‐ 11-­‐0836. Magidson, V., and Khodjakov, A. (2013). Circumventing photodamage in live-­‐cell microscopy. Methods Cell Biol 114, 545-­‐560. doi: 10.1016/B978-­‐0-­‐12-­‐ 407761-­‐4.00023-­‐3. Malenka, R.C., and Bear, M.F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5-­‐21. doi: 10.1016/j.neuron.2004.09.012. 46 Manley, S., Gillette, J.M., Patterson, G.H., Shroff, H., Hess, H.F., Betzig, E., and Lippincott-­‐Schwartz, J. (2008). High-­‐density mapping of single-­‐molecule trajectories with photoactivated localization microscopy. Nat Methods 5, 155-­‐157. doi: 10.1038/nmeth.1176. Matsuzaki, M., Honkura, N., Ellis-­‐Davies, G.C., and Kasai, H. (2004). Structural basis of long-­‐term potentiation in single dendritic spines. Nature 429, 761-­‐766. doi: 10.1038/nature02617. Mattila, P.K., and Lappalainen, P. (2008). Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9, 446-­‐454. doi: 10.1038/nrm2406. Matus, A., Ackermann, M., Pehling, G., Byers, H.R., and Fujiwara, K. (1982). High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A 79, 7590-­‐7594. Megias, M., Emri, Z., Freund, T.F., and Gulyas, A.I. (2001). Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102, 527-­‐540. Miyawaki, A., Llopis, J., Heim, R., Mccaffery, J.M., Adams, J.A., Ikura, M., and Tsien, R.Y. (1997). Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882-­‐887. doi: 10.1038/42264. Mullins, R.D., Heuser, J.A., and Pollard, T.D. (1998). The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A 95, 6181-­‐6186. Nabavi, S., Fox, R., Proulx, C.D., Lin, J.Y., Tsien, R.Y., and Malinow, R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348-­‐352. doi: 10.1038/nature13294. Ng, T., Squire, A., Hansra, G., Bornancin, F., Prevostel, C., Hanby, A., Harris, W., Barnes, D., Schmidt, S., Mellor, H., Bastiaens, P.I., and Parker, P.J. (1999). Imaging protein kinase Calpha activation in cells. Science 283, 2085-­‐2089. Nimchinsky, E.A., Sabatini, B.L., and Svoboda, K. (2002). Structure and function of dendritic spines. Annu Rev Physiol 64, 313-­‐353. doi: 10.1146/annurev.physiol.64.081501.160008. Noguchi, J., Matsuzaki, M., Ellis-­‐Davies, G.C., and Kasai, H. (2005). Spine-­‐neck geometry determines NMDA receptor-­‐dependent Ca2+ signaling in dendrites. Neuron 46, 609-­‐622. doi: 10.1016/j.neuron.2005.03.015. Oida, T., Sako, Y., and Kusumi, A. (1993). Fluorescence lifetime imaging microscopy (flimscopy). Methodology development and application to 47 studies of endosome fusion in single cells. Biophys J 64, 676-­‐685. doi: 10.1016/S0006-­‐3495(93)81427-­‐9. Okamoto, K., Nagai, T., Miyawaki, A., and Hayashi, Y. (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7, 1104-­‐1112. doi: 10.1038/nn1311. Oray, S., Majewska, A., and Sur, M. (2006). Effects of synaptic activity on dendritic spine motility of developing cortical layer v pyramidal neurons. Cereb Cortex 16, 730-­‐741. doi: 10.1093/cercor/bhj019. Ozawa, E. (2010). Our trails and trials in the subsarcolemmal cytoskeleton network and muscular dystrophy researches in the dystrophin era. Proc Jpn Acad Ser B Phys Biol Sci 86, 798-­‐821. Patterson, G.H., and Lippincott-­‐Schwartz, J. (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873-­‐1877. doi: 10.1126/science.1074952. Paunola, E., Mattila, P.K., and Lappalainen, P. (2002). WH2 domain: a small, versatile adapter for actin monomers. FEBS Lett 513, 92-­‐97. Penzes, P., and Vanleeuwen, J.E. (2011). Impaired regulation of synaptic actin cytoskeleton in Alzheimer's disease. Brain Res Rev 67, 184-­‐192. doi: 10.1016/j.brainresrev.2011.01.003. Piston, D.W., and Kremers, G.J. (2007). Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci 32, 407-­‐414. doi: 10.1016/j.tibs.2007.08.003. Pollard, T.D. (1986). Rate constants for the reactions of ATP-­‐ and ADP-­‐actin with the ends of actin filaments. J Cell Biol 103, 2747-­‐2754. Pollard, T.D., and Cooper, J.A. (2009). Actin, a central player in cell shape and movement. Science 326, 1208-­‐1212. doi: 10.1126/science.1175862. Rachlin, A.S., and Otey, C.A. (2006). Identification of palladin isoforms and characterization of an isoform-­‐specific interaction between Lasp-­‐1 and palladin. J Cell Sci 119, 995-­‐1004. doi: 10.1242/jcs.02825. Ramón y Cajal S. 1888. Estructura de los centros nerviosos de las aves. Rev. Trim. Histol. Norm. Patol. 1:1–10 Rao, J.N., Madasu, Y., and Dominguez, R. (2014). Actin cytoskeleton. Mechanism of actin filament pointed-­‐end capping by tropomodulin. Science 345, 463-­‐ 467. doi: 10.1126/science.1256159. 48 Reits, E.A., and Neefjes, J.J. (2001). From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol 3, E145-­‐147. doi: 10.1038/35078615. Rex, C.S., Gavin, C.F., Rubio, M.D., Kramar, E.A., Chen, L.Y., Jia, Y., Huganir, R.L., Muzyczka, N., Gall, C.M., Miller, C.A., Lynch, G., and Rumbaugh, G. (2010). Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67, 603-­‐617. doi: 10.1016/j.neuron.2010.07.016. Reymann, A.C., Boujemaa-­‐Paterski, R., Martiel, J.L., Guerin, C., Cao, W., Chin, H.F., De La Cruz, E.M., Thery, M., and Blanchoin, L. (2012). Actin network architecture can determine myosin motor activity. Science 336, 1310-­‐ 1314. doi: 10.1126/science.1221708. Reymann, A.C., Suarez, C., Guerin, C., Martiel, J.L., Staiger, C.J., Blanchoin, L., and Boujemaa-­‐Paterski, R. (2011). Turnover of branched actin filament networks by stochastic fragmentation with ADF/cofilin. Mol Biol Cell 22, 2541-­‐2550. doi: 10.1091/mbc.E11-­‐01-­‐0052. Riedl, J., Crevenna, A.H., Kessenbrock, K., Yu, J.H., Neukirchen, D., Bista, M., Bradke, F., Jenne, D., Holak, T.A., Werb, Z., Sixt, M., and Wedlich-­‐Soldner, R. (2008). LifeAct: a versatile marker to visualize F-­‐actin. Nature Methods 5, 605-­‐607. doi: 10.1038/nmeth.1220. Roberts, T.F., Tschida, K.A., Klein, M.E., and Mooney, R. (2010). Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature 463, 948-­‐952. doi: 10.1038/nature08759. Rust, M.B., Gurniak, C.B., Renner, M., Vara, H., Morando, L., Gorlich, A., Sassoe-­‐ Pognetto, M., Banchaabouchi, M.A., Giustetto, M., Triller, A., Choquet, D., and Witke, W. (2010). Learning, AMPA receptor mobility and synaptic plasticity depend on n-­‐cofilin-­‐mediated actin dynamics. EMBO J 29, 1889-­‐ 1902. doi: 10.1038/emboj.2010.72. Ryu, J., Liu, L., Wong, T.P., Wu, D.C., Burette, A., Weinberg, R., Wang, Y.T., and Sheng, M. (2006). A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron 49, 175-­‐182. doi: 10.1016/j.neuron.2005.12.017. Sala, C., and Segal, M. (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol Rev 94, 141-­‐188. doi: 10.1152/physrev.00012.2013. Sassa, T., Richter, W.W., Uda, N., Suganuma, M., Suguri, H., Yoshizawa, S., Hirota, M., and Fujiki, H. (1989). Apparent "activation" of protein kinases by okadaic acid class tumor promoters. Biochem Biophys Res Commun 159, 939-­‐944. 49 Sellers, J.R. (2000). Myosins: a diverse superfamily. Biochim Biophys Acta 1496, 3-­‐22. Shaner, N.C., Steinbach, P.A., and Tsien, R.Y. (2005). A guide to choosing fluorescent proteins. Nat Methods 2, 905-­‐909. doi: 10.1038/nmeth819. Shimomura, O., Johnson, F.H., and Saiga, Y. (1962). Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea. Journal of Cellular and Comparative Physiology 59, 223-­‐239. doi: 10.1002/jcp.1030590302. Shizuta, Y., Shizuta, H., Gallo, M., Davies, P., and Pastan, I. (1976). Purification and properties of filamin, and actin binding protein from chicken gizzard. J Biol Chem 251, 6562-­‐6567. Small, J.V. (1981). Organization of actin in the leading edge of cultured cells: influence of osmium tetroxide and dehydration on the ultrastructure of actin meshworks. J Cell Biol 91, 695-­‐705. Smith, M.B., Kiuchi, T., Watanabe, N., and Vavylonis, D. (2013). Distributed actin turnover in the lamellipodium and FRAP kinetics. Biophys J 104, 247-­‐257. doi: 10.1016/j.bpj.2012.11.3819. Stamatakou, E., Marzo, A., Gibb, A., and Salinas, P.C. (2013). Activity-­‐dependent spine morphogenesis: a role for the actin-­‐capping protein Eps8. J Neurosci 33, 2661-­‐2670. doi: 10.1523/JNEUROSCI.0998-­‐12.2013. Star, E.N., Kwiatkowski, D.J., and Murthy, V.N. (2002). Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5, 239-­‐246. doi: 10.1038/nn811. Straight, A.F., Cheung, A., Limouze, J., Chen, I., Westwood, N.J., Sellers, J.R., and Mitchison, T.J. (2003). Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299, 1743-­‐1747. doi: 10.1126/science.1081412. Sun, H.Q., Yamamoto, M., Mejillano, M., and Yin, H.L. (1999). Gelsolin, a multifunctional actin regulatory protein. J Biol Chem 274, 33179-­‐33182. Sun, Y., Rombola, C., Jyothikumar, V., and Periasamy, A. (2013). Forster resonance energy transfer microscopy and spectroscopy for localizing protein-­‐protein interactions in living cells. Cytometry A 83, 780-­‐793. doi: 10.1002/cyto.a.22321. Takahashi, H., Sekino, Y., Tanaka, S., Mizui, T., Kishi, S., and Shirao, T. (2003). Drebrin-­‐dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-­‐95 and dendritic spine morphogenesis. J Neurosci 23, 6586-­‐6595. 50 Tojkander, S., Gateva, G., and Lappalainen, P. (2012). Actin stress fibers-­‐-­‐ assembly, dynamics and biological roles. J Cell Sci 125, 1855-­‐1864. doi: 10.1242/jcs.098087. Tonnesen, J., Katona, G., Rozsa, B., and Nagerl, U.V. (2014). Spine neck plasticity regulates compartmentalization of synapses. Nat Neurosci 17, 678-­‐685. doi: 10.1038/nn.3682. Trachtenberg, J.T., Chen, B.E., Knott, G.W., Feng, G., Sanes, J.R., Welker, E., and Svoboda, K. (2002). Long-­‐term in vivo imaging of experience-­‐dependent synaptic plasticity in adult cortex. Nature 420, 788-­‐794. doi: 10.1038/nature01273. Tramier, M., Piolot, T., Gautier, I., Mignotte, V., Coppey, J., Kemnitz, K., Durieux, C., and Coppey-­‐Moisan, M. (2003). Homo-­‐FRET versus hetero-­‐FRET to probe homodimers in living cells. Methods Enzymol 360, 580-­‐597. Vaughn, J.E. (1989). Fine structure of synaptogenesis in the vertebrate central nervous system. Synapse 3, 255-­‐285. doi: 10.1002/syn.890030312. Vicente-­‐Manzanares, M., Ma, X., Adelstein, R.S., and Horwitz, A.R. (2009). Non-­‐ muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778-­‐790. doi: 10.1038/nrm2786. Vicente-­‐Manzanares, M., Zareno, J., Whitmore, L., Choi, C.K., and Horwitz, A.F. (2007). Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol 176, 573-­‐580. doi: 10.1083/jcb.200612043. Vishwasrao, H.D., Trifilieff, P., and Kandel, E.R. (2012). In vivo imaging of the actin polymerization state with two-­‐photon fluorescence anisotropy. Biophys J 102, 1204-­‐1214. doi: 10.1016/j.bpj.2012.01.031. Wang, S.S., Khiroug, L., and Augustine, G.J. (2000). Quantification of spread of cerebellar long-­‐term depression with chemical two-­‐photon uncaging of glutamate. Proc Natl Acad Sci U S A 97, 8635-­‐8640. doi: 10.1073/pnas.130414597. Watanabe, N., Yamashiro, S., Vavylonis, D., and Kiuchi, T. (2013). Molecular viewing of actin polymerizing actions and beyond: combination analysis of single-­‐molecule speckle microscopy with modeling, FRAP and s-­‐FDAP (sequential fluorescence decay after photoactivation). Dev Growth Differ 55, 508-­‐514. doi: 10.1111/dgd.12060. Wear, M.A., Yamashita, A., Kim, K., Maeda, Y., and Cooper, J.A. (2003). How capping protein binds the barbed end of the actin filament. Curr Biol 13, 1531-­‐1537. 51 Westphal, M., Jungbluth, A., Heidecker, M., Muhlbauer, B., Heizer, C., Schwartz, J.M., Marriott, G., and Gerisch, G. (1997). Microfilament dynamics during cell movement and chemotaxis monitored using a GFP-­‐actin fusion protein. Curr Biol 7, 176-­‐183. Wilson, C.A., Tsuchida, M.A., Allen, G.M., Barnhart, E.L., Applegate, K.T., Yam, P.T., Ji, L., Keren, K., Danuser, G., and Theriot, J.A. (2010). Myosin II contributes to cell-­‐scale actin network treadmilling through network disassembly. Nature 465, 373-­‐377. doi: 10.1038/nature08994. Yan, Y., and Marriott, G. (2003). Fluorescence resonance energy transfer imaging microscopy and fluorescence polarization imaging microscopy. Methods Enzymol 360, 561-­‐580. Yang, G., Pan, F., and Gan, W.B. (2009). Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920-­‐924. doi: 10.1038/nature08577. Yasumatsu, N., Matsuzaki, M., Miyazaki, T., Noguchi, J., and Kasai, H. (2008). Principles of long-­‐term dynamics of dendritic spines. J Neurosci 28, 13592-­‐13608. doi: 10.1523/JNEUROSCI.0603-­‐08.2008. Yuste, R., and Bonhoeffer, T. (2004). Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci 5, 24-­‐34. doi: 10.1038/nrn1300. Zhang, H., Webb, D.J., Asmussen, H., Niu, S., and Horwitz, A.F. (2005). A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci 25, 3379-­‐3388. doi: 10.1523/JNEUROSCI.3553-­‐04.2005. Zhang, W., and Benson, D.L. (2001). Stages of synapse development defined by dependence on F-­‐actin. J Neurosci 21, 5169-­‐5181. Zhou, Q., Homma, K.J., and Poo, M.M. (2004). Shrinkage of dendritic spines associated with long-­‐term depression of hippocampal synapses. Neuron 44, 749-­‐757. doi: 10.1016/j.neuron.2004.11.011. Ziv, N.E., and Smith, S.J. (1996). Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17, 91-­‐102. Zuo, Y., Lin, A., Chang, P., and Gan, W.B. (2005). Development of long-­‐term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46, 181-­‐189. doi: 10.1016/j.neuron.2005.04.001. 52