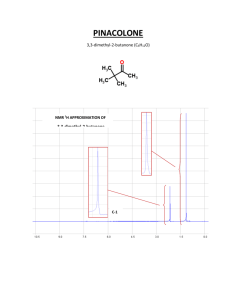
Results and Discussion
advertisement

2 A1B2 120 member library HO A2B2 OH O BL1 BL2 A2B2 A3B2 MIC = 0.2 mg/mL Set 2 BL1-BL20 BL3 BL4 A4B2 A5B2 BL5 A6B2 BL6 BL7 BL8 BL9 BL10 BL11 BL12 BL13 BL14 BL15 BL16 BL17 BL18 BL19 BL20 Results & Discussion Results and Discussion The present work describes the design and combinatorial synthesis of small parallel libraries of chalcones, peptidyl chalcones and peptidyl heterocycles, either in solution or on solid phase. The chalcone libraries, both parallel and indexed, were synthesized under Claisen-Schmidt conditions in solution phase. Peptidyl chalcones were synthesized on solid phase using phosphorane bound resin, while the derived heterocycles were synthesized in solution phase. The characterization of all the synthesized compounds was based on their physicochemical and spectroscopic data. Combinatorial chemistry is an exciting approach to chemical synthesis that enables the creation of a large number of organic compounds by linking chemical blocks in all possible combinations. Fig 2.1 shows a comparison of the conventional versus combinatorial synthesis. Conventional synthesis One compound only Combinatorial synthesis A library of nine compounds Fig 2.1. Comparison of conventional versus combinatorial synthesis. Classical synthesis i.e. the conversion of a reactant A to a final product D usually involves a multistep sequence, followed by purification and complete characterization of the products before screening. Guided by the biological activity of the previous compound, the next analogue is then designed, prepared and screened again. This process is repeated to optimize both activity and selectivity. On the contrary, combinatorial synthesis involves the synthesis of a large number of compounds; this collection may be a chemical mixture, a physical mixture, or individual pure Chapter 2 ♦Results and Discussion - 36 - compounds. The collection is then tested for biological activity and the active compound is identified finally by deconvolution and is made in quantity as a single compound.1 2.1. Combinatorial synthesis Parallel solution phase synthesis is the most straightforward method for library preparation due to its close resemblance to traditional synthesis. It involves the reaction of a single compound A with multiple reactants (B1, B2, …Bn), which gives rise to a compound library of n individual products AB1, AB2…ABn. The library is then evaluated, usually without purification and with only minimal characterization of the individual compounds, by means of a rapid-throughput screening methodology. Individual compounds are synthesized in different vessels and are available directly for analysis after purification. Parallel chemistry has the advantage that doing all the reactions at a time in different vessels saves time. However, the work-up has to be done separately for each NCE. 2.1.1. Synthesis of a parallel library of amino chalcones (1-20) A small parallel library of twenty different substituted amino chalcones (1-20) was synthesized in solution phase under Claisen Schmidt conditions using different substituted benzaldehydes and 4′-aminoacetophenone (Scheme 2.1). The selection of substituents on benzaldehyde (B ring) was made on the basis of their size, lipophilicity and electronic properties and they were changed systematically, one at a time at 2-, 3- and 4- positions to get a set of regioisomers. The reaction was carried out at room temperature and the reaction time varied from 3-8 h.146 H2N R H O NaOH/ EtOH B H2N O R A O 1-20 No. R No. R No. R No. R 1 H 6 3-OCH3 11 4-NO2 16 2-Br 2 2-OH 7 4-OCH3 12 2-Cl 17 3-Br 3 3-OH 8 3,4-(OCH3)2 13 3-Cl 18 4-CH3 4 4-OH 9 2-NO2 14 4-Cl 19 3-OH, 4-OCH3 5 2-OCH3 10 3-NO2 15 4-F 20 4-N(CH3)2 Scheme 2.1. Parallel synthesis of a library of amino chalcones (1-20). Chapter 2 ♦Results and Discussion - 37 - The synthesized chalcones were either purified by recrystallization or by flash chromatography using n-hexane and ethyl acetate as eluent. The purity was assessed through multiple TLC and the Rf values were recorded. Melting points were found to vary from 55 to 200 oC, while the yield varied from 60-87%. The characterization was made on the basis of physical parameters and spectral analysis as given in Chapter 4. In IR, amino chalcones, being the primary amines, gave two bands of medium intensity in the range of 2847-3474 cm–1 and 2555-3385 cm–1. The C=O and C=C stretching vibrations appeared in the range of 1600-1690 cm–1 and 1508-1600 cm–1, respectively. A weak peak of aromatic C–H stretching was often observed at 30413246 cm–1. A broad peak due to –OH group appeared at 3065-3600 cm–1. The stretching frequencies for different substituents on ring B e.g. C-OCH3, N-CH3, C-Br, C-Cl, C-F in different chalcones were observed in the range of 3041-3239, 2904, 736752, 842-869, 1092 cm–1. The two bands of unsymmetrical and symmetrical stretching in chalcones 9-11 for C-NO2 appeared at 1404-1514 cm–1. In an inspection of the IR stretching frequencies of different chalcones, it was observed that electron withdrawing substituents particularly at ortho and para positions on ring B shifted the stretching frequency corresponding to an amino group on ring A by a higher value e.g. the nitro group at ring B was found to shift the stretching frequencies of the 4NH2 on ring A by about 10 cm-1 i.e. p-NO2 substituted B ring resulted in the highest shift of 4-NH2 group; at 3481 cm-1 and 3385 cm-1; o-NO2 resulted in a lower shift; 3472 cm-1, 3382 cm-1, whereas the m-NO2 was found to shift these frequencies to 3469 cm-1 and 3380 cm-1. The HR-MS spectra of chalcones 1-20 showed the molecular ion peak in each case confirming the identity of these compounds. Halogen substituted chalcones (12-14 and 16-17) showed isotopic peaks corresponding to molecular ion and respective, fragments in almost 3:1 ratio for Cl35: Cl37 (12-14) and 1:1 ratio for Br79: Br81 (16-17), respectively. The mass fragmentation pattern of chalcone 1 is given in Scheme 2.2. Chapter 2 ♦Results and Discussion - 38 + H2N m/z = 66 m/z = 91 + -C2H2 + O H2N H2N H2C m/z = 222 m/z = 92 + -H H2N H2N O m/z = 223 + O m/z = 131 -CO CH m/z = 103 m/z = 120 + -CO + + C O -H -H + Scheme 2.2. Mass fragmentation pattern of amino chalcone 1. The 1H NMR data of all compounds 1-20 were recorded and a few generalizations could be made e.g. the aromatic protons of ring A H-2′, H-6′ and H-3′, H-5′ always appeared as two distinct doublets with an integration of two protons. The vinylic protons appeared as two distinct doublets, each with an integration of one proton with a coupling constant of ~14-16 Hz in all cases. 1H NMR data of chalcone 2 is given in Table 2.1 as a representative example. The most characteristic chemical shifts are those of α- and β- olefinic protons (vinylic protons). α Proton appeared upfield at 6.98 ppm, while β proton appeared at 7.78 ppm with a coupling constant of 14.8 Hz indicating a trans orientation of the double bond and hence an E configuration of the compound. The –NH2 protons appeared at 5.98 ppm. The two doublets at 8.01 ppm and 7.31 ppm (J = 8.3 Hz) were assigned to H-2′, 6′ and H-3′, 5′, respectively. Chapter 2 ♦Results and Discussion - 39 - Table 2.1. 1H NMR data of 1-(4′-aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (2). HO 3' H2N 4' β 2' 3 1 4 5 6 1' α 5' 6' 2 O 2 δ ppm Multiplicity Integration Coupling constant Assignment J (Hz) 5.98 bs 2H - NH2 6.98 d 1H 14.8 H−C(α) 7.75 d 1H 14.8 H−C(β) 8.01 d 2H 8.3 H−C(2′,6′) 7.31 d 2H 8.3 H−C(3′,5′) 7.41 d 1H 8.0 H−C(3) 7.04 d 1H 8.1 H−C(6) 6.98 t 1H 8.0 H−C(5) 6.95 t 1H 8.0 H−C(4) 9.88 s 1H - OH 2.1.2. Biological screening of a chalcone library Amino chalcones have been reported in the literature with potential antitumor, antiproliferative, antiparasitic and bactericidal activities.138-139 The synthesized amino chalcones were tested for their inhibitory potential as phosphatase inhibitors, antibacterial and cytotoxic agents, as discussed in the following sections. 2.1.2.1. Phosphatase inhibition A phosphatase is an enzyme that removes a phosphate group from its substrate by hydrolyzing phosphoric acid monoesters into a phosphate ion and a molecule with a free hydroxyl group. They act opposite to the kinases/phosphorylases, which add a phosphate group to proteins by using higher energy molecules such as ATP. The addition of a phosphate group may activate or deactivate an enzyme (e.g. Kinase signaling pathways) or enable a protein-protein interaction to occur; therefore, phosphatases are integral to many signal transduction pathways e.g. in diabetes. Their role in signal transduction is because they regulate the proteins to which they are Chapter 2 ♦Results and Discussion - 40 - attached. In order to reverse the regulatory effect, the phosphate group is removed, which occurs on its own by hydrolysis, or is mediated by protein phosphatases. Insulin is the major hormone responsible for lowering blood sugar level. The lack of insulin as a result of the degeneration of special insulin producing cells (called beta cells) in pancreas,170 or the lack of response of target cells to normal circulating insulin levels, are the key pathological features of type I and type II diabetes,171 respectively. Insulin binding to its cell surface transmembrane receptor stimulates autophosphorylation and activation of intrinsic protein tyrosinase kinase (PTK) activity and subsequent phosphorylation of insulin receptor substrates. The control of insulin receptor signaling involves the coordinated action of both positive and negative regulatory proteins. Among the negative regulatory proteins, protein tyrosinase phosphatases (PTPs) play a prominent role and their inhibition mimics several actions of insulin including the stimulation of glucose uptake.172-174 The development of drugs capable of inhibiting PTPs may allow the enhanced or prolonged activation of the insulin receptor and have therapeutic use in the treatment of type II diabetes.175-177 With the aim of finding some potential antidiabetic compounds, the synthesized amino chalcones 1-20 were tested against a large variety of fifteen protein phosphatases, e.g. Mycobacterium tuberculosis inosit phosphatase (Mtb Inosit Pptase), Mycobacterium tuberculosis phosphoglyceromutase (Mtb PGM), Mycobacterium tuberculosis aroA (Mtb aroA), Mycobacterium tuberculosis aroK (Mtb aroK), Mycobacterium tuberculosis protein tyrosinase phosphatase A (Mtb ptpA), human protein tyrosinase phosphatase 1B (hum ptp1B), human protein tyrosinase phosphatase N3 (hum ptpN3), human protein tyrosinase phosphatase N5 (hum ptpN5), human protein tyrosinase phosphatase N7 (hum ptpN7), human protein tyrosinase phosphatase RJ (hum ptpRJ), human protein tyrosinase phosphatase RK6 (hum ptpRK6), human protein tyrosinase phosphatase ROG7 (hum ptpROG7), human protein tyrosinase phosphatase RR (hum ptpRR), human protein tyrosinase phosphatase RS (hum ptpRS) and Mycobacterium tuberculosis protein tyrosinase phosphatase SHP2 (ptp SHP2). All compounds were tested at a concentration of 0.1 mM. They were found weakly active against Mtb Inosit Pptase, but were found to be very potent when tested against (Mtb PGM). The data of the activity of the tested Chapter 2 ♦Results and Discussion - 41 - compounds 1-20 represented as % inhibition against Mtb inosit Pptase is given in Table 2.2. Compounds 1, 3-9 and 13 inhibited the target enzyme only to some extent. Table 2.2. Mtb inosit Pptase inhibitory activity of chalcones 1-20. No. % Inh. No. % Inh. No. % Inh. No. % Inh. 1 21 6 4 11 - 16 - 2 - 7 26 12 - 17 - 3 32 8 3 13 7 18 - 4 21 9 14 14 - 19 - 5 8 10 - 15 - 20 - PGM is the key enzyme for isomerization of 3-phosphoglycerate to 2phosphoglycerate in the glycolysis pathway during respiration and PGM inhibitors are expected to maintain blood sugar levels for routine tasks in ischemic patients.178 The phosphoglycerate kinase step follows after isomerization step involving PGM and this results in the production of ATP, synthesis of pyruvic acid for aerobic production of energy in Krebs cycle and chimiosmosis. PGM is also involved in gluconeogenic transformation of phosphoenolpyruvate into glycogen and results in weakness after strenuous exercise. Overacting PGM may result in lower blood sugar levels in diabetic patients. PGM is found in all species but uses different variants. Chalcones 120 when tested against Mtb PGM, were found to be quite potent. The data is represented as % inhibition in Table 2.3. Chalcones 8 and 19 showed 60 % inhibition, while chalcone 3, 13 and 15 did not show any activity. Chalcones 4 and 11 with substituents 4-OH and 4-NO2 groups were found to be the lead structures of the library with 96 and 99 % inhibition, respectively. Table 2.3. Mtb PGM inhibitory activity of chalcones 1-20. No. % Inh. 1 44 2 No. % Inh. No. % Inh. No. % Inh. 6 43 11 99 16 24 17 7 44 12 47 17 17 3 - 8 60 13 - 18 48 4 96 9 45 14 41 19 60 5 15 10 34 15 - 20 41 It may be concluded that the suitable competitive reversible inhibitors of PGM e.g. the lead structures 4 and 11 are expected to be potential candidates for developing into antidiabetic drugs. Molecular docking studies on these chalcones in the active site of Chapter 2 ♦Results and Discussion - 42 - PGM providing further insight into the mechanism of their inhibition have been carried out as discussed in Chapter 3. 2.1.2.2. Antibacterial activity Chalcones are well known to have antibacterial properties.140 The members of the library (1-20) were, therefore, tested for their bactericidal activity against six bacterial strains, namely Escherichia coli, Bacillus subtilis, Enterobacter aerogenes, Staphylococcus aureus, Pseudomonas pickitti and Salmonella setubal. Each compound and the controls were tested at a concentration of 0.1 mg/mL. Roxithromycin and Cefixim were used as positive control, whereas DMSO was used as negative control. Table 2.4. Bactericidal activity of amino chalcones (1-20). No. S.aureus E.aerogens 1 0 0 0 11 2 0 10 0 11 3 0 0 0 11 4 0 0 0 11 5 0 0 0 11 6 0 0 0 11 7 0 0 0 11 8 0 0 0 0 9 0 0 9 0 10 0 0 9.5 0 11 0 0 0 0 12 0 0 0 0 13 0 0 0 11 14 0 10 0 0 15 0 0 9 0 16 0 0 9 0 17 0 0 9 0 18 0 0 9 0 19 11 12 13 0 20 0 0 0 0 Ra 26 26 19 21 b 24 26 32 21 C a Zone of Inhibition (mm) B.subtilis E.coli R= Roxithromycin (= Rulid®); b C= Cefixim®. Chapter 2 ♦Results and Discussion - 43 - It is evident from the data in Table 2.4 that all the tested chalcones did not show any significant activity against any of the tested bacterial strains. Compounds 1-7 and 13 showed weak activity against E. aerogenes, while 2 and 14 showed weak activity against B.subtilis. Compounds 9, 10, 15, 16, 17 and 18 showed very weak activities against E.coli, while chalcone 19 showed moderate activity against E.coli. The same compound was found weakly active against B.subtilis and S.aureus. 2.1.2.3. Brine shrimp lethality studies Chalcones, in general, are known to have cytotoxic potential179-180 and amino chalcones have been reported as cytotoxic agents.138-140 Studies on the cytotoxicity of chalcones 1-20 was carried out through brine shrimp lethality (BSL) assay, which is a rapid and economic benchtop assay employing the use of easily available brine shrimps; Artimisia salina as test animals. Artimisia salina The test is a pre-screen of antitumor properties.181-182 BSL testing was performed with different concentrations (150, 100 & 50 µg/mL) of the test samples prepared in DMSO and LD50 (lethal dose to kill 50% of test animals) values were recorded in µM quantities as shown in Table 2.5. Table 2.5. BSL studies on amino chalcones (1-20). No. LD50 No. LD50 No. LD50 No. LD50 1 6.68 6 3.79 11 130.56 16 8.34 2 46.50 7 0.24 12 16.22 17 83.71 3 60.54 8 13 52.30 18 150.11 4 7.10 9 160.63 14 14.61 19 - 5 2.72 10 557.02 15 614.42 20 2.59 - Chalcone 7 with methoxy substituent on ring B was found to be the LEAD structure with an LD50 value of 0.24 µM, while chalcones 1, 4-6, 16 and 20 showed strong cytotoxic behavior as reflected from their LD50 values ranging from 2.59-8.34 µM. Chalcones 2, 3, 12-14 and 17 showed good cytotoxic activity with LD50 < 100. Chapter 2 ♦Results and Discussion - 44 OCH3 H2N O 7 (LD50 = 0.24 µM) 2.2. Indexed combinatorial synthesis The synthesis of positional scanning library also called indexed libraries represents one of the most useful protocols for mixture synthesis. It is much less time intensive as compared to parallel synthesis of individual compounds and produces depository libraries for use in multiple screens with immediate deconvolution.183 Deconvolution is a multistep process where smaller libraries are successively prepared and tested to identify the individual active members of a combinatorial library. Positional scanning libraries provide lead identities in a single round of testing and the method is ideally suited for cellular assays that involve membrane-bound targets.184 The synthesis of positional scanning libraries represents one of the most useful protocols for mixture synthesis and can easily be conducted in solution phase but it is not easily adaptable to solid phase synthesis.185 Indexed combinatorial synthesis is a mixture synthesis, which involves the reaction of two components (building blocks) at a time such that one component is kept constant, while the other is used as an equimolar mixture. Alternatively, the synthesis is carried out through keeping second building block constant and varying the first one. These efforts result in the synthesis of two daughter libraries (two sets of indexed libraries, also called sub-libraries or pools). The two libraries (product mixtures) are tested and their activities are used as “indices” to the rows or columns of a 2D matrix reflecting the activities of individual compounds. Libraries can be preceded with the third building block for getting 3D indexed libraries as well and the process may continue.65 Two indexed combinatorial libraries (120 and 175 member) of chalcones were synthesized in solution phase and tested for their antibacterial, cytotoxic and antitumor properties followed by deconvolution, which led to the identification of potent antibacterial and antitumor chalcones as leads of two libraries. The reliability and success of the process was assessed by confirming the presence of all component chalcones in the indexed library (product mixture) through GC-MS analysis. Chapter 2 ♦Results and Discussion - 45 - 2.2.1. A 120-member chalcone library147 A 120 member chalcone library was designed and synthesized with a view of finding potential antibacterial chalcones through deconvolution. 2.2.1.1. Synthesis. A 120 member indexed chalcone library was designed by using six acetophenones (A1-A6) and 20 benzaldehydes (B1-B20) under Claisen Schmidt conditions (Scheme 2.3). R R' R O H NaOH/ EtOH R' O Acetophenones O Benzaldehydes R′ Benzaldehydes R R A1 C6H5 B1 Ph B11 4-NO2C6H4 A2 2′-OHC6H4 B2 2-OHC6H4 B12 2-ClC6H4 A3 4′-NH2C6H4 B3 3-OHC6H4 B13 3-ClC6H4 A4 2′,4′,5′-(OCH3)3C6H2 B4 4-OHC6H4 B14 4-ClC6H4 A5 3′,4′,5′-(OCH3)3C6H2 B5 2-OCH3C6H4 B15 4-FC6H4 A6 2′,4′-(Br)2C6H3 B6 3-OCH3C6H4 B16 2-BrC6H4 B7 4-OCH3C6H4 B17 3-BrC6H4 B8 3,4-(OCH3)2C6H3 B18 4-CH3C6H4 B9 2-NO2C6H4 B19 3-OH,4-OCH3C6H3 B10 3-NO2C6H4 B20 4-N(CH3)2C6H4 Scheme 2.3. Synthesis of a 120 member chalcone library. The library was synthesized in the form of two sets of daughter libraries (Set 1 and Set 2). Set 1 contained six daughter libraries, each containing 20 compounds, whereas Set 2 contained twenty daughter libraries, each containing 6 compounds (Fig. 2.2). Parent indexed library n=6 n = 20 ∑ R´COCH3 + ∑ RCHO R n = 120 ∑ R' O Indexed daughter libraries (Set 1) (6 libraries, each containing 20 compounds) n = 20 R´COCH3 + R n = 20 ∑ RCHO ∑ R' O Indexed daughter libraries (Set 2) (20 libraries, each containing 6 compounds) n=6 ∑ R n=6 R´COCH3 + RCHO ∑ R' O Fig. 2.2. Synthesis of indexed libraries of Sets 1 and 2. Chapter 2 ♦Results and Discussion - 46 - The presence of all the library members in each library was confirmed through GCMS analysis. The identification of some small molecular libraries through GC-MS has already been reported.186-187 For indexed library, the daughter libraries were run through a DB-5ms column containing a stationary phase of 5% phenyl and 95% dimethylarylenesiloxane with He gas. A 5973 inert MS selective detector was used. The GC-MS spectrum of AL1 library is given in Fig 2.3, whereas the mass fragments contained in each peak are given in Fig. 2.4 (a-c) and Fig. 2.5 (d-l). The GC-MS of the daughter library AL1 is discussed in detail. The library was analyzed using different GC-MS programs and the best results were found when the temperature was kept from 120 to 300 °C at a rate of 10°C/min, whereas inlet was kept at 250 °C. The 20 members of the library were found separated as 12 peaks, but the relative peak areas indicated the presence of all 20 compounds. All the compounds were eluted in 17 minutes. The first peak separated at 11.2 min with M+. peak of 226 indicating the presence of fluorochalcone. The second peak separated at 11.4 min, which contained four chalcones as indicated through the relative peak area. The presence of these chalcones was confirmed through the M+. and fragment ion peaks. The M+. peaks at 208 indicated the presence of unsubstituted chalcone. A hydroxychalcone was detected through a weak [M + H]+ peak at 226. A strong peak at 207 indicated the presence of M+. – NO2 from 2- nitrochalcone or 4-nitrochalcone. The third peak appeared at 12.75 min with a weak M+. peak at 238 and [M – CH3]+ peak at 223 indicating a methoxy chalcone. The peaks at 12.93 min, 13.10 min and 13.28 min corresponded to chlorochalcones, each giving a molecular ion peak at 242 and M+.+2 peak at 244 with relative abundance of 3:1. The 3-hydroxy-4-methoxychalcone appeared at 13.71 min giving [M – O]+ peak at 238, [M – OH]+ at 237 and [M – OCH3]+ at 223. A bromochalcone was eluted at 14.09 min giving M+. and M+.+2 peaks at 286 and 288 in 1:1 ratio. At 14.22 min, a peak eluted with a peak area indicating the presence of four chalcones. A careful analysis indicated the presence of two hydroxy- and two methoxy-chalcones. The molecular ion peaks were observed as M+. at 238 and [M + H]+ at 239 for methoxy chalcones, whereas as M+. at 224 and [M – H]+ at 223 for hydroxy chalcone. The 238 – 28 and 239 – 28 were the peaks found at 210 and 211 after CO. removal from methoxy chalcones. Similarly, 224 – CO. and 223 – CO. were observed at 196 and 195 for hydroxy chalcones. At 14.28 min, Chapter 2 ♦Results and Discussion - 47 - Fig. 2.3. The GC-MS spectrum of the library AL1 containing 20 compounds. Fig. 2.4. The mass spectra for each GC peaks (a-c). Chapter 2 ♦Results and Discussion - 48 - Fig. 2.5. The mass spectra for each GC peaks (d-l). Chapter 2 ♦Results and Discussion - 49 - a bromochalcone appeared with M+. and M+. + 2 peaks at 286 and 288. The spectrum showed very weak peaks at 256 and 258 after CO. removal from M+. and M+. + 2. The methyl chalcone also appeared in the same peak as indicated by [M + H]+ peak at 223. Furthermore, a peak at 207 indicated the [M – CH3]+ for methyl chalcone or the M+. – Br. for bromochalcone. Dimethoxychalcone and a nitrochalcone at 15.79 min were identified through the M+. peaks at 268 and 253, respectively. The 253 – O. was observed at 237 whereas 253 – NO. appeared at 223 and 253 – NO2 at 207 confirming the presence of a nitro chalcone. The last peak appeared at 16.7 min containing the N,N-dimethylaminochalcone with a molecular ion peak at 251. The M+.– CO. appeared at 223. The synthesized indexed library can be represented as a 2D matrix, wherein the x-axis has six structural variants (acetophenones), and the y-axis has 20 structural variants (benzaldehydes) leading to a 6 × 20 grid (Table 2.6). Table. 2.6. Conceptual matrix for an indexed 120 member chalcone library. Acetophenones A1 A2 A3 A4 A5 A6 B1 A1 B1 A2 B1 A3 B1 A4 B1 A5 B1 A6 B1 B2 A1 B2 A2 B2 A3 B2 A4 B2 A5 B2 A6 B2 B3 A1 B3 A2 B3 A3 B3 A4 B3 A5 B3 A6 B3 B4 A1 B4 A2 B4 A3 B4 A4 B4 A5 B4 A6 B4 B5 A1 B5 A2 B5 A3 B5 A4 B5 A5 B5 A6 B5 B6 A1 B6 A2 B6 A3 B6 A4 B6 A5 B6 A6 B6 B7 A1 B7 A2 B7 A3 B7 A4 B7 A5 B7 A6 B7 B8 A1 B8 A2 B8 A3 B8 A4 B8 A5 B8 A6 B8 B9 A1 B9 A2 B9 A3 B9 A4 B9 A5 B9 A6 B9 B10 A1 B10 A2 B10 A3 B10 A4 B10 A5 B10 A6B10 B11 A1 B11 A2 B11 A3 B11 A4 B11 A5 B11 A6B11 B12 A1 B12 A2 B12 A3 B12 A4 B12 A5 B12 A6B12 B13 A1 B13 A2 B13 A3 B13 A4 B13 A5 B13 A6B13 B14 A1 B14 A2 B14 A3 B14 A4 B14 A5 B14 A6B14 B15 A1 B15 A2 B15 A3 B15 A4 B15 A5 B15 A6B15 B16 A1 B16 A2 B16 A3 B16 A4 B16 A5 B16 A6B16 B17 A1 B17 A2 B17 A3B17 A4 B17 A5 B17 A6B17 B18 A1 B18 A2 B18 A3 B18 A4 B18 A5 B18 A6B18 B19 A1 B19 A2 B19 A3 B19 A4 B19 A5 B19 A6B19 B20 A1 B20 A2 B20 A3 B20 A4 B20 A5 B20 A6B20 Benzaldehydes Chapter 2 ♦Results and Discussion - 50 - The number of the building blocks was chosen such that the library was large enough to demonstrate the principle but small enough to verify the composition of library by analytical methods. Besides, the size of the library was kept such that it was soluble in an assay mixture for its effective screening. The chemistry of the reaction used in the preparation of library was investigated on a single compound. The choice of the reagents reflects the chemical diversity in terms of hydrophobic and lipophilic substituents. All library components share a common α,β-unsaturated carbonyl functionality. Synthesis of such a library in indexed fashion resulted in 26 daughter libraries with six members in Set 1, while 20 members in Set 2. Both sets were subjected to screening experiments followed by deconvolution and subsequent LEAD identification. The assay value of each cell is contained in the combination AxBy (x = 1–6, y = 1–20) (Table 2.6). Obviously, the examination of all the pure compounds would have required 120 experiments. Since only one cell out of 120 may possess the maximum response function, the next step involves its identification without looking at all 120 cells. The best way of doing this is to choose any B for testing with all Atype compounds; conversely, any A for testing with all B-type members. 120 Member Parent Library Indexed Libraries Set 1: Pools 1-6 Set 2. Pools 1-20 1. A1 + B1-20 2. A2 + B1-20 : 6. A6 + B1-20 1. B1 + A1-6 2. B2 + A1-6 : 20. B20 + A1-6 Screening LEAD Fig. 2.6. A general schematic diagram for library synthesis in search of the LEAD in a bioassay. Chapter 2 ♦Results and Discussion - 51 - By screening six columns and 20 rows, which are indices to the cells at their intersections, as mixtures, only 26 reactions (instead of 120) need to be carried out to find the maximum response. Therefore, a combinatorial synthesis of 26 sub-libraries was carried out. This method has the advantages over the parallel approach in that it is fast and relatively inexpensive. However, the number of compounds prepared is much greater than the number of chemical steps required 2.2.1.2. Antibacterial studies. All 26 indexed libraries were subjected to antibacterial studies against six bacterial strains; three Gram positive (B. subtillis ATTCC 6633, M. leuteus and S. aureus ATCC 6538) and three Gram negative (E. coli AATCC 1522, E. aerogenes AATCC 13048 and S. setubal ATCC 19196) using the Agar Well Diffusion method.188 Each compound was tested twice, once as a component of sublibraries of Set 1 and then as a member of sub-libraries of Set 2. Thus, a total of 26 assays were required to reveal the antibacterial activity of 120 compounds, which corresponded to a factor of 4.6 in terms of time improvement in the synthesis and data collection efficiency. The antibacterial activities of the sub-libraries AL1-AL6 and BL1-BL20 are shown in Tables 2.7 and 2.8, respectively. All libraries were tested at a concentration of 0.1 mg/mL. The libraries were found to be active against S. aureus and B. subtilis, very weekly active against E. coli, while inactive towards the other three bacterial strains i.e. M. leuteus, E. aerogenes and S. setubal. Roxithromycin (Rulid®) and Cefixime® were used as standard positive controls in these studies. Table 2.7. Antibacterial activities of Set 1 sub-libraries (pools AL1-AL6). Zone of inhibition(mm)a S. aureus B. subtilis E. coli AL1 15.1±0.15 16.0±0.55 11.5±0.50 AL2 10.9±0.15 12.0±0.26 0 AL3 AL4 a Zone of inhibition(mm) a 8.8±0.12 0 10.8±0.11 0 0 0 S. aureus B. subtilis E. coli AL5 10.7±0.20 10.7±0.16 0 AL6 0 17.8±0.20 15.1±0.05 b 26.5±1.30 32.1±0.31 11.6±0.10 c 21.3±0.70 12.4±0.18 31.9±0.17 R C mean value of three replicates, b R = Roxithromycin, c C =Cefixime®. After having synthesized the sub-libraries and carrying out their antibacterial screening, the next step is the synthesis of the library corresponding to the most active column and the most active row, and to identify the most active library and the lead structures therein by deconvolution. Chapter 2 ♦Results and Discussion - 52 - Table 2.8. Antibacterial activities of Set 2 sub-libraries (pools BL1-BL20). Zone of inhibition (mm)a S. aureus Zone of inhibition (mm)a B. subtilis E. coli S. aureus B. subtilis E. coli BL1 9.4±0.32 12±0.20 0 BL12 0 0 8.8±0.15 BL2 18.6±0.11 0 15.3±0.05 BL13 11.3±0.05 0 8.6±0.11 BL3 10.9±0.05 0 0 BL14 11.1±0.23 12.2±0.15 0 BL4 13.7±0.05 0 0 BL15 0 12.1±0.05 0 BL5 0 0 0 BL16 10.8±0.10 14.1±0.17 0 BL6 8.6±0.17 0 0 BL17 16.2±0.15 16.2±0.05 0 BL7 8.5±0.00 0 0 BL18 11.1±0.05 13.1±0.15 0 BL8 0 0 0 BL19 0 0 0 BL9 0 0 0 BL20 BL10 11.5±0.05 BL11 0 10.9±0.10 0 0 10.5±0.20 10.5±0.05 0 b 26.5±1.3 32.1±0.31 11.6±0.1 c 21.3±0.70 12.4±0.18 31.9±0.17 R 0 C 2.2.1.3. Deconvolution. Deconvolution is the process by which active compounds are identified in library mixtures. The antibacterial activities of the libraries were used as indices to the cells of columns and rows of the conceptual matrix (Table 2.9). Table 2.9. Calculated zone of inhibition (mm) of chalcones against S. aureus. Set 1 Set 2 AL1 AL2 AL3 AL4 AL5 AL6 BL1 12.2 10.1 9.1 4.7 10.0 13.6 BL2 16.9 14.8 13.7 9.3 14.7 18.2 BL3 13.0 10.9 9.8 5.4 10.8 14.3 BL4 14.4 12.3 11.2 6.8 12.2 15.7 BL5 7.6 5.4 4.4 0 5.3 8.9 BL6 11.8 9.7 8.7 4.3 9.6 13.2 BL7 11.8 9.7 8.6 4.25 9.6 13.1 BL8 7.5 5.4 4.4 0 5.3 8.9 BL9 7.5 5.4 4.4 0 5.3 8.9 BL10 13.3 11.2 10.1 5.75 11.1 14.6 BL11 7.5 5.4 4.4 0 5.3 8.9 BL12 7.5 5.4 4.4 0 5.3 8.9 BL13 13.2 11.1 10.0 5.65 11 14.5 BL14 13.1 11 9.95 5.55 10.9 14.4 BL15 7.5 5.4 4.4 0 5.3 8.9 BL16 12.9 10.8 9.0 5.4 10.7 14.3 BL17 15.6 13.5 12.5 8.1 13.4 17 BL18 13.1 11 9.9 5.55 10.9 14.4 BL19 7.5 5.4 4.4 0 5.3 8.9 BL20 12.8 10.7 9.6 5.25 10.6 14.1 Chapter 2 ♦Results and Discussion - 53 - The data was expanded on 120 cells of chalcones on the matrix against S. aureus, since the libraries were found to be the most active against this strain only. This data expansion was carried out for each cell by taking the average of the activities of the respective column and row cell, and the data were used as indices to the particular cell. This resulted in calculated antibacterial activity against S. aureus for all 120 chalcones of the matrix. For the identification of the lead compounds, the individual members of the most active column and row needed to be identified. A careful examination of the data revealed that the members of the column AL1 and the row BL2 showed maximum inhibition. Hence, parallel synthesis of the individual members of these two sub-libraries was accomplished. Parallel synthesis of active libraries AL1 and BL2. Parallel solution phase synthesis of a total of twenty five chalcones belonging to the active libraries AL1 and BL2 was carried out under standard Claisen-Schmidt conditions using a 24 parallel reactions workstation, GreenHouse Synthesizer (Scheme 2.4). R R' R H O NaOH/ EtOH O Synthesizer O R R O AL1 (A1B1-A1B20) (21 - 40) GreenHouse R' R R 21 H 28 3,4-(OCH3)2 35 4-F 22 2-OH 29 2-NO2 36 2-Br 23 3-OH 30 3-NO2 37 3-Br 24 4-OH 31 4-NO2 38 4-CH3 25 2-OCH3 32 2-Cl 39 3-OH,4-OCH3 26 3-OCH3 33 3-Cl 40 4-N(CH3) 2 27 4-OCH3 34 4-Cl HO R′ R' O BL2 (A1B2-A6B2) (2, 22, 41 – 44) R′ R′ 2* 4′-NH2 41 2′-OH 43 3′,4′,5′ -(OCH3)2 22** H 42 2′,3′,4′- (OCH3)2 44 2′,4′-(Br)2 *already mentioned in scheme 2.1. ** already mentioned above. Scheme 2.4. Synthesis of individual chalcones of active libraries AL1 and BL2. Chapter 2 ♦Results and Discussion - 54 - All the compounds were recrystallized from EtOH and identified through IR, GCMS and 1H NMR data. The purity of the compounds was checked through multiple TLC (solvent system, n-Hex: EtOAc; 9:1, 3:1) and the Rf values were recorded. The yield of chalcones varied from 61-85 % and the data is given in the Experimental Section. FT-IR spectral data of chalcones 21-44 is given in the Experimental Section. The C=O and C=C stretching vibrations appeared in the range of 1631-1690 cm–1 and 1508-1606 cm–1, respectively. A weak peak of Ar-H was observed at 3041-3246 cm– 1 . The stretching frequencies for different substituents on the B ring, derived from benzaldehydes i.e. C-OH, C-OCH3, N-CH3, C-Br, C-Cl, C-F and C-NO2 were also observed. 1 H NMR data of chalcones 21-44 has been reported in Experimental Part in detail. Generally, protons on ring A appeared as a downfield doublet for H-2′, 6′, then a triplet for H-4′ followed by a triplet for H-3′, 5′ with a coupling constant in the range of 7-8 Hz. The α-proton at the vinylic position appeared in the range of 7.39-7.74 ppm, whereas β-proton appeared at 7.73-8.17 ppm, each with a coupling constant of 14-16 Hz. 1H-F coupling was observed in compound 35 (Fig. 2.7). The 1H-F coupling for ortho position was found in the range of 6-11 Hz, whereas for meta position, it was 3-9 Hz. In 35, protons 3 and 5 appeared as a doublet of doublets (dd) at 7.98 ppm (J = 9.0, 7.0 Hz), whereas protons 2 and 6 appeared as an apparent triplet (app t) at 7.30 ppm (J = 8.5 Hz) due to H-F coupling. Fig. 2.7. 1H NMR of 1-(phenyl)-3-(4′-fluorophenyl)-2-propen-1-one (35). Chapter 2 ♦Results and Discussion - 55 - Screening of libraries AL1 and BL2. The active libraries AL1 and BL2 were tested against six bacterial strains; however, they were found active only toward two strains i.e. S. aureus and B. subtilis. Table 2.10. Antibacterial activities of individual chalcones of library AL1. Zone of inhibition (mm)a Compound a Zone of inhibition (mm)a S. aureus B. subtilis Compound S. aureus B. subtilis A1B1 10.5±0.10 9.2±0.10 A1B12 0 9.5±0.10 A1B2 10.9±0.01 9.7±0.20 A1B13 11.1±0.02 9.2±0.05 A1B3 16.6±0.05 12.5±0.10 A1B14 0 0 A1B4 10.1±0.20 11.7±0.02 A1B15 0 0 A1B5 0 0 A1B16 0 0 A1B6 0 10.7±0.03 A1B17 10.23±0.06 9.5±0.10 A1B7 0 10.4±0.07 A1B18 0 0 A1B8 0 0 A1B19 0 0 A1B9 15.1±0.10 14.9±0.10 A1B20 0 0 b A1B10 0 9.5±0.10 R 26.5±1.3 32.1±0.31 A1B11 0 0 Cc 21.3±0.70 12.4±0.18 mean value of three replicates. b R = Roxithromycin (positive control). c C =Cefixime® (positive control). Table 2.11. Antibacterial activities of individual chalcones of library BL2. Zone of inhibition (mm)a Compound a Zone of inhibition (mm)a S. aureus B. subtilis Compound A1B2 10.9±0.01 12.5±0.20 A5B2 13.2±0.05 A2B2 18.2±0.10 17.6±0.05 A6B2 9.4±0.10 b S. aureus B. subtilis 13.4±0.10 0 A3B2 9.6±0.10 11.5±0.01 R 26.5±1.3 32.1±0.39 A4B2 13.1±0.10 13±0.10 Cc 21.3±0.70 12.4±0.18 mean value of three replicates. b R = Roxithromycin (positive control). c C =Cefixime® (positive control). The results of the antibacterial studies of chalcones of sub-libraries AL1 and BL2 are given in Tables 2.10 and 2.11, respectively. The minimum inhibitory concentration (MIC) values of the members of AL1 and BL2 are given in Table 2.12. The concentration of each library member and of the control was 0.1 mg/mL in DMSO. It is evident from the data in Table 2.10 and 2.11 that chalcones A1B3 and A2B2 were the most active members of the libraries AL1 and BL2. Based on the MIC values, chalcone A1B2 and A2B2 (MIC = 0.2 mg/mL) may be regarded as the LEAD of the designed 120-member library. Chapter 2 ♦Results and Discussion - 56 - Table 2.12. Minimum inhibitory concentration (MICs)a of active chalcones (mg/mL). a Compound S.aureus B.subtilis Compound S.aureus B.subtilis A1B1 0.5 0.4 A1B9 0.4 0.3 A1B2 0.2 - A1B17 0.5 - A1B3 0.4 - A2B2 0.2 0.2 A1B4 - 0.5 A4B2 0.3 0.4 A1B6 - 0.6 A5B2 0.4 0.6 A1B7 - 0.6 MIC = minimum concentration at which no colony was observed after incubation. HO HO OH O O A1B2 (22) A2B2 (41) LEADS It is evident that starting from designing of the combinatorial library to a sequential logical identification of the lead through positional scanning protocol led to a reduction in the number of steps from 120 to 26, thereby, reducing the cost in terms of time and resources in the identification of a lead. Hence a positional scanning protocol can be considered as a cost effective technique for rapid lead discovery. This identification of the lead from the designed library through deconvolution has been shown schematically in Figs. 2.8 and 2.9. 2.2.1.4. Structure activity relationship. Based on the antibacterial activities, it is also possible to deduce some trends in structure activity relationship (SAR). The following order of decreasing activity was noticed: A2B2 > A4B2 A5B2 HO HO O O HO > A1B2 O O > A3B2 > A6B2 HO HO HO Br H2 N O O OH O > O O O O Br O The activity of the chalcones was found to be dependent on the substitution pattern on ring A. Chalcones with electron donors at ortho position of A ring were found as the most active as indicated from 2-OH and 2-OCH3 substituted chalcones A2B2 and Chapter 2 ♦Results and Discussion - 57 - A5B2. A steric bulk at ortho position of ring A appeared to be detrimental for activity as indicated from the least potent chalcone A6B2. A1B1 HO A1B2 A1B3 120 member library AL1 AL2 Set 1 AL1-AL6 AL3 O A1B2 A1B4 A1B5 MIC = 0.2 mg/mL A1B6 A1B7 AL4 AL5 AL6 A1B8 A1B9 A1B10 A1B11 A1B12 A1B13 A1B14 A1B15 A1B16 A1B17 A1B18 A1B19 A1B20 Fig. 2.8. Identification of lead through deconvolution of a 120 member library (Set 1) against S. aureus. Chapter 2 ♦Results and Discussion - 58 - A1B2 120 member library A2B2 BL1 BL2 Set 2 BL1-BL20 BL3 BL4 BL5 A3B2 A4B2 HO OH O A2B2 MIC = 0.2 mg/mL A5B2 A6B2 BL6 BL7 BL8 BL9 BL10 BL11 BL12 BL13 BL14 BL15 BL16 BL17 BL18 BL19 BL20 Fig. 2.9. Identification of lead through deconvolution of a 120 member library (Set 2) against S. aureus. Chapter 2 ♦Results and Discussion - 59 - 2.2.1. 175 Member chalcone library148 Chalcones are well known to act as antitumor agents by interacting and depolymerizing tubulin dimers in living cells.189 Tubulin is a globular protein of molecular weight 100,000, which exists in dynamic equilibrium with microtubules. Microtubules are vital components of the cell and are responsible for intracellular transport, mechanical stabilization and formation of the mitotic spindle during cell division. Vinblastine, vincristine, epithilone and taxol are known as tubulin binders that work by depolymerization of microtubules.190 Some chalcones are known to bind to tubulin dimer and halt mitosis at G2/M phase.191 Using chalcones as antitumor agents may prove quite successful in cancer chemotherapy, as spindle poisons exert their influence when mitosis is in the metaphase. Furthermore, a number of α,βunsaturated ketones have been reported to demonstrate preferential reactivity towards thiols in contrast to amino and hydroxy groups.192 The chalcones, therefore, are expected to be free from problems of mutagenecity and carcinogenecity associated with a number of alkylating agents used in cancer chemotherapy.193 Based on the well known antitumor properties of a chalcone template,106-114 the studies were expanded for antitumor evaluation of a diverse set of chalcones. Therefore, a 175 member chalcone library was designed, synthesized and evaluated for antitumor potency through potato-disc tumor inhibition (PDT) assay.148 Fig. 2.10. Mechanism of tubulin depolymerization (courtesy from Calbiochem).194 Chapter 2 ♦Results and Discussion - 60 - 2.2.2.1. Synthesis. Having two chalcones as potential candidates to be developed as antibacterials and two other chalcones as LEADS in BSL bioassay in a 120 member library, the studies were extended to a still larger library. The building blocks were chosen with a view to have diversity in terms of hydrophobicity, hydrophilicity and electronic effects. Seven acetophenones A1-A7 and 25 aldehydes B1-B25 were used to design this library. R R' R O H NaOH / EtOH R' O O Aldehyde R Aldehyde R B1 C6H5 B14 3-NO2C6H4 R′ B2 2-OHC6H4 B15 3-OH,4-OCH3C6H4 A1 C6H5 B3 3-OHC6H4 B16 4-N(CH3)2C6H4 A2 2΄-OHC6H4 B4 4-OHC6H4 B17 4-CH3C6H4 A3 3΄-OHC6H4 B5 2-OCH3C6H4 B18 2-Thienyl A4 4΄-OHC6H4 B6 3-OCH3C6H4 B19 5-Methyl-2-thienyl A5 2΄-NH2C6H4 B7 4-OCH3C6H4 B20 5-Bromo-2-thienyl A6 3΄-NH2C6H4 B8 3,4-(OCH3)2C6H3 B21 5-Nitro-2-thienyl A7 4΄-NH2C6H4 B9 2-ClC6H4 B22 2-Pyrrolyl B10 3-ClC6H4 B23 2-Pyridyl B11 4-ClC6H4 B24 3-Pyridyl B12 3-BrC6H4 B25 4-Pyridyl B13 4-FC6H4 Acetophenone Scheme 2.5. Building blocks for the synthesis of a 175 member library. The testing of this indexed library has been conceptually represented as a 2-D matrix, wherein x-axis has 7 structural variants (acetophenones), while y-axis has 25 structural variants (aldehydes) leading to a 7 × 25 grid, as discussed earlier (Fig. 2.11). The synthesis of these chalcone libraries was carried out using standard ClaisenSchmidt conditions.148 Each of the 32 row and column reaction was carried out with one specific acetophenone An and a mixture of 25 aldehydes leading to daughter libraries of Set 1. The libraries (BL1-BL25) of Set 2 were obtained likewise by the reaction of an aldehyde Bn with a mixture of seven acetophenones. To eliminate kinetics effects, all reactions were forced to completion by conducting them with a stoichiometric quantity of the unitary reagent relative to the total of the mixed Chapter 2 ♦Results and Discussion - 61 - reagents. The synthesis of library BL7 was carried out independently both under microwave irradiation and conventional heating. The presence of all the components of the mixture in BL7 was indicated by means of TLC (thin-layer chromatography). The composition of the library was then compared in two experiments and found to be the same under both conventional heating and microwave irradiation. The successful synthesis of indexed libraries was confirmed through GC-MS analysis. Set 1 Set 2 BL1 AL1 AL2 AL3 AL4 AL5 AL6 AL7 A1 B1 A2 B1 A3 B1 A4 B1 A5 B1 A6 B1 A7 B1 BL2 A1 B2 A2 B2 A3 B2 A4 B2 A5 B2 A6 B2 A7 B2 BL3 A1 B3 A2 B3 A3 B3 A4 B3 A5 B3 A6 B3 A7 B3 BL4 A1 B4 A2 B4 A3 B4 A4 B4 A5 B4 A6 B4 A7 B4 BL5 A1 B5 A2 B5 A3 B5 A4 B5 A5 B5 A6 B5 A7 B5 BL6 A1 B6 A2 B6 A3 B6 A4 B6 A5 B6 A6 B6 A7 B6 BL7 A1 B7 A2 B7 A3 B7 A4 B7 A5 B7 A6 B7 A7 B7 BL8 A1 B8 A2 B8 A3 B8 A4 B8 A5 B8 A6 B8 A7 B8 BL9 A1 B9 A2 B9 A3 B9 A4 B9 A5 B9 A6 B9 A7 B9 BL10 A1 B10 A2 B10 A3 B10 A4 B10 A5 B10 A6 B10 A7B10 BL11 A1 B11 A2 B11 A3 B11 A4 B11 A5 B11 A6 B11 A7B11 BL12 A1 B12 A2 B12 A3 B12 A4 B12 A5 B12 A6 B12 A7B12 BL13 A1 B13 A2 B13 A3 B13 A4 B13 A5 B13 A6 B13 A7 B13 BL14 A1 B14 A2 B14 A3 B14 A4 B14 A5 B14 A6 B14 A7 B14 BL15 A1 B15 A2 B15 A3 B15 A4 B15 A5 B15 A6 B15 A7 B15 BL16 A1 B16 A2 B16 A3 B16 A4 B16 A5 B16 A6 B16 A7 B16 BL17 A1 B17 A2 B17 A3B17 A4 B17 A5 B17 A6 B17 A7 B17 BL18 A1 B18 A2 B18 A3 B18 A4 B18 A5 B18 A6 B18 A7 B18 BL19 A1 B19 A2 B19 A3 B19 A4 B19 A5 B19 A6 B19 A7 B19 BL20 A1 B20 A2 B20 A3 B20 A4 B20 A5 B20 A6 B20 A7 B20 BL21 A1 B21 A2 B21 A3 B21 A4 B21 A5 B21 A6 B21 A7 B21 BL22 A1 B22 A2 B22 A3 B22 A4 B22 A5 B22 A6 B22 A7 B22 BL23 A1 B23 A2 B23 A3 B23 A4 B23 A5 B23 A6 B23 A7 B23 BL24 A1 B24 A2 B24 A3 B24 A4 B24 A5 B24 A6 B24 A7 B24 BL25 A1 B25 A2 B25 A3 B25 A4 B25 A5 B25 A6 B25 A7 B25 Fig. 2.11. Conceptual matrix for designing a 175 member chalcone library. 2.2.2.2. Antitumor studies. Crown gall is a neoplastic disease of plants, which occurs in more than 60 families of dicotyledons and many gymnosperms. Crown galls cause Chapter 2 ♦Results and Discussion - 62 - the bulging of a mass of tissues from stems and roots of woody and herbaceous plants. These tumors may be spongy or hard, and may or may not have deleterious effects on the plant. Histologically, crown-gall tumors are similar to those found in humans and animals. The causative agents of this disease are specific strains of the Gram-negative Agrobacterium tumefaciens (AT). The inhibition of crown-gall tumor (induced by A. tumefaciens in potato-disc tissue) is an assay based on antimitotic activity. This assay is capable of detecting a broad range of known and novel antitumor effects. During infection of plant material with A. tumefaciens, a tumorproducing plasmid, found in bacterial DNA, is incorporated in the chromosomal plant DNA. When plant tissue is wounded, it releases phenols and other compounds that activate the Ti-plasmid in A. tumefaciens. The Ti-plasmid causes the plant’s cells to multiply rapidly without going through apoptosis, resulting in tumor formation that are similar in nucleic acid content and histology to human and animal cancers. The relevance of the crown-gall-tumor system to the general cancer problem has been thoroughly reviewed.195-197 The validity of this bioassay is based on the observation that certain tumorigenic mechanisms are similar in plants and animals. For example, it has been observed that the inhibition of crown-gall tumor on potato discs and their subsequent growth shows good correlation with compounds and extracts active in the 3PS leukemic mouse assay.198 It has also been reported that the potato disc tumor assay is statistically more predictive in terms of 3PS activity than either the 9KB or the 9PS cytotoxicity assays199 and can be used as a fairly rapid, inexpensive, and reliable prescreen for antitumor activity.200 PDT, being a simple bench top assay has been used as a pre-screen for evaluation of antitumor potential of different compounds obtained either from nature or from synthetic world. The designed chalcone libraries of Sets 1 and 2 were, therefore, subjected to this bioassay. The screening of two sets of libraries enabled the testing of each compound twice, once as a component of the sub-libraries of Set 1, and then as a member of the sub-libraries of Set 2. Thus, a total of 32 assays were required to find the antitumor activity of 175 compounds which corresponded to a factor of ~ 5.5 in terms of time improvement in the synthesis and data collection efficiency. Chapter 2 ♦Results and Discussion - 63 - The sub-libraries of two sets, AL1-AL7 and BL1-BL25, were screened initially at a concentration of 1000 ppm and their antitumor activities were determined as shown in Table 2.13. Table 2.13. PDT inhibition studies of libraries of Sets 1 and 2 at 1000 ppm concentration. Library % Inh. Library % Inh. Library % Inh. Library % Inh. AL1 100 BL3 95 BL12 100 BL21 80 AL2 96 BL4 100 BL13 79 BL22 66 AL3 100 BL5 100 BL14 57 BL23 95 AL4 95 BL6 96 BL15 66 BL24 33 AL5 71 BL7 100 BL16 65 BL25 74 AL6 78 BL8 100 BL17 64 AL7 48 BL9 100 BL18 69 BL1 40 BL10 77 BL19 79 BL2 85 BL11 100 BL20 64 All the libraries showed significant antitumor activity. Libraries AL1, AL3, BL4-BL5, BL7-BL9 and BL11-BL12 showed 100% tumor inhibition. Further short listing was done by screening these nine libraries at lower concentrations, e.g. at 100 and 10 ppm, as shown in Table 2.14. As a result, the libraries AL1 and BL9 (corresponding to column 1 and row 9, respectively) were found to be the most active, both at 10 and 100 ppm. Table 2.14. PDT inhibition studies on the active libraries at 10 and 100 ppm concentration. Library % Inh. Library % Inh. Library % Inh. AL1 64 (88) BL5 33 (54) BL9 73 (95) AL3 60 (84) BL7 15 (29) BL11 14 (35) BL4 11 (78) BL8 23 (45) BL12 22 (41) Values in parentheses correspond to inhibition at 100 ppm. 2.2.2.3. Deconvolution. Having synthesized and screened all the sub-libraries, the identification of the lead(s) was carried out by deconvolution. This required the calculation of activities of all the members of the library using the experimentally determined values at 1000 ppm, as indices to the cells of columns and rows of the 2D matrix. The assay value of each cell is contained in the combination AxBy (x = 1-7 and y = 1-25). Since only one cell out of 175 may possess the maximum response function (Rxy), deconvolution is the identification of this particular combination Chapter 2 ♦Results and Discussion - 64 - without looking at all 175 cells. Therefore, each B (aldehyde) was tested with all A’s, (acetophenones) and each A was tested with all B’s. The data was expanded in 175 cells of chalcones on the matrix by taking the average of the activity of the respective column and row, which were used as indices to the particular cells. This resulted in calculated antitumor activities for all 175 chalcones in the 2D matrix as shown in Table 2.15. Table 2.15. Calculated antitumor activities of designed library at a concentration of 1000 ppm. Set 1 Set 2 BL1 AL1 AL2 AL3 AL4 AL5 AL6 AL7 70 69 70 67 56 59 44 BL2 92 92 92 90 78 82 67 BL3 97 96 97 95. 83 86 71 BL4 99 BL5 100 100 97 85 89 74 99 100 100 97 85 89 74 BL6 98 97 98 96 84 87 72 BL7 99 85 89 74 97 85 89 74 99 100 100 100 97 BL9 100 100 100 97 85 89 74 BL10 88 87 88 86 74 77 62 BL11 99 85 89 74 99 100 100 97 BL12 100 100 97 85 89 74 BL13 89 89 89 87 75 78 64 BL14 78 78 78 76 64 67 53 BL15 83 82 83 80 68 72 57 BL16 82 82 82 80 68 71 57 BL17 82 81 82 79 68 71 56 BL18 84 83 84 82 70 73 58 BL19 89 88 89 87 75 78 63 BL20 82 81 82 79 67 71 56 BL21 90 89 90 87 76 79 64 BL22 83 82 83 80 68 72 57 BL23 97 96 97 95 83 86 71 BL24 66 66 66 64 52 55 41 BL25 87 86 87 84 73 76 61 BL8 99 A number of chalcones showed 100% inhibition at 1000 ppm concentration (Table 2.13). The activities of nine short-listed sub-libraries (inhibiting 100% at 1000 ppm) were screened at lower concentrations i.e. 100 and 10 ppm values. The inhibitory potentials at 10 ppm concentrations were used as indices to the cells of the 2D matrix Chapter 2 ♦Results and Discussion - 65 - comprising two columns and seven rows and the data was spread over 14 cells, as shown in Table 2.16. Table 2.16. Calculated antitumor activities of the most active library members at 10 ppm. Set 1 Set 2 AL1 AL3 BL4 37.5 35.5 BL5 48.5 46.5 BL7 39.5 37.5 BL8 43.5 41.5 BL9 50.5* 48.5 BL11 39.0 37.0 BL12 43.0 41.0 * most active member. It is evident that both sub-libraries AL1 and BL9 may be predicted to be the mostactive. Thus, for the identification of the most active chalcone, parallel synthesis of the members of the most active column AL1, and the most active row BL9, was carried out by means of microwave assisted organic synthesis. Synthesis of members of active libraries AL1, AL3 and BL9. Members of the active columns AL1 and AL3 as well as active row BL9 were synthesized in parallel under microwave irradiation. The structures of the members of AL1 and AL3 are given in Schemes 2.6, 2.7, whereas those of BL9 are given in Scheme 2.8. H O R R NaOH/ EtOH M.w. O O R R R 21 Ph 32 2-ClC6H4 46 5-Methyl-2-thienyl 22 2-OHC6H4 33 3-ClC6H4 47 5-Bromo-2-thienyl 23 3-OHC6H4 34 4-ClC6H4 48 5-Nitro-2-thienyl 24 4-OHC6H4 35 4-FC6H4 49 2-Pyrrolyl 25 2-OCH3C6H4 37 3-BrC6H4 50 2-Pyridyl A1B1-A1B25 26 3-OCH3C6H4 38 4-MeC6H4 51 3-Pyridyl (21-30, 3235,37-40, 45-52) 27 4-OCH3C6H4 39 3-OH,4-OCH3C6H3 52 4-Pyridyl 28 3,4(OCH3)C6H3 40 4-N(CH3)2C6H4 30 3-NO2C6H4 45 2-Thienyl R O Scheme. 2.6. Parallel synthesis of chalcones of active column AL1. Chapter 2 ♦Results and Discussion - 66 - R 53 R C6H5 62 3-ClC6H4 71 5-Methyl-2-thienyl R 54 2-OHC6H4 63 4-ClC6H4 72 5-Bromo-2-thienyl 55 3-OHC6H4 64 3-BrC6H4 73 5-Nitro-2-thienyl 56 4-OHC6H4 65 4-FC6H4 74 2-Pyrrolyl 57 2-OCH3C6H4 66 3-NO2C6H4 75 2-Pyridyl 58 3-OCH3C6H4 67 3-OH,4-OCH3C6H3 76 3-Pyridyl 59 4-OCH3C6H4 68 4-N(CH3)2C6H4 77 4-Pyridyl 60 3,4(OCH3)C6H3 69 4-MeC6H4 61 2-ClC6H4 70 2-Thienyl OH O A3B1-A3B25 (53-77) R Scheme. 2.7. Parallel synthesis of chalcones of active column AL3. R′ Cl R' O A1B9-A7B9 (12, 32, 62, 78-81) R′ 12 4′-NH2C6H4 79 4′-OHC6H4 32 C6H5 80 2′-NH2C6H4 62 3′-OHC6H4 81 3′-NH2C6H4 78 2′-OHC6H4 Scheme. 2.8. Parallel synthesis of chalcones of active row BL9. FT-IR spectral analysis of all the chalcones A1B1-A1B25, A3B1-A3B25 and A1B9-A7B9 was carried out. The C=O and C=C stretching vibrations for the prepared compounds appeared in the range of 1631-1690 cm–1 and 1508-1606 cm–1, respectively. Aryl C-H stretches are often observed at 3041-3246 cm–1. 1 H NMR spectra of the synthesized chalcones were recorded. A detailed spectral analysis of 1-(phenyl)-3-(5-methyl-2-thienyl)-2-propen-1-one (46) has been given in Table 2.17. The most characteristic chemical shifts were those of vinylic protons H (α) and H (β); the former appeared upfield at 7.21 ppm, while the later appeared at 7.88 ppm. The coupling constant was 15.3 Hz indicating a trans orientation of the double bond. The methyl protons appeared as a singlet at 2.53 ppm. In thienyl ring, doublet (d) at 6.76 ppm with a coupling constant 3.6 Hz was assigned to H–C(3), a doublet at 7.18 ppm with 3.6 Hz was assigned to H–C(4). A doublet at 8.01 ppm with a coupling constant 7.2 Hz was assigned to H–C(2′, 6′). A triplet at 7.51 ppm with coupling constant 7.2 Hz was assigned to the H–C(3′, 5′), whereas a triplet at 7.59 ppm, again with a coupling constant 7.2 Hz was assigned to H–C(4′). Chapter 2 ♦Results and Discussion - 67 - Table 2.17. 1H NMR data of 1-(phenyl)-3-(5-methyl-2-thienyl)-2-propen-1-one (46). Me 1 3' 4' 1' 2 3 α 5' 6' 4 β 2' 5 S O 46 δ ppm Multiplicity Integration J (Hz) Assignment 2.53 s 3H - H−C(CH3) 6.76 d 1H 3.6 H−C(3) 7.18 d 1H 3.6 H−C(4) 7.21 d 1H 15.3 H-C(α) 7.51 t 2H 7.2 H−C(3′, 5′) 7.59 t 2H 7.2 H−C(4′) 7.88 d 1H 15.3 H−C(β) 8.01 d 2H 7.2 H−C(2′, 6′) Antitumor screening. The purified and characterized compounds were assessed for their antitumor potencies through PDT assay. The individual members of the active libraries AL1, AL3 and BL9 were tested for their antitumor potential at 10, 100 and 1000 ppm concentrations and the data is shown in Tables 2.18-2.20. Table 2.18. Antitumor activities of chalcones of library AL1. Chalcone a Inhibitiona Chalcone Inhibitiona Chalcone Inhibitiona A1B1 17.0, 34.1, 62.1 A1B10 43.1, 47.5, 76.8 A1B19 26.2, 37.7, 45.9 A1B2 35.3, 40.2, 80.5 A1B11 71.9, 75.7, 96.3 A1B20 54.1, 56.4, 78.7 A1B3 53.6, 58.5, 100 A1B12 64.6, 74.4, 78.2 A1B21 44.3, 54.1, 54.1 A1B4 34.1, 56.1, 64.6 A1B13 32.9, 53.6, 93.9 A1B22 40.9, 75.4, 80.3 A1B5 19.5, 52.6, 57.5 A1B14 37.8, 58.5, 69.5 A1B23 67.2, 80.3, 83.6 A1B6 34.1, 48.8, 54.8 A1B15 53.6, 54.9, 74.4 A1B24 63.9, 98.4, 90.2 A1B7 35.4, 47.7, 53.6 A1B16 10.6, 29.5, 31.1 A1B25 42.6, 49.2, 68.8 A1B8 56.1, 68.7, 70.7 A1B17 14.7, 14.7, 36.1 A1B9 100, 100, 100 A1B18 11.5, 32.7, 49.2 Percent inhibition at 10, 100 and 1000 ppm, respectively. All the chalcones were tested on twelve potato-discs at each concentration. Chapter 2 ♦Results and Discussion - 68 - Table 2.19. Antitumor activities of chalcones of library AL3. Chalcone a Inhibitiona Chalcone Inhibitiona Inhibitiona Chalcone A3B1 72.8, 100, 100 A3B10 51.0, 79.7, 100 A3B19 75.6, 80.2, 100 A3B2 100, 100, 100 A3B11 33.2, 63.6, 100 A3B20 65.4, 66.7, 82.0 A3B3 57.8, 100, 100 A3B12 53.2, 77.9, 100 A3B21 65.4, 70.9, 73.1 A3B4 35.4, 70.5, 100 A3B13 37.1, 76.6, 100 A3B22 73.8, 81.8, 100 A3B5 76.8, 100, 100 A3B14 75.6, 91.0, 96.1 A3B23 57.7, 100, 100 A3B6 25.9, 49.3, 100 A3B15 64.9, 72.7, 100 A3B24 76.9, 77.9, 83.3 A3B7 31.2, 33.8, 100 A3B16 72.3, 80.8, 100 A3B25 80.8, 89.5, 94.9 A3B8 60.2, 83.6, 100 A3B17 78.2, 84.2, 100 A3B9 38.9, 60.2, 100 A3B18 52.5, 100, 100 Percent inhibition at 10, 100 and 1000 ppm, respectively. All the chalcones were tested on twelve potato-discs at each concentration. Table 2.20. Antitumor activities of the chalcones of library BL9. a Chalcone Inhibitiona Chalcone Inhibitiona A1B9 100, 100, 100 A5B9 49.4, 59.1, 100 A2B9 100, 100, 100 A6B9 55.8, 70.1, 100 A3B9 61.0, 78.6, 98.0 A7B9 52.6, 66.8, 86.4 A4B9 70.7, 77.9, 100 Percent inhibition at 10, 100 and 1000 ppm, respectively. All the chalcones were tested on twelve potato-discs at each concentration. It is evident from the data that the most active members of the library A1B9 and A2B9 and A3B2 (cf. for structures, Scheme 2.5) displayed 100% tumor inhibition at a concentration of 10 ppm, and thus, can be regarded as leads of the designed library. It may be recalled that the calculated deconvolution led to the identification of the same chalcone, A1B9, as lead structure. However, the experimental data showed that A2B9 and A3B2 also exhibited 100% tumor inhibition at 10 ppm and, therefore, are equally important candidates for developing into highly effective chemotherapeutic agents. Cl OH HO O O A1B9 (32) A3B2 (61) Cl OH O A2B9 (78) Chapter 2 ♦Results and Discussion - 69 - The identification of the lead structure in 175 member library through positional scanning method is shown in Figures. 2.12 and 2.13. AL1 A1B1 A3B1 175 member library A1B2 A3B2 AL2 A1B3 A3B3 A1B4 A3B4 AL3 A1B5 A3B5 A1B6 A3B6 Set 1 AL1-AL7 A1B7 A3B7 AL4 A1B8 A3B8 A1B9 A3B9 AL5 A1B10 A3B10 A1B11 A3B11 A1B12 A3B12 AL6 A1B13 A3B13 A1B14 A3B14 AL7 A1B15 A3B15 A1B16 A3B16 A1B17 A3B17 A1B18 A3B18 A1B19 A3B19 A1B20 A3B20 A1B21 A3B21 A1B22 A3B22 A1B23 A3B23 A1B24 A3B24 A1B25 A3B25 OH HO Cl O O A3B2 A1B9 LEADS Fig. 2.12. Identification of antitumor leads through deconvolution of 175 member library (Set 1). Chapter 2 ♦Results and Discussion - 70 - BL1 BL2 175 member library BL3 BL4 BL5 A1B9 BL6 A2B9 BL7 Set 2 BL1-BL25 BL8 A3B9 BL9 BL9 BL10 A4B9 BL11 A5B9 BL12 BL13 A6B9 BL14 A7B9 BL15 BL16 BL17 BL18 BL19 BL20 BL21 BL22 BL23 BL24 BL25 Cl Cl OH O O A2B9 A1B9 LEADS Fig. 2.13. Identification of antitumor leads through deconvolution of 175 member library (Set 2). Chapter 2 ♦Results and Discussion - 71 - Therefore, deconvolution by means of positional scanning is a quite cost-effective protocol capable of identifying a lead compound; however subtle differences in activity within the library cannot be detected. This disadvantage, however, is more than compensated by the advantage of the ease of library synthesis. 2.2.2.4. Structure activity relationship. Following the identification of hits of the designed library by deconvolution, it is essential to highlight the structural features that contribute to tumor inhibition. Such a correlation may be performed at the library level as well as at the level of individual compounds. The order of activity within these seven sub-libraries, in terms of substituent is H > OH > NH2 or more specifically, H > 3-OH > 2-OH > 4-OH > 3-NH2 > 2-NH2 > 4-NH2 Evidently, the library derived from unsubstituted acetophenone (AL1) shows 100% tumor inhibition, while all the three libraries derived from isomeric amino acetophenones (AL5-AL7) show weak tumor inhibitory potential (Table 2.16). Moreover, the substituents at para position of ring B lower the activity to a greater extent as compared to those at ortho or meta positions. Among the libraries of Set 2 (BL1-BL25), seven sub-libraries (BL4, BL5, BL7, BL8, BL9, BL11 and BL12) showed 100% tumor inhibition at a concentration of 1000 ppm. All the sub-libraries of various analogues of methoxy benzaldehydes (BL5, BL7 and BL8) showed 100% activity at 1000 ppm, reflecting the significance of a OCH3 substituent at the aromatic ring. Regarding the tumor inhibitory potential of the individual chalcones, it is interesting to note that A1B9, A2B9 and A3B2 were found to be the lead structures, since this further confirms the above conclusion that an unsubstituted A-ring is important for tumor inhibition, whereas ortho substitution was found to be important on ring B (o-chloro and o-hydroxy substituents in lead structures). Moreover, chalcones substituted with an electron donor substituent on ring A, e.g., N(CH3)2 or 3CH3 group were found to be the least active. Varieties of chalcones were derived from heteroaryl aldehydes, but the pyridyl chalcones A1B23-A1B25 and A3B23-A3B25 were found to be significantly active. Furthermore, A3B19 and A3B22 containing methyl substituted thienyl and pyrrolyl moieties as ring B resulted in significant antitumor potencies. Chapter 2 ♦Results and Discussion - 72 - 2.3. Peptidyl chalcones and peptidyl heterocycles The standard approach of combining heterocycles and peptides is the fusion of a small-molecule fragment in the side chain of amino acid building blocks or to integrate heterocycles via standard acylation of amino groups of the peptide and the heterocycles to create hybrid molecules.201-206 Only few examples have been reported where heterocycles are integrated in the peptide backbone itself, as this requires in most cases specific, C-terminal derivatization of peptides.207-212 Current strategy describes the synthesis of peptidyl chalcones on solid phase by the use of 2phosphoranylideneacetate developed as a linker reagent on polymeric triphenylphosphine resin. The use of triphenylphosphine resin has been reported in the Wittig and Mitsunobu reactions.213-214 Polymer-supported C-acylation was reported for the first time, employing triphenylphosphine resin for the synthesis of polymeric 2-phosphoranylidene acetonitrile as a polymer reagent and an anchoring group allowing subsequent derivatization of the immobilized product.215-216 Furthermore, Cacylation of polymer-supported 2-phosphoranylidene acetates was developed as a 0linker reagent with protected amino acids yielding 2-acyl-2-phosphoranylidene acetates as flexible intermediates for the C-terminal variation of carboxylic acids. As a result, several novel peptidyl bis- and tris-electrophiles have become accessible, namely peptidyl diketoesters, peptidylketoaldehydes and peptidyl vinyl ketones (Fig. 2.14).217 Pep HN Ph Ph R1 P OR* O R1 Pep HN ∗ O O R1 ∗ R″ Pep HN R1 Pep HN ∗ ∗ ∗ ∗ O O ∗ O OR* O H O Fig. 2.14. Peptidyl bis- and tris- electrophiles obtained from 2-acyl-2-phosphoranylidene acetate. Chapter 2 ♦Results and Discussion - 73 - On the basis of above findings, a small library of peptidyl chalcones was designed, synthesized and then converted to different classes of heterocyles, namely oxazoles, pyrazolines, pyrazoles, thiazepines and diazepines.218 2.3.1. Synthesis of peptidyl chalcones The conjugation of peptides with different heterocycles based on C-acylation of peptides led to peptidyl chalcones, which are also known as peptidyl vinyl ketones. The peptidyl chalcones were synthesized on phosphorane bound polystyrene divinylbenzene (PS) support using polymer-supported carbanion equivalents. Developing carbanion equivalents on solid support is an ideal approach for establishing complex reaction sequences including C-C coupling steps in polymerassisted synthesis. Ideally, the successful method should allow reactions with easily available building blocks, smooth reaction conditions and further derivatization after the C-C coupling step. Lithiated dithioacetals have been reported as polymersupported carbanion equivalents.219-221 These acetals are strongly basic and, therefore, are not a convincing approach for the synthesis of sensitive products. The integration of a C-acylation step into the standard protocol of peptide synthesis would be especially attractive for the synthesis of peptide mimetics and in the attempts to reduce the peptide character of inhibitors, which is a common challenge in drug development programs. Polymer-supported acyl anion equivalents were first established using phosphoranePS support for synthesizing a library of norstatine isosteres, active as aspartic protease inhibitors.215 Furthermore, C-acylations of polymer-supported 2-phosphoranylidene acetates (linker reagents) with different protected amino acids yielded 2-acyl-2phosphoranylidene acetates, which are flexible intermediates for the C-terminal variation of carboxylic acids: peptidyl-2,3-diketoesters, peptidyl vinyl ketones, peptidyl-2-ketoaldehydes and 1,3-diamino-2-hydroxy-propanes.217 Based on the previous studies, a parallel library of peptidyl chalcones has been synthesized on phosphorane-PS support and then subsequently converted to a library of peptidic heterocycles. This integration of heterocycles in the peptide backbone with specific Cterminal derivatization may lead to novel peptidomimetics.218 The synthetic strategy involved the following main steps: 1. Synthesis of linker reagent. Chapter 2 ♦Results and Discussion - 74 - 2. Protection of triphenylphosphin-PS support. 3. Deprotonation leading to Wittig ylide. 4. Acylation of Wittig ylide. 5. Amide couplings leading to peptides. 6. N-Acetylation of peptides. 7. Deprotection of phosphorane. 8. Synthesis of peptidyl chalcones. 2.3.1.1. Synthesis of linker reagent DCC/ DMAP OH Br O O Br Si Si O OH 82 As a first step toward polymer-supported C-acylation, linker reagent has been introduced as a tool in solid phase synthesis combining the functions of a polymer reagent and those of an anchoring group allowing for subsequent derivatization of the immobilized product.217 2-Trimethylsilylethyl-2-bromoacetate (82) was used as linker reagent on phosphorane supported polystyrenedivinylbenzene resin. This ester could be synthesized by pyridine catalyzed esterification of an acid halide with trimethylsilyl ethanol or through Steglich esterification. In the second method, the acid part is activated using dicyclohexylcarbodiimide (DCC) and catalytic amount of 4-N,N-dimethylaminopyridine (DMAP) and then reacted with the desired alcohol.222 This method has the advantage of giving only the desired ester under controlled conditions. The synthesis was achieved in good yield by activating bromoacetic acid with DCC/DMAP followed by esterification with 2-trimethylsilylethanol. The crude product after vacuum distillation gave the desired ester, which was further used as a facile linker reagent for C-C acylations on phosphorane support. The product was characterized by 1H NMR and 13C NMR analysis. 1 H NMR analysis of 2-trimethylsilylethyl-2-bromoacetate (82) showed a singlet of nine protons at 0 ppm for trimethylsilyl group, two triplets at 1.10 and 4.21 ppm, each of two protons for two methylenes attached to Si(CH3)3 and an ester moiety respectively. The two methylene protons attached to Br appeared as singlet at 3.90 ppm (Fig. 2.15). Chapter 2 ♦Results and Discussion - 75 - Fig. 2.15. 1H NMR analysis of 2-trimethylsilylethyl-2-bromoacetate 82. 2.3.1.2. Protection of triphenylphosphin-PS support Ph O Br O P Ph Ph P Ph Br Si Toluene, Mw,15 min O O Si 83 The linker reagent [2-trimethylsilylethyl-2-bromoacetate 82] was loaded on triphenylphosphin-PS support for synthesizing a Wittig salt. The commercially available triphenylphosphin-PS support was alkylated with the synthesized linker under microwave irradiation at 100 °C for 15 min in toluene (successful alkylation can also be carried out through stirring at room temperature for 72 h). The alkylation was confirmed after recording ATR-FT-IR spectrum of the intermediate obtained (Fig. 2.16). A b s o r b a n c e Wave number cm -1 Fig. 2.16. ATR-FT-IR of triphenylphosphorane support after alkylation with trimethylsilylethyl bromoacetate. Chapter 2 ♦Results and Discussion - 76 - The infrared spectrum of alkylated support 83 showed C-H and Ar-H absorbance of polystyrene at 2851 cm-1 and 3056 cm-1, while a characteristic absorbance at 1735 cm-1 corresponded to the ester carbonyl functionality. The resultant protected triphenyl phosphin-PS support was accessible for the synthesis of a Wittig ylide through base catalyzed elimination of an acidic proton at methylene directly attached to the phosphorous atom. 2.3.1.3. Deprotonation leading to Wittig ylide Ph P Ph Br O Si Ph Ph TEA / DCM P O O Si O 84 The phosphonium salt obtained after alkylation has highly acidic proton and was easily deprotonated using triethylamine as a base. The resulting Wittig ylide (polymer-bound phosphoranylidene acetate) enjoys greater stability due to resonance stabilization with the adjacent carbonyl moiety of the ester group. The product, after thorough drying, was stored at room temperature. IR analysis of product 84 showed a peak at 1740 cm-1 that was assigned to ester C=O and this confirmed the identity of Wittig ylide. 2.3.1.4. Acylation of Wittig ylide Polymer-bound phosphoranylidene acetate i.e. Wittig ylide can serve as an equivalent analogue of polymer bound acyl anion. The acylation of the Wittig ylide 84 with different F-moc protected amino acids serves as the starting point for standard peptide synthesis. However, polymer-bound phosphoranylidene acetate, being a weak nucleophile, posed a major challenge in finding efficient acylation conditions. Some early attempts of acylation remained unsuccessful using different activating agents for enhancing the electrophilicity of the carbonyl of diisopropylcarbodiimide (DIC) with catalytic amount of the amino acid e.g. DMAP, N-ethyl-N′-(3- dimethylaminopropyl)-carbodiimide hydrochloride (EDC) with DMAP and 1[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium hexafluorophos phate-3-oxide (HATU) with diisopropylethylamine (DIPEA). Successful acylations could be performed with 1-(2-mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole (MSNT), whereas usage of fluoro-N,N,N′,N′-bis(tetramethylene)formamidiniumhexafluorophos phate (TFFH) as acylation agent destroyed labile trimethylsilyl group on linker Chapter 2 ♦Results and Discussion - 77 - reagent. Therefore, for synthesizing a library of peptidyl chalcones, MSNT and lutidine (as a base) were used for racemization-free acylation of phosphoranylidene acetate. MSNT catalysed C-acylation of phosphorane was performed with a broad choice of carboxylic acids. This reagent tolerated the standard side-chain protecting groups used in peptide synthesis such as Boc, tert-butyl, and trityl groups. Ph Ph P Ph R1 P Ph Fmoc-AA-OH O O FmocHN Si MSNT, Lutidine, DCM O O Si O 85, R1 = 86, R1 = The mechanism of this C-acylation is shown in Scheme 2.9. Lutidine (as a base) deprotonates OH of an amino acid and results in a nucleophile. This nucleophile attacks the sulphone of MSNT. As a result, triazole departs whereas the amino acid now contains a better leaving group (mesitylenesulfonate). In the following step, phosphoranylidene acetate (a weak nucleophile) attacks the carbonyl sulphonate ester resulting in the removal of sulphonate while the amino acid is loaded on 84 through acylation (Scheme 2.9). R1 Fmoc N H O O O N R1 S N N O NO2 Fmoc N H O S O O O N N N NO2 Ph P Ph RO O O S O O Fmoc N H Ph Ph R1 P OR O O Scheme 2.9. MSNT/Lutidine mediated acylation on phosphorane support. The basicity of the phosphorane reagent did not allow cleavage of detectable amounts of the Fmoc-protecting group. This was verified by the negative Kaiser test subsequent to all acylations. The coupling yield of the acylated products 85 and 86 Chapter 2 ♦Results and Discussion - 78 - was determined by spectrophotometric quantification of Fmoc group that was cleaved off a dried resin sample ~ 5 mg (Table 2.21). Table 2.21. % Yield of acetylated products obtained on solid support. Entry Amino acid Yield (%)a Conditions O FmocHN 5 equiv Fmoc-Phe, 5 equiv MSNT, OH 85 88 4.9 equiv 2,6-lutidine, DCM, RT. O FmocHN 86 5 equiv Fmoc-Val, 5 equiv MSNT, OH 84 4.9 equiv 2,6-lutidine, DCM, RT. a Calculated after standard Fmoc cleavage of ~ 5 mg sample. The IR spectrum of the acylated support 85 showed great similarity to the corresponding polymeric Wittig salt 83. Important differences were the absorption due to Si-C bond, which after alkylation could be seen at 837 cm-1, while Fmoc moiety appeared as a double peak at 742-760 cm-1. After successful acylation, the protecting group Fmoc a was removed from small samples ~ 5 mg of resin, using 20% piperidine/DMF and the cleaved adducts c and d were quantified through measuring absorbance λ1 = 267 nm, λ2 = 289 nm and λ3 = 301 nm, as shown in Scheme 2.10. The concentration of the cleaved Fmoc moiety was calculated by Beer Lambert Law (Section 4A.3.4.). HN R HN R O O O O H H a N H b N H HN R O H2N R N O CO2 NH d c Scheme 2.10. Elimination of base-labile Fmoc moiety demasking amino functionality. Chapter 2 ♦Results and Discussion - 79 - 2.3.1.5. Amide coupling leading to peptide synthesis Ph Ph R1 Ph P O FmocHN O Fmoc Si R1 O H N amide coupling Rn O Ph P O N H O n Si O n = 1, 2 87-91 The acylated products 85-86 were subjected to Fmoc deprotection using 20% piperidine/DMF (2 x 6 min). The resulting free N-terminus amino group was used as the attachment site for adding amino acid monomers thus yielding peptidyl 4-amino3-oxo-2-phosphoranylidene butanoates 87-91. The activating agents were first developed for racemization-free-amide coupling on phosphorane support through test reactions. Various coupling methods using different coupling reagents e.g., DIC, N[(1H-benzotriazol-1-yloxy)(dimethylamino)methylene]-N-methylmethanaminiumtetra fluoroborate (TBTU) with DIPEA and DIC with N-hydroxybenzotriazole (HOBT) were tested for the coupling of Fmoc amino acids. The reaction of DIC with a carboxylic acid yields a highly reactive O-acyl urea. In order to enhance the electrophilicity of the carboxylate group, the negatively charged oxygen must first be "activated" to a better leaving group. R2 Fmoc N H N C N OH R2 Fmoc O H N O N H O HN a b R2 Fmoc N H R1 H N N O O c f Fmoc N H O R2 O e OR H2N N H d N H O H N O OR R1 Scheme 2.11. Amide coupling through DIC. When DIC is used for this purpose, the negatively charged oxygen acts as a nucleophile, attacking the central carbon in DIC. Nucleophilic attack by an amino group (of amino acid loaded onto support) to the former C-terminus (carbonyl group of incoming amino acid now present as an ester b) results into amide coupling (Scheme 2.11). However, intramolecular rearrangement of b proceeds without H+ Chapter 2 ♦Results and Discussion - 80 - uptake, together with the migration of an amine moiety to carbonyl carbon of ester functionality resulting into a by product f, which was stuck to resin and was not easily removed through washings. As a result, the desired product was contaminated with incomplete peptide sequence and urea derivative f, as indicated through the LC-MS of the cleaved product obtained in the test reaction. O N N N N N N O O-Form N N N N Fmoc O H N Fmoc O O H N O R2 R2 N N N O N-Form Fmoc O H N N O base H N BF4 R2 O N N N N N N O O O-Form R2 O R2 NHFmoc N-Form NHFmoc O HN OR base H R1 R2 Fmoc N H O H N O N OR R1 N N O base H Scheme 2.12. Amide coupling through TBTU/base. TBTU containing non-nucleophilic tetrafluoroborate anion and DIPEA were tested for effective amide coupling. TBTU exists in two isomeric forms i.e. O-TBTU and NTBTU, existing in dynamic equilibrium under basic conditions. The equilibrium shifts towards N- Form in strong basic conditions such as in the presence of Et3N of DIPEA. Under basic reaction conditions, the carboxylic acid part of amino acid deprotonates. The resulting carboxylate anion initially attacks the α-carbon atom of the immonium salt (uronium (O-form) or the guanidinium N-oxide (N-form)) to form an unstable acyloxyimmonium intermediate with the release of benzotriazolyloxy anion (BtO−). Chapter 2 ♦Results and Discussion - 81 - The BtO− nucleophile, thus generated, attacks the acyloxyimmonium intermediate generating benzotriazolyl ester, which rearranges into the corresponding N-acylated isomer. The two forms of the active intermediates reacted with the free amine ending of phosphorane support resulting in the synthesis of the corresponding amide (Scheme 2.12).223 When the cleaved product was analyzed through LC-MS, the product had low enantiopurity that was expected to result from intramolecular rearrangement of acyloxyimmonium intermediate to 4H-5-oxazolone and subsequent epimerization in the desired product (Scheme 2.13). R O HN O O N BF4 N O R H O N R O N R' R' O N R O N R' O H O O R' N Scheme 2.13. Mechanism of 4H-5-oxazolone mediated epimerization. Another coupling reagent HOBT was used after pre-activation of Fmoc amino acids with DIC. HOBT together with DIC was found to be the safest with the phosphorane support and Fmoc chemistry. The reaction proceeds through nucleophilic attack of the negatively charged oxygen on the central carbon in DIC (Scheme 2.14). The yields of the amide coupling reactions using DIC/HOBT are given in Table 2.22. Fmoc R2 N C N R2 Fmoc OH N H O O N H N H HN O N H OR H2N O H N O OR H N R2 R1 O R2 N H O OH N N N O N N N Fmoc N H O N N N Fmoc O R1 Scheme 2.14. Mechanism of DIC/HOBT mediated amide coupling. The yields of amide couplings were determined after the Fmoc deprotection of small resin samples. Chapter 2 ♦Results and Discussion - 82 - Table 2.22. Yields of the standard amide coupling using DIC/HOBT. No. Amino acid Ph O 87 FmocHN Yield (%)a Product OH Ph P O FmocHN 86 O N H O Si O Ph O FmocHN O OH FmocHN N H 88 OH FmocHN N H Ph O O OH 84 Ph P O N H 84 O O Si O Ph OH H N FmocHN O a Si O O 91 82 Ph FmocHN FmocHN Si O P O 90 O O 89 FmocHN O Ph O FmocHN Ph P P O N H Ph O O 83 Si O Calculated after standard Fmoc cleavage of ~ 5 mg sample. 2.3.1.6. N-Terminal acetylation of peptides (92-95) The amino terminus of the peptides 87-89 and 91 was deprotected from Fmoc moiety using standard F-moc cleavage conditions. The resulting unprotected peptides were subjected to N-terminal amine acetylation using acetic anhydride (5 eq) in DMF resulting in acetylated peptide sequences 92-95. The reaction was repeated two times to ensure complete acetylation. The complete conversion of N-terminal amino group to N-acetyl derivatives was confirmed through the Kaiser test (Section 4A.3.11). Chapter 2 ♦Results and Discussion - 83 - Ph H R1 O H N Rm N H n Ph Ph P Ac2O/DMF O O Si O O R1 O H N Rm O N H n n = 1-2 Ph P O Si O n = 1-2 92, R1 = R2 = 93, R1 = R2 = 94, R1 = R2 = 95, R1 = R2 = R3 = 2.3.1.7. Deprotection of phosphorane support After having accomplished a successful synthesis of the desired peptide sequence, the final step was the removal of trimethylsilylethyl group. Tetrabutylammonium fluoride (TBAF) is well known for TMSE removal.224 However, it was found to destroy peptide part under basic condition. By controlling the pH in buffer medium, the removal of tetrabutylammonium ions from the resin could not be achieved even after repeated washings and always resulted in contaminated products of successive steps. Furthermore, tris(dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F) was found as a safe desilylating reagent by removing TMSE without any washing problems (Fig 2.17).225 TAS-F, a mild neutral agent is an in situ source of fluoride ion. It successfully removed trimethyl silyl moiety and resulted in subsequent decarboxylation of supported peptides (Scheme 2.12). N N S N F Si F Fig. 2.17. tris(Dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F). R2 N H Ph Ph R1 P O O O F Si R2 C2H4 (CH3)3SiF N H Ph Ph R1 P O O O Ph Ph R1 P R2 CO2 N H H O H Scheme 2.12. Hydrolysis of protected 4-amino-3-oxo-2-phosphoranylidene butanoates with TAS-F. Chapter 2 ♦Results and Discussion - 84 - It was, therefore, used for deprotection of all the peptides sequences 92-95. Ph H N O R1 O N H Rm n Ph Ph P TAS-F/DMF O O Si O O R1 O H N N H Rm n n = 1, 2 Ph P H O n = 1, 2 96-99 92-95 The successful removal of TMSE was confirmed through ATR-FT-IR of the resulting supports 96-99 by the disappearance of Si-C vibration at 837 cm-1 in the resulting products (Fig 2.18). Fig. 2.18. ATR FT-IR of 96 after removal of TMSE protection with TAS-F. 2.3.1.8. Synthesis of peptidyl chalcones (100-107) Peptidyl-3-amino-2-oxo-1-phosphoranylidene propanes 96-99 obtained were subjected to Wittig cleavage using a variety of aliphatic and aromatic aldehydes that led to the formation of peptidyl chalcones. All the reactions were performed in THF. In case of aliphatic aldehydes, the reactions were complete in 4-5 h by stirring at room temperature, whereas with aromatic aldehydes, the reaction mixture had to be heated at 50oC for 5-6 h. Ph H N O R1 O Rm N H n Ph P H N R'CHO/THF H O n = 1, 2 O R1 O Rm N H n R' O n = 1, 2 100-107 The resulting peptidyl chalcones were purified through preparative HPLC in some cases. The yields were calculated for the pure compounds, whereas the purity of the crude products was assessed through LC analysis (Table 2.23). Chapter 2 ♦Results and Discussion - 85 - Table 2.23. Synthesis of peptidyl chalcones 100-107. Entry 100 O H N O 101 N H O 102 106 N H a 74 85 65 80 72 64 78 90 74 80 71 Br Br Br O F O N H O N H O H N Cl O H N O 61 S O N H O O Cl O H N N H 68 Cl O N H 104 105 80 O H N O 70 F N H 103 O O O H N O 62 F N H H N Yield (%)b O O H N O 107 Purity (%)a Product O O N H O Purity of crude products assessed using LCMS; b isolated yield. Chapter 2 ♦Results and Discussion - 86 - The structures of the synthesized peptides 100-107 were confirmed through HR-MS, 1 H NMR and 13 C NMR analysis. In the HR-MS analysis of chalcone 100, the molecular ion peak (M+H+) was found as 421.2456. Fig. 2.19. 1H NMR (300 MHz, CDCl3) spectrum of chalcone 100. Regarding 1H NMR analysis, the methyl protons of the aliphatic chain appeared as a doublet at 0.91 ppm with a coupling constant of 7.8 Hz and an integration of six protons, while the methine CH proton appeared as a multiplet at 1.65-1.81 ppm. A three proton singlet at 1.94 ppm was assigned to protons of the acetyl group. A broad triplet at 2.07 ppm with a coupling constant of 6.1 Hz was assigned to allylic CH2 protons. Benzylic protons from two phenylalanine units were found as multiplets at 2.91-3.20 ppm with an integration of four protons. α-Protons of phenylalanine units were found as multiplets at 4.64-4.78 ppm and 4.85-5.05 ppm, each with one proton integration. The two doublets at 6.15 ppm and 6.56 ppm, each with a coupling constant of 7.3 Hz were assigned to two NH protons of the amide linkage. The doublet at 6.15, was assigned to α-vinylic protons. A large coupling constant of 15.9 Hz confirmed the trans configuration of the double bond in the synthesized chalcone. The β-vinylic proton appeared downfield as a multiplet along with aromatic protons of phenyl rings at 6.89-7.06 ppm, with an integration of 11 H′s. 13 C NMR spectrum further confirmed the identity of the product as shown in Fig. 2.20. Important signals were observed at 21.9, 22.7, 27.3 ppm for methyl, methine and methylene protons, respectively. Chiral carbons appeared at 53.9 and 56.6 ppm, respectively, whereas the three carbonyls were found at 169.3, 169.9 and 195.6 ppm. Chapter 2 ♦Results and Discussion Fig. 2.20. - 87 - 13 C NMR (100 MHz, CDCl3) spectrum of chalcone 100. 2.3.2. Synthesis of peptidyl heterocycles Molecular combinations of peptides and heterocycles have been investigated in order to blend the desirable properties of peptides with those of heterocycles to obtain bioactive and bioavailable, metabolically stable and membrane-permeable molecules.226 Peptidyl chalcone derivatives are accessible for converting into a wide variety of heterocycles. As mentioned earlier (Chapter 1), chalcones may undergo a (3+2) annulation with a variety of reagents leading to HC’s of varying size e.g., reaction of enone moiety with o-aminothiophenol and o-phenylenediamine leads to 7membered heterocycles i.e. 1,5-benzothiazepines and 1,5-benzodiazepines. Likewise, the reaction of enone with PhNHNH2 or NH2OH led to 5-membered pyrazoles and oxazoles respectively.218 N O H N O R2 O Rn N H n N NR R' N NR O N S n = 1, 2 N O Fig. 2.21. Variety of heterocycles accessible through α,β-unsaturated ketone template. Following the same strategy, peptidyl chalcones were reacted with different reagents to yield different peptidyl heterocycles, as discussed in following sections. Chapter 2 ♦Results and Discussion - 88 - 2.3.2.1. Synthesis of peptidyl oxazoles Oxazoles are the rigid scaffolds containing the elements of amide bond.227 They are therefore astute choice for developing drug-like characteristics in desired peptide sequences. They are known as adrenergic receptor agonists and bind to the dopamine receptor.228-229 Peptidyl oxazoles were prepared by reacting peptidyl chalcones with hydroxylamine (3 eq) in ethanol in the presence of AcOH and NaOAc as catalysts, whereby the products were separated as white solids.151 The structures of peptidyl oxazoles were confirmed through HR-MS, 1H NMR and 13 C NMR analysis. The purity and yield of oxazoles 108-109 are shown in Table 2.24. Table 2.24. Synthesis of peptidyl oxazoles 108-109. Entry 108 Purity (%)a Product O H N Yield (%)b O N N H O 90 85 80 68 F Cl 109 a O H N O N H N O Purity of crude products assessed using LCMS; b isolated yield. The synthesis of 108, for example, was confirmed through HRMS analysis where the molecular ion peak (M+H+) was found at 507.1830 as expected (507.1833). In the 1H NMR spectrum (Fig. 2.22), a singlet at 1.84 ppm with an integration of 3 protons was assigned to the acetyl group. Three signals, a dd, a multiplet and another dd with an integration of one, two and one proton, respectively, appeared in the range of 2.453.45 ppm indicating the presence of a CH2 group. These multiplets were assigned to benzylic protons of phenylalanines. A dd appeared at 2.73 showing geminal coupling (12 Hz) and a vicinal coupling (9 Hz). Similarly, a dd at 3.14 ppm appeared with the coupling constants of 15 Hz and 8 Hz, respectively. The two dd’s at 5.39 and 4.52, each with an integration of one proton were assigned to two α- hydrogens at two Chapter 2 ♦Results and Discussion - 89 - chiral centers; dd’s showed a vicinal coupling constant of 6 Hz and 15 Hz, respectively, indicating the cis and trans placement of two vicinal protons with Fig. 2.22. 1H NMR (300 MHz, CDCl3) spectrum of oxazole 108. respect to the chiral hydrogen. Two amide NH protons appeared as singlets at 6.70 and 6.90 ppm, whereas a multiplet in the range of 7.03-7.22 ppm with an integration of 14 protons was assigned to aromatic protons of phenyl and oxazole rings. Fig. 2.24. In 13 13 C NMR (100 MHz, CDCl3) spectrum of 108. C NMR spectrum of oxazole 108 (Fig. 2.24), peaks for all carbons were observed. The most upfield signal at 22.58 ppm was assigned to the methyl carbon of terminal acetyl group. The peaks at 36.98 ppm and 37.64 ppm were assigned to methylene carbons of two benzyl groups. The methine carbons of the chiral centers appeared downfield at 43.57 ppm and 53.90 ppm. The other important signals Chapter 2 ♦Results and Discussion - 90 - corresponded to the carbons of oxazole ring. C-5 attached to nitrogen of oxazole appeared at 170.90 ppm, whereas the one attached to oxygen (C-3) appeared at 157.56 ppm. The other C-4 was found at 115.4 ppm. The two carbonyl carbons appeared at 169.15 and 171.05 ppm, respectively, while the aromatic carbons appeared in the range of 115.35 ppm to 159.15 ppm. 13 C-F19 couplings were observed in the cases where peaks were separated. The carbon as a doublet at 157.65 ppm and a 1J of 296 Hz was assigned to one attached to fluorine atom. The ipso carbon of 2-chloro-6fluorophenyl ring attached to oxazole ring appeared as a doublet at 139.64 ppm with a 2 J coupling as 96 Hz. The carbon attached to chlorine atom appeared at 129.45 with a 3 J coupling of ~ 12 Hz. 2.3.2.2. Synthesis of peptidyl pyrazolines Pyrazolines are important N-containing five-membered heterocyclic compounds used as antitumor, antibacterial and antituberculotic agents.230 Peptidyl pyrazolines were prepared by reacting peptidyl chalcone with hydrazines (Table 2.25).153 Table 2.25. Synthesis of peptidyl pyrazolines 110-114. Entry 110 O N H 111 H N H N N H 114 N H H N 95 65 98 61 95 66 65 53 89 67 N N O N H N N O N H O H N Yield (%)b N NH O O O N H a H N O N H N H O O 112 O O O N H 113 Purity (%)a Product N N O O Purity of crude products; b isolated yield. N H N N H Chapter 2 ♦Results and Discussion - 91 - A mixture of AcOH and NaOAc was used as catalyst in pyrazoline synthesis. The synthesized compounds were separated as white solids. The peptidyl pyrazolines 110114 were oxidized to the respective pyrazoles without further confirmation. 2.3.2.3. Synthesis of peptidyl pyrazoles Peptidyl pyrazolines 110-114 were oxidized to the corresponding pyrazoles 115-118 by their reaction with dichlorodicyanoquinone (DDQ). The residue was subjected to purification by preparative HPLC and pyrazoles were separated as white solids. The resulting products were characterized through NMR analysis. Yield and purity of the synthesized products are given in Table 2.26, while the characterization was done on the basis of LC-MS and 1H NMR data. Table 2.26. Synthesis of peptidyl pyrazoles 115-118. Entry 115 O N H 116 H N H N N H a H N N H H N Yield (%)b 89 >95 84 >95 85 >95 60 >95 N NH O N N O N H O O N H N H O O 117 O O O N H 118 Purity (%)a Product N N O O N H N N Purity of crude products; b isolated yield. In 1H NMR analysis of peptidyl pyrazole 115 (Table 2.27), the most upfield doublet at 1.22 ppm with an integration of 3 Hs and a coupling constant of 7.9 Hz, was assigned to CH3 group of alanine, whereas the singlet at 1.73 ppm was assigned to methyl of acetyl group. A singlet of 3 Hs appeared at 1.98 ppm was assigned to methyl group at C-5 of the pyrazole. Two multiplets at 2.65-2.73 ppm and 2.91-2.95 ppm, each with Chapter 2 ♦Results and Discussion - 92 - an integration of two protons were assigned to the benzylic protons of two phenylalanines. The three chiral centers present in the peptide part of the molecule appeared in the range of 4.20-4.24 ppm, 4.46-4.49 ppm and 4.99-5.05 ppm, each with a multiplicity of one proton. The aromatic hydrogen of pyrazole ring (H-4) was found as a singlet at 5.31 ppm, whereas the NH hydrogen of pyrazole appeared as a singlet at 5.86 ppm. The protons of three amide NH were found at 6.64 ppm, 8.02 ppm and 8.20 ppm as doublets, each with a coupling constant of ~ 8 Hz. The aromatic protons corresponding to two phenyl rings appeared at 7.12-7.25 ppm as multiplet of ten protons. Table 2.27. 1H-NMR analysis (300 MHz, DMSO-d6) of peptidyl pyrazole 115. δ ppm Multiplicity Integration Coupling constant J (Hz) Assignment 1.22 d 3H 7.9 H-Cβ(Ala) 1.73 s 3H - H-C(CH3CO) 1.98 s 3H - H-C(CH3 pyrazole) 2.65-2.73 m 2H - H-Cβ1,β1′(Phe, Benzyl) 2.91-2.95 m 2H - H-Cβ2,β2′(Phe, Benzyl) 4.20-4.24 m 1H - H-Cα(Ala) 4.46-4.49 m 1H - H-Cα(Benzyl) 4.99-5.05 m 1H - H-Cα(Phe) 5.31 s 1H - H-C(CH pyrazole) 5.86 s 1H - NH(pyrazole) 6.64 d 1H 8.0 NHI 7.12-7.25 m 10H - H-C(arom.) 8.02 d 1H 8.1 NHII 8.20 d 1H 8.0 NHIII 2.3.2.4. Synthesis of peptidyl benzothiazepines Benzothiazepines exhibit diverse biological activities. They are known as calcium antagonist,231 anticonvulsant,232 anticancer,233 antihypertensive234 and antithrombotic.235 Peptidyl benzothiazepines 119-123 were synthesized by reacting peptidyl chalcones with o-aminothiophenol at reflux temperatures under an inert N2 Chapter 2 ♦Results and Discussion - 93 - atmosphere.146 The residue was subjected to purification by preparative HPLC to afford benzothiazepines as off-white to pale yellow solids. The resulting products were confirmed through 1H NMR, 13 C NMR and HRMS analysis. The data of the purity and yield of peptidyl benzothiazepines is given in Table 2.28. Table 2.28. Synthesis of peptidyl benzothiazepines 119-123. Entry Purity (%)a Product Yield (%)b F O 119 O 120 O H N H N N H S N Cl 80 52 60 63 78 75 64 71 50 55 O N H S N F O H N O N H 121 S N Cl Cl O H N O N H 122 S S N Cl F O 123 H N O N H S N Cl Cl a Purity of crude products assessed through LCMS. b Isolated yield. Spectroscopic analysis of 119 is discussed as a representative example. Its synthesis was confirmed through HRMS analysis, wherein molecular ion peak was found as (M+H+) at 600.1842. In the 1H NMR spectrum (Fig. 2.25), a singlet for acetyl protons appeared at 1.95 ppm. One of the methylene protons of benzothiazepine ring appeared Chapter 2 ♦Results and Discussion - 94 - as a multiplet along with β protons of the benzyl rings with an integration of five protons at 2.84-3.14 ppm. The second methylene proton of benzothiazepine ring appeared as a dd at 3.55 ppm with an integration of one proton and coupling constant, 9.7 Hz and 6.0 Hz, respectively. The methine proton of benzothiazepine moiety appeared as a dd at 4.65 ppm with coupling constants of 10.2 Hz and 6.0 Hz. The two chiral carbons of the peptide part were found as doublet of doublet at 5.04 and 5.39 ppm, each with an integration of one proton and coupling constants of ~ 13 Hz and ~ 6 Hz, respectively. Aromatic protons of phenyl rings gave a multiplet of 16 protons in the range of 7.06-7.58 ppm, whereas one aromatic proton appeared as a doublet at 7.58 ppm with a coupling constant of 7.3 Hz. The two amide hydrogens were found as singlets at 6.18 and 8.08 ppm, respectively. Fig. 2.25. 1H NMR (300 MHz, CDCl3) spectrum of 119. In 13 C NMR spectrum (Fig. 2.26), three protons of acetyl group appeared at 22.22 ppm. The important peaks of methane and methylene carbons of benzothiazepine ring were found at 35.31 ppm and 37.73 ppm, respectively. Furthermore, methylene protons corresponding to two phenylalanine moieties were found at 45.02 ppm and 48.91ppm. The two peaks for chiral α- carbons appeared at 54.92 and 59.47 ppm. The characteristic peaks for the two carbonyl carbons were found at 169.53 ppm and 173.23 ppm. The carbon corresponding to C=N part of benzothiazepine ring appeared at 163.14 ppm, while the other peaks in a range of 114.45 ppm to 158.54 ppm were assigned to aromatic carbons. 13C-19F coupling was observed for the ipso C attached to F, resulting in a doublet at 156.9 ppm with a coupling constant of 320 Hz. Chapter 2 ♦Results and Discussion Fig. 2.26. - 95 - 13 C NMR (100 MHz, CDCl3) spectrum of 119. The identity of other benzothiazepines 120-123 was also confirmed on the basis of their spectral data. 2.3.2.5. Synthesis of peptidyl benzodiazepines Benzodiazepine is a potent scaffold for the treatment of cardiovascular disorders,236 epilepsy237 and HIV.238 Peptidyl benzodiazepines 124-125 were synthesized through a base catalyzed reaction of peptidyl chalcones with 1,2-phenylenediamine under an atmosphere of nitrogen. Crude products were subjected to purification by preparative HPLC. Yield and purity of the isolated peptidyl benzodiazepines are given in Table 2.29. Table 2.29. Synthesis of peptidyl benzodiazepines 124-125. Entry Purity (%)a Product Yield (%)b F O H N O N H 124 NH Cl N 80 48 62 50 F 125 a O H N O N H N NH Cl Purity of crude products assessed through LCMS; b isolated yield Chapter 2 ♦Results and Discussion - 96 - The identity of synthesized benzodiazepines was confirmed spectroscopically. The HRMS analysis of the peptidyl benzodiazepine 124 showed a M+H+ peak at 533.2140. In the 1H NMR (Table 2.30), a doublet at 1.15 ppm with an integration of six protons for two methyl protons of i-Pr group, whereas a multiplet at 1.21 ppm1.30 ppm with an integration of one proton was assigned to methine proton of i-Pr group. A three proton singlet at 1.90 ppm was assigned to N-terminal acetyl group. A multiplet of two protons at 2.75 ppm-2.80 ppm was assigned to methylene protons of phenylalanine. A multiplet at 2.95-3.10 ppm with an integration of two protons was assigned to methylene protons of diazepine ring, whereas a one proton multiplet at 3.40-3.43 ppm was assigned to NH of diazepine ring. The methine proton at the chiral center of phenylalanine part appeared as a multiplet with one proton integration at 4.17 ppm-4.24 ppm. Another multiplet at 4.75-4.81 ppm with an integration of one proton was assigned to the chiral carbon attached to isopropyl group. Furthermore, the methine proton of diazepine ring was assigned to a one proton multiplet at 4.98-5.04 ppm. The aromatic protons of benzene rings and the two amide NHs appeared as a multiplet at 6.80-7.91 ppm showing an integration of fourteen protons. Table 2.30. 1H-NMR analysis (300 MHz, CDCl3) of peptidyl benzodiazepine 124. δ ppm Multiplicity Integration Coupling constant J (Hz) Assignment 1.15 d 6H 7.9 CH3 isopropyl 1.21-1.30 m 1H - CH isopropyl 1.90 s 3H - H-C(CH3CO) 2.75-2.80 m 2H - H-Cβ1,β2(Phe) 2.95-3.10 m 2H - CH2 (diazepin) 3.40-3.43 m 1H - NH (diazepin) 4.17-4.24 m 1H - H-Cα(Phe) 4.75-4.81 m 1H - H-Cα(CH isopropyl) 4.98-5.04 m 1H - H-C(CH diazepin) 6.8-7.91 m 14H - H-Carom., NHII,III 13 C NMR (Table 2.31) of benzodiazepine 124 showed characteristic peaks for all carbons where 13C-19F coupling was observed for carbon attached to F atom, resulting in the splitting of the parent peak into two in its appearance as a doublet at 152.3 ppm Chapter 2 ♦Results and Discussion - 97 - with a JCF of 311 Hz. Similarly, the ipso carbon attached to diazepine ring appeared as a doublet at 140.7 ppm with 2J as 80 Hz. Table 2.31. δ ppm 13 C-NMR analysis (100 MHz, CDCl3) of peptidyl benzodiazepine 124. Assignment δ ppm Assignment δ ppm Assignment 18.6 CH3 36.0 CH2 111.6-141.1 aromatic 18.7 CH3 37.1 CH 151.8, 152.7 Aromatic C-F 22.9 CH3 (acetyl) 42.6 CH (chiral) 165.1 C=N 28.6 CH (isopropyl) 54.2 CH (chiral) 172.8, 184.4 C=O The present work led to establishment of a new strategy for construction of Cterminal derivatized peptidyl heterocycles using novel bis-electrophiles (peptidyl chalcones) established after successful C-acylation of phosphorane support. The results are being compiled in the form of a publication.218 All the synthesized peptidyl chalcones and derived heterocycles have been submitted for bioevaluation against different targets.