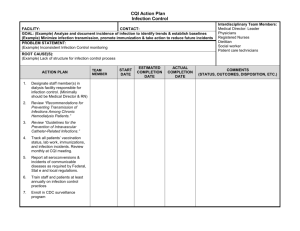
Paediatrics Life Support Algorithm
advertisement

Year 4: Paediatrics Revision
Normal Infant Feeding and Weaning
Nutrition
Growth
Development
Neonates
Birth injuries/problems
Preterm
Jaundice
Respiratory Distress
Infection
Neonatal seizures
Craniofacial disorders
Newborn examination
Childhood infections/immunity
Genetic Conditions
Systems
Haematology
Respiratory
Cardiovascular
Gastrointestinal Tract
Liver
Endocrine
Neurological
Renal/Urinary
Dermatology
Orthopaedic
Malignancy
Chronic disability in children
Extras
Child Accident prevention
Child Abuse
Child Psychiatry
Paeds Surgery
Paeds Emergency
2
8
20
21
24
24
24
25
25
25
26
21
27
44
54
61
67
75
82
91
98
104
114
115
124
144
130
138
130
147
151
-1-
Normal Infant Feeding and Weaning
Nutrition
Nutritional Requirement
Infants and children are more vulnerable to poor nutrition than adults. There are a number of
reasons for this:
-
-
-
Low nutritional stores
o Newborn, and especially those premature babies, have poor stores of fat and
protein. Body composition of preterm differs from that of term infants, 1 year
old children and adults:
Preterm (1.0kg)
85% water
8% protein
1% fat
Term (3.5kg)
65% water
12% protein
18% fat
1 year old (10.5 kg)
55% water
15% protein
23% fat
Adult (70kg)
55% water
20% protein
18% fat
High nutritional demands for growth
o A rapid rate of growth in infancy demands the greatest nourishment
requirement, per unit body size:
At 4 months 30% of infants energy intake is used on growth
At 1 year 5% of infants energy intake is used on growth
By 3 years only 2% is used.
Rapid Neuronal development
o The growth of the brain is rapid during the last trimester and first 2 years of life.
Undernourishment appears to have an affect on the rate of growth of
interneuronal connections.
Infant Feeding
Breast Feeding
“There is no doubt, that breast feeding is best for your baby”.
[http://www.smanutrition.co.uk/feedingweaning/breastfeeding.htm]
Advantages
Free
Reduces chance of gastrointestinal
infection via contaminated water
Reduced incidence of necrotising
enterocolitis in preterm breast fed
infants
Establishing maternal-infant bond
May have long-term neurological
advantages
Reduced incidence of Inflammatory
Bowel disease and diabetes mellitus
Disadvantages
Often difficult to measure the infants
milk intake
Difficulty
Infection risk in developing countries
(CMV, hepatitis, HIV)
Time
Breast milk jaundice
Vitamin K Deficiency
-2-
Reduced risk of breast cancer in those
mothers that breast-feed
Contains: Antivirals, bifidus factor,
Secretory IgA, Lymphocytes,
macrophages, lactoferrin, protein,
breast milk lipase, long-chain
polyunsaturated fatty acids
Contraceptive effect
Less flexible
Emotional upset of unsuccessful feeding
(frustration)
Speaking to many patients on the wards, breast feeding had its disadvantages. Many mothers
had found it difficult to breast-feed, were worried about the amount of milk there babies were
actually taking, and appreciated the ease of formula feeding. However breast feeding is by far
superior, it offers free milk, containing antivirals and white blood cells aiming to fight infection
where immune system hasn’t fully developed. It also has medical advantages for both the
baby and the mother. Formula feeding however is common on the ward.
Formula feeding
Alleviates many of the disadvantages of breast-feeding and designed for 0-6 months, 6-12
months, and milks with introduction of solid foods.
SMA Gold is a popular choice on the ward, information for which can be found at
www.smanutrition.co.uk
Cow’s Milk
Breast and/or formula feeding should be maintained until at least the age of 12 months, and is
often continued till around 18 months of age. Cow’s milk should not be used in the young
infant due to its deficiency of iron, and vitamins A, C and D.
Problems:
- Iron deficiency anaemia
- Constipation
- Cholestasis
Soya Formulae
A formula used instead of cow’s milk, in the belief that it may help prevent atopy (eczema and
asthma). It may also be used in those patients with cow’s milk protein intolerance.
Problems:
- ‘Lactose’ intolerance (cow’s milk intolerance) in 30% patients when fed soya milk
Lactose-Intolerance
There are a variety of different types of lactose intolerance, in addition to which there are
other causes of intolerance to lactose.
Lactase normally catalyses the cleavage of lactose to glucose and galactose.
Lactulose intolerance may be divided into three groups:
- Congenital lactose intolerance
o Congenital lactose intolerance is characterised by severe diarrhoea, abdominal
pain and distension that appear soon after birth when the diet begins to contain
lactose. The symptoms disappear if milk is withdrawn from the diet.
- Familial lactose intolerance
o Familial lactose intolerance is a rare and severe disorder, characterized by onset
of vomiting after the intial feeding of milk or during the first few days of life. The
activity of intestinal lactase is normal. There has yet to be an enzymatic defect
identified.
- Late onset lactose intolerance
o The onset of this condition is several years after birth. This condition is more
common in some races than others, for example 10% of white Americans but
70% of black Americans develop moderate symptoms of lactose intolerance in
adulthood when challenged with oral lactose. Clinical features may vary widely
-3-
but in the most severe instances include chronic diarrhoea, abdominal pain, and
distension after ingestion of lactose.
Treatment:
Lactose free diet. Nutramigen II; Iron Fortified Protein Hydrolysate Formula (Lactose-free
formula)
Weaning
This is encourage between the ages of 3 months and 6 months. Milk feeds may contain their
nutritional requirement but some infants have increased irritability representing hunger and
need for solid food.
At 6 months breast milk becomes nutritionally inadequate and deficient in calories, some
vitamins and iron.
Neonatal Requirements
All healthy newborn infants are expected to lose approx. 10% of their birthweight during the
first week of life. They regulate this themselves with their oral intake. When patients are NPO
and we are responsible for their fluid management, we still need to keep expected physiologic
processes in mind. Most non-surgical neonates on IV fluids therefore receive a graduated
regimen of IVF. Typically, the expected fluid needs are:
Day#1: 60ml/kg/day
Day#2: 80ml/kg/day
Day#3: 100ml/kg/day
Thereafter 100-150 ml/kg/day.
Fluids in Older Children
The maintenance fluid requirements of older children are also based on ml/kg/day.
1-10 kg
100
11-20 kg
1000 ml +
50 ml/kg above 10 kg/d
1500 ml +
20 ml/kg above 20 kg/d
21-30 kg
> 30 kg
1700 ml +
ml/kg/d
10 ml/kg above 30 kg/d
Malnutrition
Failure to thrive
A term used to describe suboptimal weight gain
Causes:
Non-Organic
Feeding problems – insufficient breast milk or
poor technique
Maternal stress: Inadequate food consumed,
lack of volume awareness, social exclusion
and financial difficulties
Lack of stimulation and undernutrition
Munchausen’s by proxy – Deliberate
or growth in infants and toddlers.
Organic
Inability to feed: Mechanical problem, lack of
coordination
Poor retention of food: vomiting, GORD
Illness-induced anorexia
Impaired nutrient absorption
Increased energy requirements e.g. CF,
malignancy
-4-
underfeeding
Metabolic: Hypothyoridism, CAH, aa and
organic acid disorders
Misc: Chromosomal disorders, syndromes and
congenital infection
Consider:
- Anaemia, infection, inflammation, immune deficiency
- Renal failure, renal tubular acidosis, metabolic disorders
- Liver disease, malabsorption, metabolic disorders
- Hypothyroidism
- Inflammation e.g. Crohn’s disease
- Iron deficiency anaemia
- Immune deficiency
- Coeliac disease:
o
Coeliac disease is clinically very variable and so is defined pathologically as a
permanent gluten-sensitive enteropathy.The mucosal lesions seen on upper GI
biopsy are the result of an abnormal, genetically determined, cell-mediated
immune response to gliadin, a constituent of the gluten found in wheat. A similar
response occurs to comparable proteins found in rye and barley. Gluten is not
found in oats, rice and maize.
o Investigations:
Haematology: Usually dominated by effects of iron deficiency, such as
hypochromia, anisocytosis
Immunology: Gold standard Anti-endomysial antibodies positive
Biochem: Hypoalbuminaemia in severely ill patients, ferritin, Vit D,
calcium reduced, carbohydrate malabsorption
Jejunal biopsy: demonstrates a charactersistic histological lesion e.g.
partial/subtotal villous strophy
Imaging: Barium swallow, may show mucosal oedema and thickened
jejunal folds
IgA antibodies to gliadin
Rectal gluten challenge
- UTI, renal disease
- Intestinal infection, parasites
- Turner’s syndrome
- CF
Malnutrition
Nutritional assessment
- Arthropometry
o Weight
o Height
o Mid-arm circumference
o Skinfold thickness
- Laboratory
o Low plasma albumin
o Low concentrations of specific minerals and vitamins
- Food intake
o Dietary recall
o Dietary diary
- Immunodeficiency
o Low lymphocyte count
o Impaired cell-mediated immunity
Weight/age %
Weight/height %
Normal
100
100
Wasted
70
70
Stunted
70
100
-5-
Height/age %
100
100
84
Marasmus
Severe protein-energy malnutrition in children usually leads to marasmus, with a weight less
than 60% of the mean for age, and a wasted, wizened appearance. Oedema is not present.
Skinfold thickness and mid-arm circumference are markedly reduced, and affected children are
often withdrawn and apathetic
Kwashiorkor
Another manifestation of severe protein malnutrition, in which body weight is 60-80% of
expected and oedema is present. In addition there may be:
- a ‘flaky paint’ skin rash, with hyperkeratosis and desquamation
- A distended abdomen and hepatomegaly
- Angular stomatitis
- Hair which is sparse and depigmented
- Diarrhoea, hypothermia, bradycardia, and hypotension
- Low plasma albumin, potassium, glucose and magnesium
Rickets
Derived from the old English word ‘wrickken’, meaning to twist, is used to describe the clinical
syndrome arising from an excess undermineralised bone matrix in growing bone. It usually
results from deficient intake or defective metabolism of vitamin D. It can also occur from a
nutritional deficiency of calcium, particularly in developing countries, or, in preterm infants,
from a deficiency of phosphate in breast milk or unsupplemented cow’s milk formulae.
Mineralisation of osteoid is impaired, and the epiphyseal growth area becomes disorganised
and hypertrophic.
Clinical Features:
- Misery
- FTT/Short stature
- Frontal bossing
- Craniotabes
- Delayed closure of anterior fontanelle
- Delayed dentition
- Rickety rosary – An expansion of the costochondral junctions
- Harrison’s sulcus
- Expansion of metaphyses (especially wrists)
- Bowing of weight-bearing bones
- Hypotonia
- Seizures (late)
-6-
Vitamin A (Retinol)
In developed countries, biochemical Vitamin A deficiency is seen as a complication of fat
malabsorption when supplementation is inadequate. Clinical manifestations under these
circumstances are rare, except for impaired dark adaptation.
Vitamin A deficiency is common in many developing countries, where it causes eye damage
from corneal scarring and acceleration of malnutrition from impairment of mucosal function
and immunity.
Obesity
About 1 in 5 children in the UK and USA are now overweight.
Complications:
- Orthopaedic
o Slipped femoral epiphysis
o Tibia vara
- Benign intracranial hypertension
o Headaches
o Blurred optic disc margins
- Hypoventilation syndrome
o Daytime somnolence
o Sleep anoea
o Snoring
o Hypercapnia
o Heart failure
- Gall bladder disease
- Polycystic ovary disease
-7-
-
Non-insulin dependent Diabetes mellitus
Hypertension
Abnormal blood lipids
Psychological sequelae
Dental Caries
Dental destruction occurs as a result of exposure to organic acids produced by bacterial
fermentation of carbohydrate, particularly sucrose.
Growth
Four phases of human growth:
Foetal
o Fastest period of growth
o Size at birth is determined by the size of the mother and by placental nutrient
supply
o Dependent on:
Nutrition
Placenta
- Infantile
0-18 months
o Also largely dependent on nutrition
o Inadequate rate of weight gain during this period is ‘failure to thrive’
o Dependent on:
Nutrition
Good health and happiness
- Childhood phase
18 months – 12 years
o Determinant of growth:
Largely based on Growth hormone secretion acting to produce insulin-like
growth factor 1 at the epiphyses
thyroid hormone, vitamin D and steroids also effect cartilage cell division
and bone formation
o Profound chronic unhappiness can decrease growth hormone secretion and
accounts for psychosocial short stature
- Pubertal phase
o Sex hormones (mainly testosterone and oestradiol), causes the back to lengthen
and boost GH secretion
Same sex steroids cause fusion of the epiphyseal growth plates and a cessation of growth.
-
Measurement
Normal measurements:
Measurements of preterm, infants, and young child up to adulthood can be plotted on the Child
Growth Foundation charts (www.healthforallchildren.co.uk) available on most wards and
outpatient departments. Available graphs include centile lines for height, weight and head
circumference on Pre-Term, 01-1 year, 1-5 years and 0-20 years graphs. It is important to
accurately plot these measures. Single one off plot’s are insignificant. Several plots are
required to conclude normal or abnormal growth patterns.
Important considerations:
- What is normal for the child? They may run in the 0.4th or 99.6th centile as normal for
them
- The child’s parents height, weight and head circumference
- Syndromes or disease: Down’s Syndrome and Turners Syndrome have their own
growth charts
-8-
-
The equipment used; Community scales often differ somewhat from hospital electronic
scales.
Weight
An infant should be weighed nude on a self-zeroing, electronic basin scales with maximum
divisions of 20g (community) and 10g (hospital); stand-on scales should have maximum
divisions of 100g with the child weighed in the minimum clothing.
Weight on the 50th Centile of the growth chart
Age
Newborn
4 weeks
8 weeks
12 weeks
6 months
8 months
10 months
12 months
15 months
18 months
24 months
3 years
4 years
5 years
Boys
3.5kg
4.5kg
5.3kg
6.1kg
8.1kg
8.8kg
9.6kg
10.1kg
11.0kg
11.5kg
12.5kg
14.6kg
16.5kg
18.6kg
Girls
3.4kg
4.1kg
4.9kg
5.6kg
7.5kg
8.3kg
8.9kg
9.4kg
10.2kg
10.9kg
12.0kg
14.2kg
16.2kg
18.3kg
Height
Supine Length
- Portable
o Starter’s Measure mat
o Raven Rollameter
- Incubator measure only
o Pedobaby 2
- Heavy duty
o Raven Kiddimeter
An infant’s length should be measured supinely by two people, with one holding the head
against the headboard and the other person measuring the length by bring the footboard into
contact with the heels
Standing Height
- Portable
o Leicester Height Measure
o Raven Minimeter
- Wall mounted
o Raven Magnimeter
The feet should be together with the heels, buttocks, and should blades touching the vertical
measure with the head positioned in the Frankfurt plane (an imaginary line from the centre of
the ear hole to the lower border of the eye socket). To ensure a true height, upward pressure
on the mastoid process may be considered. The discrepancy on the growth chart at the age of
two, is due to changing the measurement of children from the supine position to the standing
position. In the standing position the gravitational force has a slight impact on the final
measure.
-9-
Height on the 50th Centile of the growth chart
Age
Newborn
4 weeks
8 weeks
12 weeks
6 months
8 months
10 months
12 months
15 months
18 months
24 months
3 years
4 years
5 years
Boys
51.0cm
54.2cm
57.5cm
60.2cm
67.6cm
70.5cm
73.2cm
75.5cm
79.5cm
82cm
87cm
95cm
102cm
109cm
Girls
50cm
53.5cm
56.5cm
59.0cm
66.0cm
68.8cm
71.5cm
74.0cm
77cm
80cm
86cm
94cm
101.5cm
109cm
Head circumference
Measurement from the occipital prominence to most prominent part of frontal bone.
Appropriate thin metal or plastic tape should be used.
Head Circumference on the 50th Centile of the growth chart
Age
Newborn
4 weeks
8 weeks
12 weeks
6 months
8 months
10 months
12 months
15 months
18 months
24 months
3 years
4 years
5 years
Boys
35cm
37.5cm
39.4cm
41cm
44.5cm
45.8cm
46.8cm
47.5cm
48.5cm
49cm
50cm
51.5cm
52cm
53cm
Girls
34.5cm
36.5cm
38.5cm
40cm
43.5cm
44.5cm
45.5cm
46.5cm
47.5cm
48cm
49cm
50.5cm
51cm
51.5cm
Height velocity
Calculated as a measure of centimetre/year. It can improve the
speed of referral for patients with abnormal slowing growth. Where
a patient on the 50th centile may not get spotted for referral as
quick as a patient on the 2nd centile, and they both have slowing
growth they would have the same height velocity, which would be
noted.
Bone age
A measure of skeletal maturation. An x-ray is taken of the wrist. The bone
age is calculated as by observing and measuring the ossification of the carpal
bones, the presence and absence of certain bones and growth plates.
- 10 -
Where a child is showing slow growth, and deviation from the centile line, bone age can prove
to be useful. If the bone age is less than that of the patient; this shows that the patient still
has a growth ‘spurt’ ahead of them.
Decimal Age
Although most growth charts are plotted using weeks, months and years, it is important to know
how to calculate a decimal age. The decimal ages are often used in endocrine clinics to obtain a
more accurate measure of age. It is basically your year plus months, and days worked as a decimal,
e.g. 1 year and 3 months, would be 1.25. The below table is used to calculate decimal ages:
The date of birth, as a year plus decimal, is taken from todays date as a year plus decimal.
E.g.
D.O.B 07.12.83
Todays date 10.01.2006
2006.025 - 1983.932 = 22.093
- 11 -
Puberty
Normal Boys
First feature – enlargement of testis
4 ml testes = Tanner stage 2
Mean age 12 years
Growth spurt ~ 2 years after puberty
Normal Girls
First feature – breast budding
- 12 -
Mean age 10-11
Growth spurt ~12 years
Menstruation is late feature
Non-Pathological factors
Gender
Nutrition
Familial
Racial – Afro-Caribbean’s
Psychological stress
Secular trends
Physical activity
Light-darkness rhythms – Blind go into puberty earlier
Precocious Puberty aka Premature sexual maturation (PSM)
2º Sexual characteristics developing before 8 years in girls, 9 years in boys
Boys < Girls
Much more pathological in Boys > 50% CNS
Concerns – Parental
- Pathology
- Psychologically unacceptable
- Long term sequelae
o Get early growth spurt, so miss out on normal growth, so possibly smaller
Central Precocious puberty
- Gonadotrophin
Gonadotrophin independent precocious puberty
- Sex hormone driven
Premature thelarche – breast development
Premature adrenarche - Androgen secretion
Central Precocious puberty
- Idiopathic – often familial
- CNS tumour
- Hydrocephalus
Gonadotrophin independent precocious puberty
- Adrenal tumour
- CAH – Boys
- Ovarian tumour
- Testicular tumour
Delayed puberty
No signs
14 years girls
15 years boys
Causes:
- Familial
- Chronic ill health
- Anorexia
- Athletes
- Turner’s Syndrome – infertility?
- 13 -
Short Stature – Is this normal?
Usually defined as a height below the second or 0.4th centile (-2.6 SD)
Short and thin – Chronic illness,
Short and chubby – Endocrine, chromosome
Disproportionate – Skeletal dysplasia
Familial
Constitutional
Short
-
stature vs. Growth failure?
Normal rate f growth
Tailing off centiles
Height velocity
History
- Previous heights and weights
- Pubertal growth
- General health, significant illnesses
o Turners – Middle ear disease (often present to ENT)
- Appetite
- Symptoms of malabsorption (celiac)
- Drug History – Long term INH steroids
Family
-
History
Parental heights
Calculate mid-parental centile
Parental pubertal timing
Siblings heights
Short family members
Illnesses
Mid-Parental
Average mum and dad
- 7cm girls
+ 7cm boys
Final mid-parental centile
Target centile range ±10cm
On Examination
Height
Propionate?/Disproportionate?
Can get a sitting height – Should be on same centile as height
Specific syndromes
Growth Hormone Deficiency
Pubertal staging
Investigations
FBC, Ferritin, ESR, U&E
Sweat test
Chromosomes
, Creatinine
Anaemia in coeliac or Crohn’s
disease, Creatinine raised in
Chronic renal failure
Cystic fibrosis (CF)
Turner’s Syndrome 45XO
- 14 -
Endomysial and gliadin antibodies
TFTs – Thyroid Stimulating hormone
Bone age – Wrist carpel bones
IGF-1 (Insulin-like growth factor-1)
CRP (acute phase reactant)
MRI scan
Cortisol
Growth hormone stimulation test
Coeliac disease
Hypothyroidism
Some delay in constitutional delay
of growth and puberty
Raised in crohn’s disease
Craniopharyngioma or intracranial
tumour
Growth hormone deficiency
Growth hormone stimulation test:
- Pulsatile release of growth hormone
- Insulin stimulation test (hypoglycaemia release GH)
- Was the gold standard
- Several deaths
- Glucagon stimulation test (Stimulates ACTH, Cortisol and GH)
- Clonidine/Arginine (Directly stimulate GH)
- Exercise
Growth Hormone Use:
- Turners
- GH def
- Prader-Willi
- IUGR with failure to capture growth by 4 years
- Chronic renal failure
Long term cancer risk? You are giving a drug that causes hypertrophy in a person with
immature metabolism /physiology
Tall stature – Is this normal?
A less common presentation to clinic than short stature. Most parents are more concerned with
short children than tall stature.
Causes of tall stature:
- Familial – The most common cause of tall stature
- Obesity – Obesity in childhood extends puberty so as to advance the final height
- Secondary
o Hyperthyroidism
o Excess sex steroids
o Excess adrenal androgen steroids – Congenital adrenal hyperplasia
o Gigantism – Excess GH secretion
- Syndromes
o Long-Legged
Marfan’s syndrome
Homocystinuria
Klinefelter’s syndrome (47XXY)
o Proportionate
Maternal diabetes
Primary hyperinsulinism
Beckwith’s syndrome
o Sotos syndrome
Syndrome associated with large head, characteristic facies and learning difficulties
- 15 -
Abnormal head growth
Microcephaly
Head circumference below the second centile.
Causes:
Familial
Autosomal recessive condition
Caused by a congenital infection
Acquired after an insult to the developing brain
Macrocephaly
Head circumference above the 98th centile
Causes:
Tall stature
Familial
Raised intracranial pressure – Hydrocephalus, Chronic subdural haematoma, cerebral tumour
Neurofibromatosis
Cerebral gigantism (Sotos syndrome)
CNS storage disorders
Asymmetric heads
Can occur through unequal growth of coronal, sagittal or lambdoid sutures.
- Craniosynostosis
o This is premature fusion of sutures causing abnormalities of the head shaped. In
the normal infant the sutures close around 12 years of age. This condition may
be picked up on palpation or via CT or skull x-ray. It may be treated for cosmetic
reasons or because of raised intracranial pressure.
- Plagiocephaly
o Asymmetrical, usually due to lying on one side. This is a parallelogram-shaped
head, and is seen with increased frequency due to the “back to sleep” SIDS
compaig. This condition improves with time as the infant becomes more mobile.
It is seen more frequently in babies with hypotonia, due to increased periods of
time lying on one side.
- Breech – Often causes flat on top head, with prominent occiput.
Growth Problems
-
-
Puberty
o Early
o Delayed
Short stature
o Coeliac disease
o Crohn’s disease
o Chronic renal failure
o Turner’s Syndrome: 45XO
o Noonan’s Syndrome:
Noonan's syndrome describes a Turner's syndrome-like phenotype in an
individual with a normal genotype. There are however a number of
phenotypic differences from Turner's syndrome. This syndrome may
affect either sex.
Clinical features include:
o short stature with abnormal body proportions
o cubitus valgus
o webbed neck
Features different from Turner's syndrome include the
characteristic facies:
o Broad forehead
o ptosis
- 16 -
hypertelorism
epicanthic folds
down slanting eyes
low-set ears
flat nasal bridge
pectus carinatum & kyphoscoliosis
congenital heart disease on the right side (compare to the
left sided lesions of Turner's syndrome)
o cryptorchidism
Associated with:
o Noonan's syndrome is associated with right-sided heart
defects - in particular pulmonary artery stenosis, but also
tetralogy of Fallot. By comparison Turner's syndrome is
associated with left-sided heart defects
o There may be hypertrophic cardiomyopathy
o Mental retardation may occur in this condition, but is by no
means a mandatory feature
Prognosis: There is variable fertility in individuals affected by this
condition. The long term prognosis is mainly determined by the
cardiac problems.
o Down’s Syndrome
o Russell-Silver Syndrome
Russell-Silver syndrome is an inherited cause of short stature and a
number of other characteristic features.
After a provocation test there is a surge in growth hormone; there is an
initial response to growth hormone treatment which may not be
maintained.
There is an association with Wilm's tumour.
Clinical features:
low birthweight
short stature
frontal bossing and a triangular face - pixie-like features
a relative macroencephaly
body asymmetry - hemihypertrophy in 50%
small hands and short 5th finger in 75%
mental retardation in 20%
cryptorchidism
primary gonadal failure
hypospadias
o Short-Limbed Dysplasia
o Achondroplasia
Achondroplasia is inherited as an autosomal dominant disorder. It is
diagnosed in the first years of life and affects 1 in 26,000 births; the
majority of cases are sporadic
This is the classic circus dwarf with disproportionate shortness of the
limbs and large head, but apparently normal trunk
Surgical manipulation of leg length may improve stature.
o Storage Disorders
Tall stature
o Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia is a metabolic disorder related to
enzymatic defects in the biosynthesis of cortical steroids. Typically the
defects are inherited in an autosomal recessive manner, and within a
particular family all inherit the same enzyme deficiency.
Two principal effects result from the enzyme deficiency:
o deficient cortisol and/or aldosterone production
o excess precursor steroid
In response, there is increased ACTH secretion from the anterior pituitary
producing adrenocortical hyperplasia.
o
o
o
o
o
o
o
-
- 17 -
At least eight distinctive clinical syndromes are recognised but the
majority of cases are attributed to:
21-hydroxylase deficiency (95%)
Clinical features
Females are virilised at birth with clitoral hypertrophy and variable
labial fusion of the labia minora. They may be mistaken for male
babies with bilateral cryptorchidism and hypospadias. Hirsutism
develops in 10% of patients, usually, at adolescence. Postpubertal amenorrhoea is common.
The condition is less easy to recognise in newly born males and
may go undiagnosed until there is an adrenal crisis in the first
weeks of life.
There may be hyperpigmentation; in males this may be
particularly apparent in the scrotum.
An adrenal crisis is characterised by salt loss and circulatory
collapse. It usually occurs in the first few days of life and may be
preceded by vomiting and poor weight gain. Cortisol and
aldosterone are low or absent. Serum ACTH and 17hydroxyprogesterone, and urinary pregnanetriol and 17oxosteroids are elevated.
11-hydroxylase deficiency produces virilisation, salt retention and
hypertension. Initial growth may be excessive but the advanced
bone age may limit final adult stature.
Males born with 3-beta hydroxysteroid dehydrogenase deficiency
fail to develop normal external genitalia due to a lack of
testosterone. Females are slightly masculinised from
overproduction of DHEA-S.
Life-threatening hypoglycaemia may accompany cortisol deficiency
due to omission of steroids or intercurrent illness.
True gigantism
This is the result of pituitary acidophil cell hypersecretion.
When this condition occurs in childhood then there is stimulation of
epiphyseal growth. Patients are excessively tall; and often there is
associated sexual immaturity and mental retardation.
Slipping of the upper femoral epiphysis may occur.
Marfan’s Syndrome
Marfan's syndrome is a connective tissue disease with an autosomal
dominant inheritance and an incidence of 4-6 per 100 000.
People with Marfan's syndrome used to have a life expectancy reduced by
50% but this is now changing because of improved treatment of
cardiovascular abnormalities.
Clinical features: Three systems are predominantly affected.
Ocular:
upward lens dislocation
retinal detachment
Skeletal:
arachnodactyly
tall with disproportionately long legs and arms - the span of the
arms is greater than the height
pectus excavatum
spinal abnormalities - spondylolisthesis, scoliosis
increased incidence of slipped upper femoral epiphysis
generalised joint laxity with predisposition to flat feet or dislocation of
patella or shoulder
Cardiovascular - affecting the aortic and mitral valves and the ascending
aorta:
dilatation of the aorta may be noted at any age, beginning at the
aortic valve and usually confined to the ascending aorta
o
o
- 18 -
o
o
o
aortic insufficiency may result from stretching of the aortic valve
ring and a dissecting aneurysm of the aorta may be a terminal
event. Rarely, it occurs during pregnancy
mitral insufficiency results from redundant cusps and chordae
tendineae
other cardiac malformations have occasionally been reported
Mental development is normal.
The average lifespan of an affected individual is 40 to 50 years.
Klinefelter’s Syndrome 45XO
Sotos syndrome
Sotos' syndrome presents in childhood with characteristic facial
appearance, developmental delay and tall for age.
Clinical features
Developmental delay, with
Variable learning difficulties
Speech and language delay
Gross motor delay
Impaired fine motor activity, with poor coordination
Increased growth, which starts prenatally, and is particularly rapid in the
first two or three years, stabilising after five years, and reaching normal
adult height:
Large hands and feet
Long arms
Macrocephaly
Advanced bone age
Precocious puberty
Dysmorphic facial features:
Dolicocephaly
Hypertelorism
High palate
Prognathism
Other developmental abnormalities:
Cardiac
Genitourinary
Scoliosis
Beckwith (-Wiedemann) Syndrome
A baby with Beckwith-Wiedemann syndrome is born large, macrosomic,
and grows into a gigantic infant. Another name for this association is the
EMG syndrome because of the triad of classical features:
Exomphalos
Macroglossia
Gigantism
There may also be:
Organomegaly - especially the kidneys
Naevus flammeus of the forehead
Ear lobe grooves - a typical crease to the earlobe
Hemihypertrophy
Neonatal hypoglycaemia; hypoglycaemia may also occur later in
life
Beckwith-Wiedemann has been associated in some cases with a
duplication of chromosome 11p15; this group is more prone to
malignancy.
The dysregulation of genes at the 11p15 locus may lead to
overproduction of insulin-like growth factor.
- 19 -
Development
Gross Motor
Newborn
Head Lag
6-8 weeks
Raises head
above 45
Fine Motor and
vision
Follows face in
midline
Follows moving
ovjects or face
by turning head
3 months
Language and
hearing
Startles to loud
noise
Social
Smiles
responsively
Vocalises alone
or when spoken
to, coos and
laughs
6 months
Sits unsupported
with round back
Sits unsupported
with straight
back
8 months
8-9 months
10 months
12 months
15 months
Crawling
Walks around
furniture
Walks unsteadily
Walks alone
steadily
18 months
2 years
Jumps
2.5 years
3 years
hops
4 years
4.5 years
5 years
Palmar grasp,
transfer
Scissor grip
7: Turns to soft
sounds out of
sight
7-10: dada
mama
Pincer grip
2 to 3 words
other than dada
and mama
Puts food in
mouth
Waves bye-bye,
plays peek-a-boo
Drinks from a
cup
Scribbles
Tower of three
10 words. Shows
four parts of the
body
Tower of six
Uses 2 or more
words to make
simple phrases
Talks constantly
in 3-4 word
sentences
Tower of eight,
or train with four
blocks
Circle, Bridge
from a model
Cross, steps
after
demonstration
Square
Triangle
Holds a spoon
and gets food
safely to mouth,
Symbolic play
Dry by day. Pulls
off some clothing
Interactive play.
Takes turns.
Key Milestone
No Responsive smile
Not achieved good eye contact
Not reaching for objects
Not sitting unsupported
Not walking unaided
Not saying single words with meaning
No 2 or 3 word sentences
8/52
3/12
5/12
9/12
18/12
18/12
30/12
- 20 -
What
-
-
does normal development require?
Normal/near normal neurological status
Normal/near normal functioning body parts (ears/eyes/4 limbs etc.)
Generally good health and nutrition
Appropriate stimulation and childrearing practices, preferably within a loving family
Causes of developmental delay
Motor delay
Familial
Chronic illness
Cerebral palsy
Muscle disorders
Orthopaedic
Neglect
Language delay
Familial
Hearing loss
Neurological problems
Neglect
Learning difficulties
Autism
Global delay
Learning difficulties
Chromosomal abN
Genetic syndromes
Brain malformations
Brain injury
Congenital infection
Metabolic problems
Neonates
o
o
o
o
o
o
o
o
o
o
o
Birth Asphyxia
Birth injuries
Preterm
Jaundice
Respiratory Distress
Infection
Neonatal seizures
Hypoglycaemia
Craniofacial disorders
GI disorders
Newborn Examination
Birth
-
-
-
Make a note of the gestation
e.g. SVD 33 + 4 = Spontaneous vaginal delivery 33 weeks and 4 days
Birth weight, 3.4 kg is normal
Apgar scores (score in 10, taken at 1 min, 5 min, 10 min)
o Appearance (colour)
o Pulse (Heart rate)
o Grimmace
o Activity (muscle tone)
o Resp
Maternal Medical history
o Diabetes
o Infection e.g. Group B Strep
o Time from rupture of membranes to delivery
Previous pregnancies, and deliveries
o E.g. G7 P3+4, = 7 pregnancies, 3 births and 4 others (TOP, miscarriage etc)
Newborn Examination
Head to feet:
-
General observation of the baby’s appearance, posture and movements. Bent back??
Meningitis
Head circumference (OCF), Frontal bone, to occipital prominence. Abnormal head
may be due to Breech presentation (flat on top, prominent occiput), or Plagiocephaly
(asymmetrical head shape), and craniosynostosis (premature fusion of sutures).
- 21 -
-
-
-
-
The fontanelle and sutures palpated. A tense fontanelle when not crying may be due
to raised intracranial pressure
o Posterior fontanelle – Open till 1 month
o Anterior fontanelle – Open till 12/14 months
Facies observed, look for abnormalities related to syndromes
If plethoric or pale, haemocrit should be checked to identify polycythaemia or anemia.
Central cyanosis, which always needs urgent assessment, is best seen on the tongue.
This may be a sign of cyanotic congenital heart disease.
Jaundice within 24 h of birth requires further evaluation
The eyes are checked for cataracts (red reflex) and other abnormalities, e.g. white
reflex
The palate needs to be inspected, including posteriorly to exclude a posterior cleft
palate
Breathing and chest wall movement are observed for signs of RDS
On auscultating the heart, normal rate is 110-150 beats/min in term babies
On palpating the abdomen the liver normally extends 1-2cm below the costal
margin, the spleen tip may be palpable as may the kidney on the left side.
Genitalia and anus are inspected. If ambigious, then examination, chromosome study
and USS (may pick up uterus, ovary etc). Be careful with assigning gender.
Femoral pulses are palpated. Their pressure is:
o Reduced in coarctation of the aorta
o Increased if there is a PDA
Muscle tone is assessed
The whole of the back and the spine is observed
Hands are check for polydactly, overlapping of fingers (Edwards Trisomy 18) simian
creases
Hips are checked for DDH (Ortolani and Barlow)
Legs – Abnormalities, deformities, lymphoedema (?Turner’s)
Feet are check for abnormalities e.g. Sandal gap
Reflexes
Moro
This is an example of a primitive reflex.
It is elicited by:
placing the infant on a flat surface in the supine position.
steady the infant by placing one hand under the infant's shoulder.
the head is flexed on the body by the other hand.
when the infant is moderately relaxed and symmetrical the head is released. This will
result in the abduction phase when the infant will abduct his arms at the shoulders and
flex his arms at the elbows and wrists. The hands open but the fingers remain curved.
There may be adduction at the hips. Abduction of the arms may be followed by
adduction.
the baby usually cries after the reflex is elicited.
A moro reflex is normally present at birth and disappears by the age of about five months.
An absent moro reflex at birth implies brain dysfunction. Persistence of a moro reflex suggests
a brain lesion.
Startle Reflex
This is an example of a primitive reflex.
If a neonate is startled e.g. by a sudden loud noise, then the neonate flexes his elbows (not
extends them as in the moro reflex) with his hands remaining closed.
Sucking Reflexes
This is present in full-term babies and most pre-term babies. This can be tested by introducing
a teat or finger into the mouth which will initiate vigorous sucking. If this reflex is absent in a
full-term baby then this is suggestive of a developmental defect.
- 22 -
Rooting Reflexes
This is present in normal full-term babies. If a baby comes into contact with the mother's
breast then he 'roots' for milk.
It is examined by lightly touching the corner of the baby's mouth. In response the baby lowers
the bottom lip on the same side and his tongue moves towards the point of stimulation.
Parachute Reaction
This is an example of a reflex that appears at six to nine months and persists throughout life.
If the child is held in the ventral position and then suddenly lowered towards the ground then
the child extends his arms as if to protect him from falling.
This reflex is absent or incomplete in children with cerebral palsy because of the strong flexor
tone in these children.
Walking Reflexes
This is a primitive reflex that disappears in normal children by 5 to 6 weeks of age. The baby is
held upright over a table so that the sole of the foot presses against the table. The baby then
simulates walking by initiating reciprocal flexion and extension of the legs.
Lesions in newborn, which resolve spontaneously:
-
-
-
Peripheral cyanosis of the hands and feet
Traumatic cyanosis from a cord round the baby’s neck or from a face or brow
presentation. May show petechiae.
Swollen eyelids and distortion of the shape of the head
Subconjunctival haemorrhages – occur during delivery
Erythema toxicum neonatorum (ETN) is a benign, self-limited,
asymptomatic skin condition that only occurs during the neonatal period
V Shape on head – As a result of forceps.
Circular lesion on scalp – Ventouse? Rx – Analgesia
Small white pearls along the midline of the palate (Epstein’s pearls)
Cysts of the gum (epulis) or floor of the mouth (ranula)
Breast enlargement
White vaginal delivery – Represents a withdrawal bleed
Capillary haemangioma or ‘stork bites’. V mark on front of face.
Strawberry naevus – haemangioma, which presents at birth, may proliferate up to 1st
year. Should start regressing at this point and resolve by 5 years.
Neonatal urticaria (erythema toxicum)
Milia – White pimples on the nose and cheeks, from retention of keratin and sebaceous
material in the pilaceous follicles
Mongolian blue spots – Important to note because of the issue of child abuse, should
fade pre-school
Umbilical hernia – Refer if persistent beyond 18 months. More common in conditions
such as Down’s syndrome and hypothyroidism
Positional talipes
Harlequin colour change – when lying sideways there is reddening down one half of
the body with sharply demarcated blanching down the other side, lasting a few
minutes. It is thought to be due to vasomotor instability
Caput succedaneum
Sternocleidomastoid tumour – Tumour in neck, due to pressure on muscle during
delivery. Diagnosed with clinical and condirmed with USS. Should improved with time.
May result in torticollis.
Bruised scrotum and genitalia – Common in breech presentation. Consider analgesia
Newborn Investigations
- TORCH
o Toxoplasmosis
o Other viruses
- 23 -
-
-
o Rubella
o CMV
o Herpessimplexvirus
Guthrie Test – Heel prick, on day 5-9 of life
o PKU
o Hypothyroidism
o Screening for galactosaemia, maple syrup urine disease and homocystinuria can
be performed
FISH (Fluorescent in situ hybridisation)
o On those patients with predicted chromosomal abnormalities
Birth Asphyxia
This condition remains an important cause of hypoxic brain damage in children. Asphyxia or
suffocation, results in a decreased oxygen delivery to cells. Clinical features in the newborn are
difficult to assess but include; abnormality of fetal cardiotocograph (CTG), a metabolic acidosis
in the foetal blood sample or cord blood analysis, a low apgar score at 1 and 5 mins, being a
reflection of delayed onset of respiration and circulation failure at birth.
Hypoxic-Ischaemia Encephalopathy (HIE) is defined as the clinical manifestation of brain injury
immediately or at most 48 hours after asphyxiation has occurred. Asphyxia can occur
antenatally, intrapartum or postnatally. HIE can be described as; Mild (infant irritable, staring
eyes, and hyperventilating), Moderate (Lethargy, reduced movements), and severe (No
spontaneous movements, no response to pain, prolonged seizures)
Management
Identifying and resuscitating those infants with asphyxia is crucial. Respiratory support,
anticonvulsants, and treatment of renal failure, Hypotension and electrolyte disturbance may
be required.
Prognosis
For those infants who have suffered moderate to severe hypoxic-ischaemic Encephalopathy,
prognosis is poor, and may result in permanent neurological developmental disabilities.
Birth asphyxia or at least birth hypoxic events is associated with cerebral palsy.
Birth injuries
As a result of malposition, reduced pelvic outlet with proportion to foetal size, or forceps,
Ventouse delivery.
May include:
- Caput Succedaneum – Bruising and oedema and the presenting part of the infant,
resolves in a few days
- Cephalohaematoma – Bleeding below the periosteum. Usually involves the parietal
bone. Bleeding Consider Jaundice
- Chignon – Bruising oedema as a result of Ventouse delivery
- Subaponeurotic haemorrhage – Fluid accumulation under the scalp as a result of
bleeding breakdown, mobile fluid filled ‘sac’, limited by the frontal suture.
- Tear of the falx cerebri or tentorium cerebelli
- Nerve Palsies
o Upper nerve root (C5 and C6) results in Erb’s Palsy (Waiter’s Tip). Erb’s palsy is
a brachial plexus injury, as a result of pulling of the arm. Most recover.
o Lower toot – Klumpke’s Palsy – Weakness of the wrist extensors and intrinsic
muscles of the hand
o Facial Nerve palsy (VII) – On the side of lesion. Usually as a result of forceps
Should resolve in 95% of cases.
- Fractures
- 24 -
o
o
Clavicle – Usually as a result of should dystocia.
Humerus/Femur – Occurring in breech deliveries, usually mid-shaft fracture
Preterm Infant
Dependent on gestational age at birth. A Preterm infant is one born before 37 completed
weeks of pregnancy. In SCBU if asymptomatic, they stay to their due date.
Causes:
- Pre-eclampsia
- Antepartum haemorrhage
- Placenta praevia
- Hypertension
- Infection
- Cardiac disease
- Renal disease
- Age below 20 years old
- Socioeconomic status
- Multiple pregnancies
- Prolonged rupture of membranes
- Idiopathic
Characteristics:
- Length = number of weeks x 11/4 (32 weeks = 40 cm in length)
- Head large in comparison to body
- Sutures and fontanelles widely spaced
- Skin is red due to lack of s/c fat and may be transparent
- Skin is covered with lanugos
- Limbs are thin and nails are soft
- Small genitalia
- Weak cry
- Poor or absent suck reflex
- Poor muscle tone
- Umbilical cord (two arteries and one vein) is low set and covered in Wharton’s Jelly
Problems include:
- Very thin, transparent, Dark red skin
- Shapeless, soft ears
- Undescended tests
- Poor muscle tone
- Hypotension
- Patent Ductus Arteriosus
- Chronic Lung Disease
- Jaundice
- Necrotising Enterocolitis
- Hydrocephalus – Secondary hydrocephalus as a result of bleeding
Jaundice
Physiological – Due to immaturity of liver – Can’t conjugate bilirubin
- >48 hours age
- Commoner if breast fed or low birth weight
- Baby physically well
- Worsened by excessive bruising or cephalohaemotoma
- Typically gone with in a week
Pathological
- Excess bilirubin production e.g. Haemolysis (Common in Rhesus, Mother –ve, Baby
+ve), Bruising
- Delayed conjugation e.g. Hepatitis, metabolic
- 25 -
When
-
Delayed enterohepatic excretion
E.g. atresia – stenosis or absent of biliary tree, would be a conjugated jaundice
Treatment – Light box
to Check a bilirubin level
< 48 hrs old, suggests pathological
Symptomatic e.g. XS wt loss, poor feeding
When the midwife says so!
Check Conjugated bilirubin
- Prolonged >2 weeks bottle fed, 3 weeks if breast fed
Symptomatic, e.g. Pale stools, dark urine
Respiratory Distress
-
-
-
-
Hyaline Membrane disease aka Respiratory Distress Syndrome
o May not have surfactant
o ‘Ground glass’ appearance of lungs (see right)
Transient Tachypnoea of the newborn (TTN)
o Most disease of term babies
o Lungs full of fluid
o Looks similar to RDS, look for fluid, usually resolves within 24 hours
Bronchopulmonary dysplasia/Chronic lung disease
o Defined as when, oxygen is still required in a prem baby, after it has reached the
due date
Pneumothorax – Usually asyptomatic
Congenital pneumonia
o Risk factors: Prolonged Water’s breaking, chorioamnionitis, and low birth weight
Infection
At risk of infection from:
- Maternal birth canal
- Nosocomial (hospital –acquired)
- Catheterisation
Infections include:
- Strep B
- Gram negative e.g. E-Coli
- Conjuctivitis e.g. Chlamydia Trachomatis
- Listeria Monocytogenes
- Umbilical
- Herpes Simplex
- Hepatitis B
Neonatal seizures
Extent of seizure may vary from tonic, clonic, or more subtle. EEG may detect the seizures.
Causes include:
- Hypoxic-Ischaemic Encephalopathy
- Birth trauma
- Septicaemia
- Metabolic
- Drug withdrawal
- Congenital infection
- 26 -
Hypoglycaemia
Associated with intrauterine growth retardation, in the first 24 hours, who are preterm, born to
diabetic mothers, hypothermic, polycythaemic, are large-for-dates, or are ill for any other
reason.
Craniofacial disorders
Cleft Lip and Palate
Cleft Lip may be unilateral or bilateral, as a result of failure of fusion of the frontonasal and
maxillary processes.
Cleft Palate is a result of failure of fusion of the palatine processes and the nasal septum.
Pierre-Robin Sequence
Association of:
- Micrognathia
- Glossoptosis (Posterior displacement of the tongue)
- Midline Cleft of the soft palate
Gastrointestinal Disorders
Oesophageal atresia
Small bowel obstruction
Large bowel obstruction
Childhood infections/Immunity
Common Childhood infections
Measles
Effective vaccine since 1968
Exposure – Droplet spread, highly infectious during viral shedding
Incubation 10-14 days
Illness – 5.5 days of viral shredding
‘A miserable, coughing, pyrexial child with red, watering eyes – is it measles?’
Clinical features:
- Respiratory
o Pneumonia
o Secondary bacterial infection and otitis media
o Tracheitis
- Neurological
o Febrile convulsions
o EEG abnormalities
o Encephalitis
Occurs in only 1 in 5000, about 8 days after the onset of the illness.
Initial symptoms are headache, lethargy and irritability, proceeding to
convulsions and ultimately coma. Mortality is 15%. Seriouslong-term
sequelae include seizures, deafness, hemiplegia and severe learning
difficulties affecting up to 40% of survivors
o Subacute sclerosing panencephalitis (SSPE)
A rare but devastating illness manifestating, on average, 7 years after
measles infection in about 1 in 100 000 cases. Most children who develop
SSPE have primary measles infection before 2 years of age. SSPE is
- 27 -
caused by a variant of the measles virus which persists in the CNS. The
disorder presents with loss of neurological function, which progresses
over several years to dementia and death. The diagnosis is essentially
clinical, supported by finding high levels of measles antibody in both
blood and CSF and by characteristic EEG abnormalities. Since the
introduction of measles immunisation, it has become extremely rare.
-
-
Other
o
o
o
o
o
Koplik
o
Rash
o
Diarrhoea
Hepatitis
Appendicitis
Corneal ulceration
Myocarditis
spots
White spots on buccal mucosa, seen against bright red background.
Pathognomonic but difficult to see
Spreads downwards, from behind the ears to the whole of the body. Discrete,
maculopapular rash initially, becomes blotchy and confluent. May desquamate in
second week
Treatment:
Treatment for measles is symptomatic. Children who are admitted to hospital should be
isolated. In immunocomprised patients, the antiviral drug ribavirin may be used. Vitamin A,
which may modulate the immune response, should be given in developing countries.
Mumps
Mumps usually occurs in the winter and spring months. It is spread by droplet infection to the
respiratory tract where the virus replicates within epithelial cells. The virus gains access to the
parotid glands before further dissemination to other tissues.
Clinical features:
- Incubation
o 14-21 days
- Onset of illness is with:
- Fever, malaise and parotitis, but in up to 30% of cases the infection is subclinical
- Only one side may be swollen initially, but bilateral involvement usually occurs over the
next few days
- The parotitis is uncomfortable and children may complain of earache or pan on eating
or drinking
- Examination of the parotid duct may show redness and swelling
- Occasionally, parotid swelling may be absent
- The fever usually disappears within 3-4 days.
- Plasma amylase levels are often elevated, and, when associated with abdo pain, may
be evidence of pancreatic involvement.
- Infectivity is for up to 7 days after the onset of parotid swelling. The illness is generally
mild and self-limiting.
- Although hearing loss can follow mumps it is usually unilateral and transient
Viral Meningitis and CNS involvement
Lymphocytes are seen in the CSF in about 50%, meningeal signs are only seen in 10% and
encephalitis in about 10 in 5000. The common clinical features are headache, photophobia,
vomiting and neck stiffness
Orchitis
Most feared complication. Usually unilateral. Some evidence of sperm count reduction but
infertility is rare. Rarely, oophoritis, mastitis and arthritis may occur.
Rubella (German Measles)
- 28 -
Generally a mild disease in childhood. It occurs in winter and spring. It is an important
infection as it can cause severe damage to the foetus.
Incubation period is 14-21 days.
It is spread by the respiratory route, frequently from a known contact. The prodrome is usually
mild with a low-grade fever or none at all.
The maculopapular rash is often the first sign of infection, appearing initially on the face and
then spreading centrifugally to cover the whole body. It faeds in 3-5 days.
Unlike in adults, the rash is not itchy. Lympadenopathy, particularly the suboccipital and
postauricular nodes, is prominent. Complications are rare in childhood but include arthritis,
encephalitis, thrombocytopenia and myocarditis.
Clinical differentiation from other viral infections is unreliable. The diagnosis should be
confirmed serologically if there is any risk of exposure of a non-immune pregnant woman.
There is no effective antiviral treatment. Prevention therefore lies in immunisation.
The human herpesviruses
There are currently eight known human herpesviruses. The hallmark of the herpesviruses is
that, after primary infection, latency is established and there is long-term persistence of the
virus within the host, usually in a dormant state. After certain stimuli, reactivation of infection
may occur.
1. Herpes Simplex infections
Usually eneters the body through the mucous membranes or skin. The incubation
period for primary infection is 3-5 days. After the neonatal period, the HSV1 infections
predominate. The prevalence of HSV2 increases in early adulthood. HSV1 is transmitted
in body fluids such as saliva, while HSV2 is mainly transmitted through the transfer of
genital secretions. Treatment is with acyclovir, a viral DNA polymerase inhibitor, which
may be used to treat severe symptomatic skin, ophthalmic, cerebral and systemic
infections.
- Asymptomatic – Most herpes simplex infections are very common and are mostly
asymptomatic
- Gingivistostomatitis – This is the most common form of primary HSV illness in children.
It usually occurs from 10 months to 3 years of age. There are vesicular lesions on the
lips, gums and anterior surfaces of the tongue and hard palate, which often progress to
extensive, painful ulceration with bleeding. There is a high fever and the child is very
miserable. The illness may persist for up to 2 weeks. Eating and drinking are painful,
which may lead to dehydration. Management is symptomatic, but sever disease may
necessitate IV fluids and acyclovir.
- Skin Manifestations – Mucocutaneous junctions and amaged skin are particularly prone
to infection. ‘Cold sores’ are recurrent HSV1 lesions on the lip margin.
- Eczema herpeticum – In this serious condition, widespread vesicular lesions develop on
eczematous skin. This may be complicated by secondary bacterial infection, which may
result in septicaemia.
- Herpetic Whitlows – These are painful, erythematous, oedematous white pustules on
the site of broken skin on the fingers. Spread is by autoinoculation from
gingiovostomatitis and infected adults kissing their children’s fingers. In sexually active
adolescents, HSV2 may be the cause.
- Eye disease – Eye disease may cause blepharitis or conjuctivits. It may extend to
involve the cornea, producing dendritic ulceration. This can lead to corneal scarring and
ultimately loss of vision. Any child with herpetic lesions near or involving the eye
requires ophthalmic investigation of the cornea by slit lamp examination.
- Central Nervous System Infection
o Aseptic meningitis: This is rare in children, but may occur in sexually active
adolescents. It is usually a complication of HSV2 infection, occurring within 10
days of a primary infection. It resolves without sequelae
o Encephalitis: By contrast, this is a very serious condition with mortality of more
than 70% if untreated. It may follow either primary or recurrent infection. The
clinical features and management are described below.
- 29 -
o
o
Neonatal infection: The infection may be focal, affecting the skin or eyes, may
cause encephalitis or may be widely disseminated. Its morbidity and mortality
are high
Infection in the immunocompromised host: Infection is severe. Cutaneous
lesions may spread to involve adjacent sites, e.g. Oesophagitis and proctitis.
Pneumonia and disseminated infections involving organs are serious
complications.
2. Varicella Zoster Virus(VZV)
a. Chickenpox
Shares many features with HSV, as both produce a vesicular rash. In contrast to
HSV, however, varicella zoster is spread by the respiratory route, progressing
via the blood and lymphatics to cause vesicular lesions in the skin. More than
90% of primary VZV infections are clinically symptomatic with a vesicular rash.
Exposure: Spread by resp droplets. Highly infectious during viral shedding
Incubation: 10-23 days (mean 14)
Illness: ~7 days
Complications: Bacterial Superinfection (staph, strep, toxic shock, necrotising
enterocolitis), CNS (cerebellitis, generalised encephalitis, aseptic meningitis),
immunocomprised (haemorrhagic lesions, pneumonitis, progressive and
disseminated infection, DIC) stroke (increased incidence in VZV) and typical
vesicular rash (200-500 lesions starting on head and trunk progressing to
peripheries)
Treatment: Human varicella zoster immunoglobulin (ZIG) is recommended for
high-risk immunosupressed individuals with deficient T lymphocyte function
b. Shingles
Latent varicella zoster can reactivate, causing a vesicular eruption in the
distribution of sensory nerves (shingles). It occurs most commonly in the
thoracic region, although any dermatome, can be affected. Children, unlike
adults, rarely suffer neuralgic pain with shingles. Shingles in childhood is more
common in those who had primary infection in the first year of life. Recurrent
shingles may be a manifestation of underlying immune suppression, e.g. HIV
infection. In the immunocompromised, reactivated infection can also
disseminate to cause severe disease
3. Epstein-Barr Virus
Major cause of infectious mononucleosis syndrome, but it is also involved in the
pathogenesis of Burkitt’s Lymphoma, lymphoproliferative disease in
immunocompromised hosts and nasopharyngeal carcinoma. The virus has a particular
tropism for B lymphocytes and epithelial cells of the pharynx. Transmission usually
occurs by oral contact and the majority of infections are subclinical.
Infectious mononucleosis (glandular fever):
Fever, malaise, tonsillopharyngitis, lymphadenopathy
Petechiae on the soft palate, Splenomegaly (50%), hepatomegaly (10%), a
maculopapular rash (5%) and jaundice.
Diagnosis is supported by:
Atypical lymphocytes (numerous large T Cells seen on blood film), a positive Monospot
test, the presence of heterophil antibodies (i.e. antibodies that agglutinate sheep or
horse erythrocytes but which are not absorbed by guinea pig kidney extracts – this test
is often negative in young children with the disease), seroconversion with production of
IgM and IgG to EBV antigens.
4. Cytomegalovirus
Usually transmitted via saliva, genital secretions or breast milk, and more rarely via
blood products, organ transplants and transplacentally. CMV causes mild or subclinical
infection in normal host. As with EBV, CMV may cause a mononucleosis syndrome.
Pharyngitis and lymphadenopathy are not usually as prominent as in EBV infections.
5. Human Herpesvirus 6 (HHV6)
Most children are infected with HHV6 by the age of 2, usually from the oral secretions
of a family member. The classic infectious syndrome associated with HHV6 is
- 30 -
exanthema subitum, where there is a high fever with malaise lasting a few days,
followed by a generalised macular rash which appears as the fever wanes. Many
children have a febrile illness without rash, and some subclinical infection.
Primary HHV6 infection is a common cause of febrile convulsions. It is associated with
up to a third of febrile convulsions in the first year of life.
6. Human herpesvirus 7 (HHV7)
Closely related to HHV6, causing similar clinical disease, including exanthema subitum
and febrile convulsions. Again, most children are infected with HHV7 in the first few
years of life
7. Human herpesvirus 8 (HHV 8)
This virus is associated with Kaposi’s Sarcoma (KS), a tumour which occurs in
immunocomprised patients as well as certain populations in Africa and around the
Mediterranean. HHV8 does not appear to be associated with disease in otherwise
healthy children. Primary HHV8 infection may be associated with a mild febrile illness
and infection is usually transmitted from saliva. Up to 40% of African teenagers have
antibodies to HHV8, whereas <5% are seropositive in the UK.
Parvovirus B19
Parvovirus B19 causes erythema infectiosum or fifth disease (so-named because it was the
fifth disease to be described of a group of illnesses with similar rashes), also called slapped
cheek syndrome. Infections can occur at any time of the year, although outbreaks are most
common during the spring months. Transmission is via respiratory secretions from viraemic
patients, by vertical transmission from mother to foetus and by transfusion of contaminated
blood products. Parvovirus infects the erythroblastoid red cell precursors in the bone marrow.
Parvovirus causes a range of clinical syndromes:
- Asymptomatic infection
o Common; about 5-10% of preschool children and 65% of adults have antibodies
- Erythema infectiosum
o The most common illness, with a viraemic phase of fever, malaise, headache
and myalgia followed by a characteristic rash a week later on the face (‘slapped
cheek’) progressing to a maculopapular, ‘lace’-like rash on the trunk and limbs;
complications are rare in children, although arthralgia or arthritis is common in
adults
- Aplastic crisis
o The most serious consequence of parvovirus infection; it occurs in children with
chronic haemolytic anaemias, where there is an increased rate of red cell
turnover (e.g. sickle cell disease or thalassaemia); and in immunodeficient
children (e.g. malignancy) who are unable to produce an antibody response to
neutralise the infection
- Foetal disease
o From maternal parvovirus infection may lead to foetal hydrops and death due to
severe anaemia, although the majority of infected foetuses will recover.
Enteroviruses
Human enteroviruses, of which there are many (including the Coxsackie viruses, echoviruses
and polioviruses), are a common cause of childhood infection. Transmission is primarily by the
faecal-oral route. Following replication in the pharynx and gut, the virus spreads to infect other
organs. Infections occur most commonly in the summer and autumn. Over 90% of infections
are asymptomatic or cause a non-specific febrile illness, but characteristic clinical syndromes
exist and are listed below. An effective vaccine is available against the polioviruses.
- Asymptomatic or non-specific febrile illness
o Over 90% of infections
- Herpangina
o Vesicular and ulcerated lesions on the soft palate and uvula causing anorexia,
pain on swallowing and fever
- Hand, foot and mouth disease
- 31 -
Painful vesicular lesions on the hands, feet, mouth and tongue. Systemic
features are mild. The disease subsides within a few days.
Meningitis/encephalitis
o Aseptic meningitis is caused by many of the enteroviruses. There may be a skin
rash, which can be petechial and therefore difficult to differentiate clinically from
meningococcal infection. Complete recovery can be expected.
Pleurodynia (Bornholm’s disease)
o An acute illness with fever, pleuritic chest pain, and muscle tenderness. There
may be a pleural rub but examination is otherwise normal. Recovery is within a
few days.
Myocarditis and pericarditis
o Heart failure associated with a febrile illness and ECG evidence of myocarditis
Poliovirus infection
o Clinical disease is now very rare, due to successful immunisation programmes. It
falls into four main categories:
>90% are asymptomatic
5% have a poliomyelitis ‘minor illness’ – fever, within 4 days of exposure
and recovery is uneventful
2% of patients progress to CNS involvement with aseptic meningitis;
there is stiffness of the back, neck and hamstrings from meningeal
irritation.
In <1% of cases, classical paralytic polio occurs about 4 days after the
minor illness has subsided – cortex leads to varying degrees of paralysis
which may recover completely or be permanent; involvement of the
muscles of respiration may be fatal.
Infection in the immunocompromised host
o Eneteroviruses can cause severe disease in immunocomprised individuals.
Echovirus can cause a persistent and sometimes fatal CNS infection in
agammaglobulinaemic patients. Live attenuated vaccine strains of poluo can
cause fatal infections in children with severe combined immunodeficiency or
agammaglobulinaemia, and in these patients inactivated vaccine should be used.
o
-
-
-
-
Staphylococcal and Streptococcal infections
Usually caused by direct invasion of the organisms. They may also cause disease by releasing
toxins which act as superantigens. Whereas conventional antigens stimulate only a small
subset of T cells which have a specific receptor, superantigens bind to a part of the T cell
receptor which is shared by many T cells and therefore stimulates massive T cell proliferation
and cytokine release. Other disease are immune-mediated. The wide range of disease caused
by the organisms are:
Staphylococcus aureus
Steptococcus pyogenes
Mode of action
Direct effect
Disease caused
Abscess, Impetigo,
cellulites, pneumonia,
osteomyelitis, septic
arthritis, orbital
cellulitis
Toxin-mediated
(indirect)
Toxic shock syndrome,
Food poisoning,
Kawasaki’s disease
Toxin-mediated (direct)
Toxic epidermal
necrolysis (scalded skin
syndrome)
Tonsillitis, cellulitis,
osteomyelitis,
septicaemia,
Direct effect
- 32 -
erysipelas, impetigo,
otitis media, septic
arthritis, necrotising
fasciitis
-
-
-
-
-
-
Toxin-mediated
Toxic shock-like
syndrome, scarlet fever
Post-infectious
Glomerulonephritis,
rheumatic fever,
erythema nodosum,
arthritis
Impetigo
o This is a localised, highly contagious, staphylococcal and/or streptococcal skin
infection, most common in infants and young children. It is more common where
there is pre-existing skin disease e.g. atopic eczema. Lesions are usually on the
face, neck and hands and begin as erythematous macules which become
vesicular. Rupture of the vesicles with exudation of fluid leads to the
characteristic confluent honey-coloured crusted lesions. The lesions are
sometimes are bullous. Infection is readily spread to adjacent areas and other
parts of the body by autoinoculation of the infected exudate. Topical antibodies
are effective for mild cases. Systemic antibiotics are need for more severe
infections. Affected children should not go to nursery or school until the lesions
are dry. Nasal carriage is an important source of infection which can be
eradicated with a nasal cream containing mupirocin or chlorhexidine and
neomycin.
Boils
o These are infections of hair follicles or sweat glands, usually caused by
Staphylococcal aureus. Treatment is with systemic antibiotics and occasionally
surgery. Recurrent boils are usually from persistent nasal carriage in the child or
family acting as a reservoir for reinfection. Only rarely are they a manifestation
of immune deficiency.
Periorbital cellulitis
o There is fever with erythema, tenderness and oedema of the eyelid. It is almost
always unilateral. In young, unimmunised children it may also be caused by a
Haemophilus influenzae b infection, which may also cause infection at other
sites, e.g. meningitis. It may follow local trauma to the skin.
Scalded skin syndrome
o This is caused by an exfoliative staphylococcal toxin which causes separation of
the epidermal skin through the granular cell layers. It affects infants and young
children, who develop fever and malaise and may have a purulent, crusting,
localised infection around the eyes, nose and mouth with subsequent widespread
erythema and tenderness of the skin. Areas of epidermis separate on gentle
pressure (Nikolsky’s sign) leaving denuded areas of skin, which subsequently dry
and heal withouscarring. Management is with IV antistaphylococcal antibiotic
and monitoring of fluid balance.
Necrotising fasciitis/necrotising cellulites
o This is a severe S/C infection involving tissue planes from the skin down to
fascia and muscle. The skin surface involved may enlarge rapidly, leaving poorly
perfused necrotic areas of tissue, usually at the centre. There is severe pain and
systemic illness which may require intensive care. The invading organism may
be Staphylococcus or group A Streptococcus, with or without another synergist
anaerobic organism. IV antibiotic therapy alone is not sufficient in this condition.
Without surgical debridement of necrotic tissue, the infection will continue to
spread. Clinical suspicion of necrotising fasciitis warrants urgent surgical
consultation and intervention
Toxic shock syndrome
- 33 -
o
Toxin-producing staphylococci and streptococci can produce this syndrome. The
toxin, release from infection at any site, including small abrasions, acts a
superantigen. It causes a systemic illness with a high fever, a diffuse macular
rash, hypotension and shock. There may be redness of the mucous membranes,
vomiting or diarrhoea, severe mylagia, altered consciousness,
thrombocytopenia, coagulopathy, and abnormal hepatic and renal function.
About 1-2 weeks after the onset of illness, there is desquamation of the palms,
soles, fingers and toes. Intensive care support is required to manage the shock.
Areas of infection should be surgically debrided. IV Immunoglobulin may be
given to neutralise circulating toxin.
Kawasaki’s disease
(Acute Febrile Mucocutaneous Syndrome)
Although, uncommon, Kawasaki’s disease is an important diagnosis to make, as aneurysms of
the coronary arteries are an important potential complication. Prompt treatment reduces their
incidence.
Kawasaki’s disease mainly affects children of 6 months to 4 years old, with a peak at the end
of the first year. The disease is much more common in children of Japaense and, to a lesser
extent, Afro-Caribbean ethnicity than it is in Caucasians. Young infants tend to be more
severely affected than older children. The cause is unknown, but the many clinical and
immunological similarities with the staphylococcal and streptococcal toxic shock syndromes
have led to the suggestion that it is also caused by a bacterial toxin acting as a superantigen.
The diagnosis is made on clinical findings.
Clinical features:
- Fever >5 days
- 4 of the 5 other features of:
o Conjuctival infection
o Mucous membrane changes (pharyngeal injection red, dry, cracked lips,
strawberry tongue)
o Cervical lymphadenopathy
o Rash (polymorphous)
o Extremities: red and oedematous palms and soles, peeling of fingers and toes
- Complications of CV signs<4th- 8th week>: Gallop rhythm, myocarditis, pericarditis,
coronary and peripheral aneurysms, sudden death (mortality is 1-2%)
Investigations:
- Raised WBC (neutrophils)
- Raised ESR/C-reactive protein
- Raised platelets (500-3000x109/L)
Treatment:
IV immunoglobulin given within the first 10 days has shown to lower the risk of coronary
artery aneurysms. Aspirin is used to reduce the risk of thrombosis. Used at a high antiinflammatory dose until the fever subsides and continued at a low anti-platelet dose when
there is an abnormality of the coronary arteries.
Meningitis
Inflammation of the meninges with an increased number of white cell in the CSF
Aetiology
- Viral infection - enteroviruses, mumps
- Bacterial infection – Neisseria meningitdis (meningococcus: gram –ve diplococcus),
Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae type B (gram –ve
coccobacilli), listeria, TB
- Fungal infection - cryptococcal (usually immunocompromised)
- (Protozoal infection - amoebic (not Entamoeba histolytica which causes amoebic
dysentery)
- (Helminth infection - certain geographic areas e.g. Thailand)
- 34 -
-
Non infective e.g. neoplastic, drugs - NSAIDs
Influence of age on bacterial meningitis
Age
neonates
1 month -6
years
>6 years
Adults
elderly
Common organisms
Group B streptococci, E. coli,
listeria – organisms from the
female genital tract transferred
during delivery
Neisseria meningitides, Strep
pneumoniae, HiB
Neisseria Mengitides, Strep
pneumoniae
meningococci, pneumococcus
pneumococcus
* Haemophilus influenzae type B was common in children aged <5 years prior to HiB
immunisation
Pathogenesis of acute infective meningitis
Organisms reach the meninges through 3 main routes:
- Via the bloodstream (most common)
- From parameningeal infection
- By direct inoculation
Bloodstream e.g. meningococcal, pneumococcal, HiB
This usually occurs after colonisation of the upper respiratory tract. Many causes of
bacteraemia are rare causes of meningitis e.g. viridans group streptococci - there are virulence
factors related to the capsular polysaccharide in the organisms that regularly cause meningitis
enabling them to cross the blood-brain barrier.
Parameningeal infection
Severe sinusitis or middle ear infection may lead to meningitis usually with osteomyelitis of the
nearby bone. S pneumoniae is the most common organism involved.
Direct inoculation
Head injury, skull fracture - especially if fracture goes into paranasal sinuses, usually S
pneumoniae because of upper respiratory colonisation. Open injury (or following cranial
surgery) skin organisms such as staphylococci. The presence of CSF rhinorrhoea indicates a
defect in the cribriform plate - high risk of meningitis
Immunocompromise and meningitis
Advanced HIV infection- cryptococcal meningitis
Deficiencies of terminal complement can lead to recurrent meningococcal meningitis
Neonates are relatively immunocompromised due to immaturity of the immune system
Symptoms and signs:
- Headache, neck stiffness, photophobia, Kernig's sign (these signs together can be
referred as meningism)
- Fever
- 35 -
-
Rash (characteristic in meningococcal)
Drowsiness/confusion
Convulsions
Focal neurological signs
Shock (usually due to accompanying septicaemia)
Problems in diagnosis
-
Severely ill patients may not have fever or even have subnormal body temperature
Neonates, elderly, immunocompromised may have non-specific or atypical symptoms
Headaches are common in febrile illness and photophobia common in severe headaches
Neck stiffness and positive Kernig's sign are not specific
Neck stiffness
To be significant in meningitis it must be stiffness in AP flexion/extension - limitations in lateral
flexion or rotation often due to musculoskeletal problems
Kernig's
A positive sign is indicated by pain in the back on extension of the knee when the hip is
flexed. False positive sign occurs in back disease e.g. OA spine, prolapsed intervertebral disk.
Investigations
Immediate
FBC, clotting
urea, electrolytes
blood cultures
Later (often after empirical therapy has started)
consider CT brain scan
this must be done before LP if signs suggest raised intracranial pressure as LP may lead to
coning in such cases
Symptoms suggesting raised IC pressure include:
- Papilloedema (late sign so often not there in acute meningitis)
- Focal neurological signs
- Coma
- BP or pulse abnormal
Lumbar puncture
CSF for:
- Cell count, protein, glucose
- Stains - gram, AAFB, India ink
- Culture - bacteria, viral, TB, fungal
- Detection e.g. PCR for meningococci
Throat swabs - bacterial and viral culture
Faeces for viral culture
(CSF cytology for neoplastic disease)
- 36 -
CSF results in meningitis
Test
Normal
bacterial
viral
tuberculous
fungal
<5
>1000
<500
<1000
<500
<5
-
++
++
++
0
+++
+/-
+/-
+/-
glucose
>60% blood
glucose
very low
normal
low
normal or low
protein
<0.4 g/l
high
normal or
slightly raised
very high
high
white cells
lymphocytes
neutrophils
Treatment:
Antimicrobial therapy
In acute meningitis it is important to start treatment as soon as possible - as delay will
increase mortality in meningococcal or pneumococcal disease. We encourage GPs to give an
injection of penicillin before sending the patient to A&E, or it can be started immediately after
taking routine bloods and blood cultures. This potentially affects culture results from the CSF
but risks of delay outweighs disadvantages
Empirical treatment (in children and adults) needs to cover the commonest bacteria i.e.
pneumococci and meningococci. Most of these are sensitive to penicillin but some pneumococci
are resistant so treatment is with a 3rd generation cephalosporin such as cefotaxime.
Most patients reporting a rash with penicillin can still be given cephalosporins but those with
anaphylactic reaction to penicillin or cephalosporins may be given chloramphenicol
Other antimicrobials are given according to CSF results.
Viral meningitis requires symptomatic treatment alone
Steroids
These have been shown to reduce morbidity in pneumococcal meningitis and TB meningitis
Prevention
- HiB immunisation has been highly successful in almost eradicating this cause of
meningitis in the under 5s
- Meningococcal type C immunisation is effective but there is no vaccine against type B
which is common in UK
- Antibiotic prophylaxis (usually rifampicin) is given to close contacts of meningococcal
and haemophilus meningitis
Encephalitis/Encephalopathy
Whereas in meningitis there is inflammation of the meninges, in encephalitis, there is
inflammation of the brain substance, although the meninges are often also affected. Can be
caused by:
- 37 -
-
Direct invasion of the cerebrum by a neurotoxic virus
Delayed brain swelling following a disordered neuroimmunoological response to an
antigen, usually a virus (post-infectious encephalopathy), e.g. following Chicken pox
Slow virus infection such as HIV infection or subacute sclerosing panencephalitis (SSPE)
following measles.
Clinical features: See meningitis
Cause:
- Underlying causative organism only detected in up to 50%
- UK: Enteroviruses, respiratory viruses, and herpesviruses
- Worldwide: Mycoplasma, Borrelia burgdorfii, Bartonella Henselae (cat scratch disease),
Rickettsial infections (rocky Mountain spotted fever) and arboviruses.
Tuberculosis
Background
The incidence of tuberculosis in the UK has decreased over the last 100 years - even before
the advent of chemotherapy (1950s) probably due to improved living conditions.
However the notifications are showing an upward trend over the last decade
The organism
Mycobacterium tuberculosis (occasionally M bovis, M africanum)
-
Slow growing, lipid-rich aerobic bacillus, weak Gram-positive
Ziehl-Neelsen stain - acid (and alcohol) fast bacilli [hence AFB or AAFB]
Grows in 4-6 weeks on traditional media eg Lowenstein-Jensen
Newer more rapid techniques using radiometric system - result in 2 weeks
PCR to detect mycobacterial DNA under development
Mode of spread
- The main source of infection is from droplet nuclei created by a coughing patient with
active pulmonary disease
- The source patient's sputum usually shows AAFB on microscopy - known as (sputum)
smear positive disease or 'open' TB
Most at risk:
Other risk factors:
immigrants from countries where TB is common
alcoholics
household contact of open TB
immunosuppressed patients especially HIV infected
elderly and the very young
poorly ventilated housing, overcrowding, poor nutrition, poverty
Pathogenesis and clinical syndromes
Primary TB
- non-immune host
- often subclinical
3-8 weeks after exposure
- In lungs - focus of inflammation with few mycobacteria surrounded by a granuloma =
Ghon focus
regional lymph nodes involved + Ghon focus = primary complex
May spread by the bloodstream to distant organs e.g. meninges, kidneys, pericardium
Occasionally this produces miliary TB but more often heals to form latent TB which may
reactivate later
- 38 -
Post-primary TB
Reactivation of latent TB or reinfection in person previously exposed
-
Intense immunological response with granulomata and central necrosis
Necrosis also called caseation or caseous necrosis (cheesy material)
Abscesses are ‘cold’ not hot like other pyogenic infection
Symptoms depend on site of reactivation or infection
Lungs - productive cough, haemoptysis
Meninges - headache, neck stiffness, cranial nerve lesions, coma
Bone - pain, paraplegia (if spinal)
Lymph nodes - glandular enlargement, cold abscess
Together with systemic illness with fever, night sweats, anorexia, weight loss
Investigations
General: FBC, PV/ESR, CRP, U&E, LFTs
Specific:
- Chest Xray
- Sputum microscopy and culture (best to send 3 early morning specimens ‘for AFB’)
- Gastric washings - to collect swallowed sputum can be used in children
- Bronchial washings at bronchoscopy for AFB
- Biopsy & culture of affected tissue
- Skin tests - Heaf, Mantoux - limited use, see below
- HIV Testing is recommended as HIV predisposes to TB
Tuberculin tests
Intradermal injection of tuberculin protein detects cellmediated immunity to TB
Heaf - six pronged gun through tuberculin dropped onto
skin - read at 3-7 days
recorded in grades 1 to 4
Mantoux - 0.1 ml intradermal injection of 1 or 10 unit
tuberculins – read at 2-3 days
recorded as mm of induration: strong positive = >6 mm
unit test) or >15 mm (10 unit test)
(1
Heaf grade 3 or 4 or strongly positive Mantoux =
suspicious (but not proving) active TB
Heaf grade 1 or 2 or positive Mantoux may indicate
previous exposure of prior BCG
People with previous BCG or past exposure but no
active disease do not usually have any response to a 1 unit Mantoux test so it is best
to use this dose if the test is being used in a workup for patients who might have
active TB
Clinical condition and history of vaccination or exposure has to be taken into account to
interpret tuberculin tests
Negative tests can be seen in active TB especially if:
- disseminated or miliary disease
- malnourished patients
- immunosuppressed patients
- malnourished patients
- patients on steroids
- sarcoidosis
BCG = Bacillus Calmette-Guerin
- Live attenuated vaccine from M bovis
- About 70% protective
- Used in 10-14 year olds with negative Heaf tests (grade 0 or 1)
- 39 -
-
Also in - healthcare staff, contacts of open TB, immigrants from high risk countries,
infants of immigrant groups
Contraindicated in: HIV infection, immunosuppression, lymphoproliferative disorders,
steroid therapy, pregnancy
Treatment of TB
Antituberculous chemotherapy
Different drug combinations are available
Short course treatment suitable for pulmonary disease but not all TB
Intensive phase for 2 months - rifampicin, isoniazid, pyrazinamide, ethambutol
Maintenance phase for 4 months - rifampicin, isoniazid
if single drugs are used then resistance quickly arises - at least 2 drugs for the whole
course
4 drugs given at first in case of resistance
maintenance decided once sensitivities are known
short course treatment only if rifampicin and isoniazid are used - otherwise 12-18 months
Some non-pulmonary TB may require more prolonged treatment e.g. TB meningitis usually
treated for 12 months
Compliance/adherence needs monitoring
e.e. examining urine for the bright orange colour caused by rifampicin, tablet counts,
prescription records
DOTS (directly observed therapy, short course) should be considered
Drug side effects
Rifampicin - orange urine, purple tears - stains soft contact lenses, hepatitis, liver enzyme
induction interfering with other drugs eg oral contraceptive pill, steroids
Isoniazid - hepatitis, peripheral neuropathy (reversed by pyridoxine - vitamin B6, so it is usual
to prescribe pyridoxine as prophylaxis against this)
Pyrazinamide - hepatitis, gout
Ethambutol - optic neuritis - so visual acuity is recorded before starting and patients told to
report any visual problems
The most common adverse effect is liver dysfunction
Almost all patients will have slightly increased liver transaminases on therapy
In patients with normal LFTs it is reasonable to check LFTs shortly after starting treatment and
repeating if the patient becomes symptomatic - such as malaise, vomiting, jaundice
Patients with pre-existing liver disease should have more frequent monitoring of LFTs
If transaminases are >5 times normal, all treatment is usually stopped until they have
improved; treatment is then gradually restarted to try and find the culprit drug
Multidrug resistant TB
Defined as resistance to rifampicin and isoniazid with or without other drugs
'Reserve' antituberculous agents are needed e.g. amikacin, clarithromycin, ofloxacin,
protionamide, cycloserine
- 40 -
at least 5 drugs used in an initial phase until cultures negative followed by at least 3 drugs for
up to 2 years
Other treatment
- Nutrition
- Steroids in some types of TB to diminish scarring due to the immune reaction which
might cause problems itself e.g. pericarditis to prevent constrictive pericarditis,
meningitis to reduce the risk of hydrocephalus, ureteric to reduce the risk of obstructive
uropathy
- Immunotherapy to boost cell medicated immunity being investigated - gammainterferon
All cases must be notified by law to the MOEH/CCDC - to organise contact tracing
Contacts are usually seen in clinics organised by TB specialist nurses, often based in the local
chest clinic. Contacts of all cases are screened, not just those with open TB and may reveal the
person who transmitted the disease to the patient as well as those at risk from the patient.
Screening involves a combination of Heaf skin tests and/or chest Xrays depending on the
contacts age.
Lyme Disease – Borrelia Burgdorferi
First recognised in 1975 in a cluster of children with arthritis in Lyme, Connecticut.
Spirochaetal Infections:Spirochaets are thin, spiral and motile organisms.
Incubation: 4-20 days
Host: Deer and moose
Diagnosis: Based on clinical and epidemiological features and serology.
Borrelia Burgorferi and spread by tick-bite. At the site of the tick-bite usually a limb, a slowing
expanding erythematous ring develops. Arthritis (joint disease occurs in about 50%, and varies
from brief migratory arthralgia to acute asymmetric mono- and oliogoarthrtis of the large
joints), neurological (cranial nerve palsies) and cardiac disease (myocarditis and heart block)
may follow. Rx – Penicillin or tetracycline
Tropical Infections
See MedSurg Revision document
Malaria
Typhoid
Dengue Fever
Gastroenteritis and dysentery
Viral Haemorrhagic fever
Immunodeficiency disorders
Immunodeficiencies may be primary or secondary. In primary disorders there is an intrinsic
defect in the immune system. Secondary immune deficiency is more common and may occur
with malignant disease, immunosuppressive therapy, HIV infection, malnutrition, splenectomy,
nephritic syndrome, and many bacterial and viral infections.
Clinical features:
- Many of the primary immunodeficiencies are inherited as X-linked or autosomal
recessive disorders. There may be a family history of parental consanguinity and
unexplained death, particularly in boys. Children with an immunodeficiency will usually
- 41 -
have a history of infections which are recurrent, persistent or unusual. There may also
be evidence of protein-losing enteropathy and FTT.
SCID
Major immunoglobulin
deficiencies
Minor immunoglobulin
deficiencies
Defects of bacterial
phagocytic function
Defects in leucocyte
function
Opsonisation deficiencies
Associated syndromes
A heterogeneous group of inherited disorders
characterised by profoundly defective cellular and
humoral immunity. Presents in the first 6 months of
the life with unusual and severe infections and severe
FTT. Defects which lead to SCID include purine
metabolism abnormalities (adenosine deaminase
deficiency), abnormal lymphocyte maturation and IL2 receptor gamma chain deficiency (X-Linked SCID)
The most common is X-linked agammaglobulinaemia,
caused by abnormalities in the Burton tyrosine kinase
(BTK) gene. This leads to profound failure of B-cell
development and immunoglobulin production.
Presents in the first 2 years of life with severe
bacterial infections particularly with sinopulmonary
infections. In the hyper IgM syndrome the B cells are
prevented from switching from IgM production to
more specific IgG and IgA. Presents with pyogenic
infections and Pneuomcytis carinii infections.
Abnormal production of one or more IgG subclasses
(IgG 1-4), IgA or poor antibody responses to specific
infections. Diseases of variable severity. Prophylactic
antibiotics or rarely IV Ig therapy may be indicated.
Chronic granulomatous disease – an inherited
disorder where phagocytic cells fail to produce
superoxide after ingestion of microorganisms due to a
lesion in a membrane-associated NADPJ oxidase.
Results in recurrent and chronic bacterial and fungal
infections, particularly involving the skin, lymph
nodes, lungs, liver and bones. Prophylactic antibiotics
are give; interferon gamma may be beneficial
Leucocyte adhesion deficiency presents with delayed
separation of the umbilical cord, delayed wound
healing, chronic skin ulcers and deep-seated
infections
Complement and mannose binding lectin deficiencies
causes susceptibility to bacterial infection, particularly
Neisseria Meningitides. Complement deficiencies may
also lead to autoimmune disease (SLE)
Ataxia telangiectasia – defect in DNA repair
Wiskott-Aldrich syndrome – Immunodeficiency,
thrombocytopenia, and eczema
DiGeorges’ syndrome – Immunodeficiency is
associated with maldevelopment of the 5th branchial
arch causing heart malformations, palatal defects,
absence of the thymus and hypocalcaemia (deletion
of section of chromosome 22)
Duncan’s Syndrome (X-linked lymphoproliferative
disease) – A syndrome where patients cannot make a
normal response to EBV infection and either second to
the infection or develop a secondary malignancy.
HIV infection
Reduction of vertical transmission of HIV
Can now be reduced to under 2%
- 42 -
-
Avoidance of breast feeding
Use of antenatal, perinatal and postnatal antiretroviral drugs to reduce viral load, e.g.
Zidovudine or nevirapine
Avoidance of labour and contact with the birth canal by elective C-section delivery
HIV-infected children
- May remain asymptomatic for months or years before progressing to severe disease
and immunodeficiency
- Clinical presentation varies with the degree of immunosuppression.
o Children with mild immunosuppression may have lymphadenopathy or parotitis;
o If moderate, they may have recurrent bacterial infections, candidiasis, chronic
diarrhoea, and lymphocytic interstitial pneumonitis
o Severe AIDS diagnoses: include opportunistic infections, e.g. Pneumocytis carinii
infection, severe FTT, encephalopathy, and malignancy, which is rare in children.
Treatment:
- Prophylaxis against primary pneumocystis pneumonia (PCP) with cotrimoxazole is
prescribed for infants who are HIV-infected, for infants whose infection has not yet
determined and for older children with low CD4 counts.
- Be careful with immunisation schedules, avoid BCG in HIV-positive children
- Antiretroviral therapy:
o The nucleoside analogue reverse transcriptase inhibitors e.g. ziodvudine,
didanosine, lamivudine
o The non-nucleoside analogue reverse transcriptase inhibitors e.g. nevirapine,
efavirenz
o The protease inhibitors e.g. ritonavir, indinavir, nelfinavir
Immunisation
HOW IT IS
GIVEN
WHEN TO IMMUNISE
WHAT IS GIVEN
2, 3 and 4 months old
Diphtheria, tetanus, pertussis (whooping cough), polio and Hib
(DTaP/IPV/Hib)
One injection
MenC, BCG if at risk
One injection
Around 13 months old
Measles, mumps and rubella (MMR)
One injection
3 years and 4 months to 5
years old
Diphtheria, tetanus, pertussis (whooping cough) and polio
(dTaP/IPV or DTaP/IPV)
One injection
Measles, mumps and rubella (MMR)
One injection
Diphtheria, tetanus, polio (Td/IPV)
One injection
13 to 18 years old
NHS Immunisation Information http://www.immunisation.org.uk/article.php?id=97
WHO Vaccinations
http://www.who.int/vaccines/
- 43 -
Genetic Conditions
Why learn genetics?
- Taking family histories to aid diagnosis. E.g
-
Offering patients support for genetic conditions, and reassurance, or care for offspring,
genetic counselling.
Down’s Syndrome (Trisomy 21)
The most common autosomal trisomy. Incidence is 1 in 600 in live-born infants. Usually
suspected at birth because of the baby’s facial appearance, but it can be difficult to be certain
when relying on clinical characteristics alone. Chromosome analysis takes several days and
sufficient certainty about the diagnosis is needed even to take blood for karyotyping. Down’s
occurs due to a variety of cytogenetic reasons:
- Non-disjunction (94%):
o Most cases result from an error at meiosis.
o The pair of chromosomes 21 fail to separate, so that one gamete has two
chromosomes 21 and one has none
o Incidence of translocation increases with maternal age (30 incidence 1 in 900,
44 incidence is 1 in 37)
- Translocation (5%):
o A chromosome 21 is translocated onto a chromosome 14, or more rarely onto
chromosome 15, 22 or 21.
- Mosaicism (1%):
o In mosaicism some of the cells are normal and some have trisomy 21. This
usually arise after the formation of the zygote, by non-disjunction at mitosis.
The phenotype may be milder in mosaicism.
The chromosomes of a baby with Down’s syndrome must always be examined. If there is free
trisomy 21, parental chromosomes need not be examined. If the baby has a translocation,
then the parents’ chromosomes should be studied, and if one carries a balanced translocation
other relatives should also be offered genetic counselling.
Clinical Features:
- Typical facial appearance
o Round face
- 44 -
-
-
o Epicanthic folds
o Brushfield spots in iris
o Protruding tongue
o Small ears
Other abnormalities
o Flat occiput
o Abnormal creases on palms and soles
o Hypotonia
o Congenital heart defects
o Duodenal atresia
Later medical problems
o Severe learning difficulties
o Small stature
o Recurrent respiratory infections
o Hearing impairment
o Visual impairment from cataracts and squints
o Increased risk of leukaemia
o Hypothyroidism
o Alzheimer’s disease
Edward’s Syndrome (Trisomy 18)
1 in 8000 live births
Clinical Features:
- Small chin
- Overlapping of fingers
- Rocker-bottom feet
- Cardiac and renal malformations
- Prognosis – Poor. 50% do not survive a week of life. Many spontaneously abort, or
early death after birth (3 months – 1 year)
Patau’s Syndrome Trisomy 13
1 in 14 000 live births
Clinical Features:
- Structural defect of birth
- Scalp lesions
- Small eyes
- Polydactyly
- Cardiac and renal malformations
- Many midline defects: Encephalocele, spina bifida
- Median survival – 3 days
Turner’s Syndrome (45, X)
Most result in early miscarriage
1 in 2500 live female births
Clinical features:
- Lymphoedema of hands and feet in neonate
- Short stature – Usual presentation to clinic, parental concern of height
- Neck webbing
- Wide carrying angle
- Widely spaced nipples
- Congenital heart defects
- Ovarian dysgenesis resulting in infertility
- Normal intellectual development
Klinefelter’s Syndrome 47, XXY
1-2 in 1000 live male births
- 45 -
Clinical Features:
- Infertility
- Hypogonadism – Small testes
- Pubertal development
- Gynaecomastia in adolescence
- Tall stature
- Intelligence – Usually normal, but may have some educational and psychological
problems
Autosomal Dominant
An affected individual carries the abnormal gene in a heterozygous state on one of a pair of
autosomes (1-22). Male and female offspring each have a 1 in 2 (50%) chance of inheriting
the abnormal gene from an affected parent.
Achondroplasia
Inherited autosomal dominant disorder
1 in 26 000 live births
Usually sporadic mutations
Clinical Features:
- Short stature
- Rhizomelia
- Lordosis
- Bradydactyly
Ehler’s-Danlos Syndrome
Abnormalities of collagen production, which results in bruising, wide scars, joint laxity, and
hyperelasticity of the skin. 1 in 150 000 in UK incidence.
Clinical Features:
- Premature rupture of membranes
- Herniae
- Bleeding from the GI tract
- Dissection aortic aneurysms
- Reduced lifespan if marked vascular fragility.
Familial hypercholesterolaemia
Autosomal dominant disorder of Chromosome 19.
Clinical features:
- Xanthomata
- Xanthelasma
- Corneal Arcus
- Ischaemic Heart disease
Huntington’s
Huntington’s Chorea shows autosomal dominant trait. 1 in 20 000 prevalence in UK.
Clinical Features:
- irritable and aggressive behaviour
- Schizophreniform psychosis may develop
- Chorea – develops to affect walking and speech
- Dementia
- Eventually develop akinesia and rigidity
- Prognosis – 15 years after onset of symptoms
Marfan’s Syndrome
Connective tissue disease, autosomal dominant inheritance. 4-6 in 100 000 incidence.
Clinical Features:
- Upward lens dislocation
- Retinal detachment
- Arachnodactyly
- 46 -
-
Tall stature
Pectus excavatum
Joint laxity
Dilatation of aorta, dissection aneurysm of aorta
Mitral insufficiency
Myotonic Dystrophy
Autosomal dominance. Incidence 5 in 100 000
Clinical features:
- Frontal baldness
- Myopathy in face
- Sternocleidomastoid wasting
- Inability to let go of a shaken hand
- Percussion myotonia
- Distal wasting and weakness
- Testicular atrophy in males
- Mental impairment in 30% of cases
Neurofibromatosis (NF-I and NF-II)
Von Recklinghausen’s Disease. Autosomal dominant inheritance, 50% chance of passing to
offspring, plus additional risk of sporadic mutation. NF-I, is associated with peripheral clinical
features, and the problem has been located on Chromosome 17. NF-II is associated with
bilateral acoustic neuromata and sometimes a cerebellopontine angle syndrome
Clinical Features of NF:
- Axillary freckles
- Neurofibromatas
- Six or More café-au-lait patches
- Deafness
- Hypertension
- Phaeochromocytoma
- Gliomatous change
Noonan’s Syndrome
Osteogenesis Imperfecta
Polyposis Coli
Otosclerosis
Tuberous Sclerosis
Autosomal Recessive
An affected individual is homozygous for the abnormal gene, having inherited an abnormal
allele from each parent, both of whom are unaffected heterozygous carriers. For two carrier
patients, the risk of each child, make or female being affected is 1 in 4 (25%), 50% carriers.
All offspring of affected individuals will be carriers.
In this inheritance check consanguinity.
Most common
Europeans: CF
Black: Sickle cell
Congenital Adrenal Hyperplasia
Autosomal recessive inheritance. The effects of enzyme deficiency are a result of: deficient
cortisol and/or aldosterone production or as a result of excess precursor steroids.
Clinical Features:
- Hyperpigmentation
- Hypoglycaemia
- Virilsation of females
- Macropenis
- Adrenal crisis
- 47 -
-
Hirsutism
Short stature
Amenorrheoa
Cystic Fibrosis
Characterised by chronic suppurative lung disease, and chronic exocrine pancreatic
insufficiency.
Clinical features:
- Recurrent respiratory infection
- 85% pancreatic insufficiency
- 10% suffer meconium ileus
- Prone to cirrhosis and male infertility
Friedreich’s Ataxia
Friedreich's ataxia is the most common inherited ataxia with a prevalence of 1 in 50000.
The mode of inheritance is autosomal recessive.
There is progressive gait and limb ataxia, loss of proprioception, pyramidal weakness and
dysarthria.
Extra-neurological involvement includes:
Hypertrophic cardiomyopathy in most patients
Diabetes mellitus in 10%
Onset is usually during adolescence.
Clinical features:
- Males and females equally affected
- An ataxic gait is an early sign:
o Upper limbs become ataxic in the later stages, often with an intention tremor
o Dysarthria is usually a late feature
- Features of corticospinal tract involvement:
o Limb weakness
o Absent abdominal reflexes
o Extensor plantars
- Features of posterior column involvement:
o Loss of vibration and joint position sense in the extremities
- Dorsal root and peripheral nerve involvement:
o Loss of lower limb reflexes: ankle before knee
o Axonal sensory peripheral neuropathy
- Musculoskeletal abnormalities present in 80% of cases - pes cavus and kyphoscoliosis
- Cardiomyopathy with resultant cardiac failure or dysrhythmias
- Nystagmus is common; optic atrophy and retinitis pigmentosa may occur
- Diabetes mellitus occurs in 10%
- No mental retardation
Galactosaemia
Galactosaemia is an autosomal recessive condition caused by the absence of galactose-1phosphate uridyl transferase, which results in intracellular accumulation of galactose-1phosphate which is highly toxic. It has an incidence of 1/60,000 in the UK.
Affected infants are normal at birth but upon commencement of milk feeds the majority suffer:
Jaundice
Vomiting
Diarrhoea
Failure to thrive
Clinical features:
The affected infant presents following the introduction of milk into the diet with:
- Jaundice
- Diarrhoea
- Failure to thrive
- Vomiting
- 48 -
- Psychomotor retardation
- Hepatosplenomegaly
- Renal disease
- Cataracts
- Increased susceptibility to infection
- Hypoglycaemia after exposure to galactose in the diet
There is:
- Galactosaemia
- Galactosuria
- Aminoaciduria (Fanconi syndrome)
Glycogen Storage diseases
These mostly recessively inherited disorders have specific enzyme defects which prevent
mobilisation of glucose from glycogen. There is abnormal storage of glycogen. There are nine
main enzyme defects, some of which are shown below. The disorder may predominantly affect
muscle (e.g. types II, V), leading to skeletal muscle weakness. In type II (Pompe’s disease)
there is generalised intralysosomal storage of glycogen. The heart is severely affected, leading
to death from cardiomyopathy. In other types (e.g. I, II) the liver is the main organ of storage,
and hepatomegaly and hypoglycaemia are prominent. Long term complications of type I
include hyperlipidaemia, hyperuricaemia, the development of hepatic adenomas and CV
disease.
Type
Enzyme
defect
Glucose-6phosphatase
Onset
Liver
Muscle
Comments
Infant
+++
-
Type II
(Pompe’s)
Lysosomal
alphaglucosidase
Infant
++
+++
Type III
(Cori’s)
Amylo-1, 6glucosidase
Infant
++
+
Type V
(McArdle’s)
Phophorylase
Child
-
++
Enlarge liver
and kidneys,
growth failure,
hypoglycaemia.
Good prognosis
Hypotonia nd
cardiomegaly
at several
months. Death
from heart
failure
Milder features
of type I, but
muscles may
be affected.
Good prognosis
Temporary
weakness and
cramps in
muscles after
exercise.
Myoglobinuria
in later life
Type I (von
Gierke’s
disease)
Hurler’s Syndrome (MPS I)
Hurler syndrome is an autosomal recessive lysosomal storage disorder affecting
mucopolysaccharide metabolism, the underlying defect being a deficiency of alpha-Liduronidase. Hurler syndrome is also called gargoylism.
- 49 -
The patient presents before the age of 2 with coarse facial features and mild developmental
delay. The child's development ceases and a gradual intellectual decline occurs with death in
the second decade.
Clinical features:
In Hurler syndrome the infant appears normal at birth, but after the first year features become
apparent as follows:
- coarse features
- large head with dolichocephaly and frontal bossing
- kyphosis
- protuberant abdomen
- umbilical herniation
- hepatosplenomegaly
- corneal clouding - differentiates Hurler from Hunter
- cardiac abnormalities:
o thickening of the valves
o narrowing of the arteries
o increased myocardial rigidity
o heart failure
- proportionate dwarfism - the limbs and trunk are equally affected
- mental retardation
Oculocutaneous albinism
In oculocutaneous albinism the defect in melanin production is expressed in the hair, skin and
eyes.
There are more than ten forms of oculocutaneous albinism - all are inherited in an autosomal
recessive manner.
In some forms there are associated defects in other organ systems e.g. haematopoiesis.
Clinical features:
- milky white skin
- white hair
- blue eyes
- photophobia
- pendular nystagmus
PKU
Phenylketonuria is an inborn error of metabolism where phenylalanine hydroxylase is deficient.
Thus there is an inability to convert phenylalanine into tyrosine which results in the
accumulation of phenylalanine and its metabolic products in body fluids.
The condition results in mental retardation, neurologic manifestations, light pigmentation and
mousy odour.
The disease is inherited as an autosomal recessive disorder of chromosome 12 and has an
incidence of approximately 1 per 10,000 births in the UK.
Carrier detection and prenatal diagnosis is possible using DNA analysis.
Clinical features:
- Children affected by this condition are normal at birth and in the neonatal period
- Developmental delay becomes apparent at about 3 months when the child fails to sit,
walk, or talk at the appropriate times
- The IQ of untreated patients is usually under 50. Behavioural abnormalities and
microencephaly is common, and seizures occur in about 25% of patients.
- Other possible clinical features of the disease include light pigmentation, blue eyes, fair
hair, eczema and mousy odour, unless treated by low phenylalanine diet.
- 50 -
Sickle Cell Disease
Aplastic sickle cell crises occur when the bone marrow temporarily shuts down. There is an
acute fall in haemoglobin levels - by up to 1 g per dl per day - and there is a complete absence
of reticulocytes in the blood film.
The characteristics of aplastic crises are that they often occur in children, they occur in
epidemics and in families, and they have a very stereotyped clinical course and recovery if
treated appropriately.
The causative agent is usually human parvovirus B19. Because of the shortened red cell
survival marrow shutdown leads to profound anaemia over a period of a few days.
Parental education is very important so that they recognise this condition early and act to
prevent fatality.
Tay-Sach’s Disease
Tay-Sachs disease is a condition caused by an inborn error of metabolism where there is a
deficiency in the enzyme hexosaminidase A, which splits the terminal N- acetylgalactosamine
residue from GM2 ganglioside.
The disease is particularly common in the Ashkenazi Jewish population, where it has an
incidence of 1 in 3 - 4000 births. It is a hundred times less frequent in other racial groups.
Inheritance is autosomal recessive with the disrupted locus on chromosome 15.
Clinical features:
- The first sign of Tay-Sachs disease is usually an abnormal startle reflex to sound
consisting of extension and myoclonus in the first weeks of life.
- Early development is normal but between the ages of 4 and 6 months the child does
not sit up, loses head control and takes no interest in the environment.
- The infant is blind by the end of the first year. Fundoscopy reveals a cherry-red spot at
the macula. Retinal artery occlusion and Niemann-Pick disease should be excluded in
these circumstances.
- After approximately 3 years the child becomes demented, blind, cachexic, and adopts a
decerebrate posture.
Thalassaemia – <See systems>
The thalassaemias are genetic disorders in globin chain production. The production of either
the alpha or the beta chains of haemoglobin may be reduced, resulting in alpha and beta
thalassaemia respectively.
The thalassaemias are the commonest genetic disorders in the world - 150 million people carry
the beta-thalassaemia gene - and occur in a very high frequency in a tropical belt extending
from Africa, throughout the Mediterranean region, the Middle East, the Indian subcontinent,
and throughout South West Asia. They occur sporadically in every population. It is possible
that the heterozygote state provides protection against falciparum malaria, thus explaining its
high carrier rate in certain populations.
Werdnig-Hoffman Disease
Werdnig-Hoffman disease is an early infantile form of spinal muscular atrophy, with an
autosomal recessive inheritance and is seen with an incidence of 1 in 20,000 births. The
earliest feature may be decreased fetal movements during late pregnancy.
Diagnosis is based on the history and examination, and findings on electromyography and
muscle biopsy.
The condition is progressive and the infant usually dies before the age of 18 months due to
respiratory failure.
Clinical features:
- The Mother of a Werdnig-Hoffman disease child may have noticed a lack of foetal
movements. Despite being alert, the infant is floppy and weak at birth. The weakness is
maximal in the shoulder and hip girdle muscles
- There may be muscle fasciculation, especially of the tongue. Other possible features
include deformed chest with see-saw respiration, the holding of limbs in the frog
position, and absent reflexes
- Occasionally there may be swelling of a limb; this is secondary to the inability of the
infant to move it when lying on it.
- 51 -
X-linked recessive disoders
-
All males are affected
Females can be carriers byt are usually healthy
Occasionally a female carrier shows mild signs of the disease.
Each son of a female carrier has a 1 in 2 (50%) risk of being affected
Each daughter of a female carrier has a 1 in 2 (50%) risk of being a carrier
Daughters of affected males will all be carriers
Sons of affected males will not be affected, since a man passes a Y chromosome to his
son.
Colour-blindness (red-green)
Duchenne’s and Becker’s muscular dystrophies
Fragile X Syndrome
1 in 4000 live births
There is a gap (fragile) site in the X chromosome (distal part of long arm). Caused by an
unstable mutation which trinucleotide repeat expansion. The normal copy of the gene contains
fewer than 50 copies of the CGG trinucleotide repeat sequence and is stable when transmitted
to offspring. Genes with per-mutation contain 55-199 copies of the repeat sequence. This
expansion has no phenotypic effect in male or female carriers, but is unstable and may
become larger during transmission through females. Genes with the full mutation contain more
than 200 copies of the repeat sequence. This affects the gene function, causing the clinical
features of fragile X syndrome in virtually all males around half of the female carriers.
Clinical Features:
- Macrocephaly
- Macro-orchidism
- Learning difficulties – Moderate
- Appearance – Long face, large everted ears, prominent mandible and large forehead
Glucose-6-Phosphate dehydrogenase (G6PD) deficiency
This X-linked disorder is the most common type of haemolytic anaemia due to an intrinsic red
cell enzyme defect.
Males who inherit an abnormal gene are invariably affected. Heterozygote females usually
have approximately 50% G6PD enzyme activity; the random Lyonisation of X chromosomes
means that rarely carrier females may be severely affected.
Clinical features:
- It is most common in the Mediterranean, the Middle East, South East Asia and West
Africa. It is rare among Caucasians.
- Presentation is usually with an acute episode of intravascular haemolysis on exposure
to certain drugs, infection or acute illness.
- Mediterranean forms may present with neonatal jaundice.
- The condition is also known as favism as sudden haemolysis may be prepipitated by the
ingestion of fava beans - Vicia faba. The deficiency is generally less severe in Africans
and favism is rare in this population.
Haemophilia A and B
These are a group of genetic disorders characterized by a deficiency of one of the coagulation
pathway factors.
Haemophilia A is characterized by a deficiency of one subunit of factor VIII (factor VIII C).
Haemophilia B is the result of a deficiency of factor IX.
Von Willebrand's disease is a result of a variable deficiency of subunits of factor VIII (factor
VIII RAG and factor VIII RiCoF) with a relatively normal factor VIII C level.
- 52 -
Hunter’s Syndrome
Hunter syndrome is an X-linked recessive mucopolysaccharidosis where the underlying defect
is in iduronosulphate sulphatase.
This syndrome is caused by a new mutation in about a third of cases. Thus this syndrome is
generally seen in males.
Typically a patient survives until later childhood. Occasionally there is survival into the fourth
decade.
Clinical features:
There is a wide spectrum of disease but in general the severity is less than MPS I:
- coarse facial features
- short stature
- hepatosplenomegally
- joint stiffness
- herniae
- mental retardation
- deafness
- kyphosis
Hunter and Hurler syndromes may be differentiated:
- nodes over the scapula are seen in Hunter but not Hurler
- corneal clouding is seen in Hurler but not Hunter
X-Linked dominant inheritance
Although dominantly inherited X-linked disorders occur, e.g. a variant of Vitamin D-resistant
rickets, they are rare. Both females and males are affected.
Y-Linked inheritance
No serious genetic disorders, apart from some rare forms of intersex and a gene for
azoospermia, have been located to the human Y chromosome, which seems reasonable given
that half the population live happily without a Y chromosome.
Dysmorphology
Malformation
Deformation
Disruption
Dysplasia
Birth Defects (p95)
Recognised by ‘Gestalt’
Noonan’s Syndrome
Caused by faulty autosomal dominant gene, chromosomes are normal
Clinical Features:
- Characteristic facies
- Short webbed neck with trident hair line
- Short stature
- Pectus excavatum
- Congenital heart disease e.g. Pulmonary stenosis, Atrial Septal Defect
- Occasional learning difficulties
Williams’ Syndrome
Most commonly sporadic mutation
Clinical Features:
- Short stature
- Characteristic facies
- Congenital heart disease e.g. Supravalvular aortic stenosis
- Learning difficulties
- Transient neonatal hypercalcaemia in some patients
- 53 -
Prader-Willi Syndrome
Clinical Features:
- Characteristic facies
- Hypotonia
- Obesity
- Hypogonadism
- Delay in developmental
Russell-Silver Syndrome
Clinical features:
- Short stature
- Low birth weight
- Frontal bossing and triangular face
- Relative macroencephaly
- Body asymmetry
- Mental retardation
- Hypospadias
- Primary Gonadal failure
- Cryptorchidism
Associations
VATER – Vertebral anomalies, Anal atresia, Tracheo-oEsophageal, Radial defects)
CHARGE – Coloboma of the eye, Heart defect, Atresia choanae (Choanal atresia), retardation
of growth and development, Genital abnormalities, and Ear abnormalities.
Systems
Haematology
Normal Ranges
Age
Hb (g/dl)
Birth
14.5-21.5
2 weeks
13.4-19.8
8 weeks
9.4-13.0
1 year old
11.3-14.1
2-6 years
11.5-13.5
6-12 years
11.5-15.5
12-18 years
Adult Male
13.0-16.0
Adult Female
12.0-16.0
MCV (fl)
100-135
88-120
84-105
71-85
75-87
77-95
WBC (x109/L)
10-26
6-21
6-18
6-17.5
5-17
4.5-14.5
Platelets
150-450
150-450
150-450
150-450
150-450
150-450
78-95
78-95
4.5-13
4.5-13
150-450
150-450
Anaemia
Anaemia is defined as a Haemoglobin volume under the expected range. Please note that the
normal ranges vary from newborn to adolescence.
Reasons for anaemia:
- Severe anaemia in the newborn – Haemorrhage, foetal maternal transfusion, or as a
result of placental abruption. Haemolysis (usually the babies presenting or having
jaundice) may occur as a result of rhesus isoimmunisation.
- 54 -
-
Blood Loss – Parasites, Nose bleeds (epistaxis), coagulopathies, Gastro-oesophageal
reflux disease
Bone marrow failure – Transient Erythroblastopenia of childhood (TEC), Acquire red cell
aplasia (Diamond-Blackfan), and Aplastic anaemia (Fanconi’s)
Infection – Chronic infection
Inflammation – Coeliac disease and other malabsorption problems and chronic
inflammatory disorders, for example juvenile idiopathic arthritis
Malignancy
Haemolysis – Red cell enzyme deficiency (G6PD), Spherocytosis (RBC membrane
defects) and autoimmune haemolytic anaemia
Haemoglobinopathies – Sickle cell diseased and thalassaemia
Haemopoietic factor deficiency – Iron (diet), Folate (diet), and/or B12 deficiency
(terminal ileum malabsorption)
Microcytic Anaemia
Causes of Microcytic hypochromic:
- Iron deficiency – Blood loss, diet, malabsorption
- Anaemia of chronic disease
- Sideroblastic anaemia
- Thalassaemia
Iron Deficiency
Iron deficiency is the most common cause of anaemia in the world. Due to body’s limited
ability to absorb iron and the frequent loss of iron due to haemorrhage.
Iron def occurs where there is inadequate iron for haemoglobin synthesis.
Causes:
- Blood loss
- Increase demands such as growth and pregnancy
- Decreased absorption (e.g. postgastrectomy)
- Poor intake
Clinical Features (additionals):
- Brittle nails
- Kolionychia
- Atrophy of the papillae of the tongue
- Angular stomatitis (inflammation of the mucous lining of the mouth)
- Brittle hair
- A syndrome of dysphagia and glossitis (Plummer-Vinson or Paterson-Brown-Kelly
syndromes)
Anaemia of chronic disease
Patients with chronic infections such as infective endocarditis and Tuberculosis and
osyeomyelitis in developing countries, chronic inflammatory diseases such as RA, SLE and
polymyalgia rheumatica, and patients with malignant disease.
There is a decreased release of iron from the bone marrow to developing erythroblasts, an
inadequate erythropoietin response to the anaemia, and decreased red cell survival. Exact
mechanism unsure but possibly due to inflammatory cytokines such as IL-1, Tumour necrosis
factor and interferons.
Rx
Patients do not generally respond to iron therapy but treatment is that of the underlying
cause.
- 55 -
Sideroblastic Anaemia
Sideroblastic anaemia’s are inherited or acquired disorders characterised by a refractory
anaemia, a variable number of hypochromic cells in the peripheral blood, and excess iron and
ring sideriblasts in the bone marrow. Presence of ring sideroblasts is diagnostic feature.
Lead Poisoning
Characteristic haematological features include:
-
Sideroblastoic anaemia, due to inhibition by lead of several enzymes involved in haem
synthesis, including ALA synthetase
Haemolysis, which is usually mild, resulting from damage to the red cell membrane
Punctate basophilia
Normocytic Anaemia
Normocytcic, normochromic anaemia is seen in anaemia of chronic disease, in some endocrine
disorders (e.g. hypopituitarism, hypothyroidism and hypoadrenalism) and in some
haematological disorders (e.g. aplastic anaemia and some haemolytic anaemias). In addition,
this type of anaemia is seen acutely following blood loss.
Macrocytic Anaemia – Megaloblastic and Non-megaloblastic
Megaloblastic
Characterised by the presence in the bone marrow of erythroblasts with delayed nuclear
maturation because of defective DNA synthesis (megaloblasts). Megaloblasts are large and
have large immature nuclei.
Megaloblastic changes occur in:
- Vit B12 def or abnormal vit B12 metabolism
- Folic acid def. or abnormal folate metabolism
- Other defects of DNA synthesis, such as congenital enzyme deficiencies (e.g. orotic
aciduria) or resulting from therapy with drugs interfering with DNA synthesis (e.g.
hydroxycarbamide (hydroxyurea), azathioprine, azidothymidine – AZT)
- Myelodysplasia due to dyserythropoiesis
Vitamin B12 Deficiency
The most common cause of Vit B12 deficiency in adults in pernicious anaemia. Malabsorption of
vit B12 because of pancreatitis, celiac disease or treatment with metformin is mild and does not
usually result in significant vitamin B12 deficiency.
Causes:
- Low dietary intake – Vegans
- Impaired absorpion
o PA
o Gastrectomy
o Congenital def. of intrinsic factor
o Ileal disease or resection
o Bacterial overgrowth – Small bowel
o Tropical sprue – Small bowel
o Fish Tapeworm (Diphyllobothrium latum)– Small Bowel
- Abnormal metabolism
o Congenital transcobalamin II def
o Nitrous Oxide (inactivates B12)
PA
- 56 -
Is a condition in which there is atrophy of the gastric mucosa with consequent failure of
intrinsic factor production and vit B12 malabsorption.
Folate Deficiency
Main cause of which is poor intake, which may occur alone or in combination with excessive
utilisation or malabsorption.
Causes:
- Nutritional
o Poor intake
o Old age
o Poor social conditions
o Starvation
o Alcohol excess
o GI disease
o Cancer
- Antifolate drugs
o Anticonvulsants – Phenytoin, and Primidone
o Methotrexate
o Pyrimethamine
o Trimethoprim
- Excessive utilisation
o Pregnancy
o Lactation
o Prematurity
o Malignancy
o Inflammatory disease
o Haemodialysis
o Malabsorption
Macrocytic Non-Megaloblastic
Common physiological cause is pregnancy, and a newborn may also suffer.
Common pathological causes are:
- Alcohol excess
- Liver disease
- Reticulocytosis
- Hypothyroidism
- Some haematological disorders
Drugs (e.g. cytotoxcs – azathioprine)
Classification of anaemia
Red cell
appearances
Small cell
(microcytes)
Large cells
(macrocytes)
Normal sized cells
Indices
Low MCV
(<80fl)
High MCV
(>96fl)
Normal MCV
- 57 -
Appearance
of Bone
marrow
Diagnosis
Iron
deficiency
Thalassaemia
Anaemia of
chronic
disease
Sideroblastic
anaemia
Megaloblastic
Normoblastic
Vitamin B12
or folate
deficiency
Alcohol
Reticulocytes
e.g.
haemolysis
haemorrhage
Liver disease
Acute blood loss
Anaemia of
chronic disease
Renal failure
Connective tissue
disease
Marrow
infiltration/fibrosis
Endocrine disease
Haemolytic
anaemia’s
Haemolytic Anaemias
May be for a number of reasons, but usually has a genetic origin. Haemolysis leads to a
number of clinical manifestations: anaemia, hepatomegaly and splenomegaly, increased level
of unconjugated bilirubin (released on haemolysis), excess urinary urobilinogen (attempting to
reduced bilirubin) and often abnormalities on the blood film.
Red Cell enzyme Deficiencies
Glucose-6-Phosphate dehydrogenase deficiency
Most common enzyme deficiency
Clinical Features:
- Neonatal Jaundice
- Drug-induced haemolysis for example Antimalarials, some antibiotics, dapsone
- Favism
Pyruvate kinase deficiency
Autosomal recessive inheritance. Much rarer
Haemoglobinopathies
Sickle Cell Disease
inheritance of two beta-Globin genes with a substitution of a single amino acid (homozygous
state). The clinical manifestations are a result of the deformation of the red cell in its
deoxygenated state. Clinical features vary greatly between patients but include:
- Anaemia
- Crises – Vaso-occlusive. In the infant a “glove and stocking” distribution of
inflammation is common (dactylitis). Other presentations include, bone and limb pain,
and cerebral and pulmonary infarction. Haemolytic, aplastic and sequestration crises
may also follow
- Infection – Increased risk of infection due to autosplenectomy
- Priapism
- Splenomegaly
- Long term problems – As a result of hypoxia to tissues and vaso-occulsion, can include
leg ulcers and delayed puberty
Sickle Cell-Haemoglobin C disease
Heterozygous inheritance of sickle cell results in carrier status with markedly reduced clinical
manifestation of above features
- 58 -
Thalassaemia
Inherited defects in the globin chain-synthesis.
α-Thalassaemia – Common in the Asian population. Mild-moderate haemolysis, some patients
are asymptomatic
β- Thalassaemia – With a Mediterranean, Middle eastern distribution. Where β-Thalassaemia
occurs, the body reacts by having persistence foetal-Hb. The condition is divided into
Thalassaemia major, Thalassaemia intermedia and Thalassaemia minor (carrier).
Clinical Features:
- Jaundice
- Pallor
- Splenomegaly
- Maxillary overgrowth and skill bossing
- A requirement for the necessity of blood transfusions
Red Cell Membrane defects
Spherocytosis
The most common cause of hereditary haemolysis in the UK {ref}, caused by abnormalities in
skeletal proteins.
Clinical Features:
- Jaundice
- Anaemia
- Splenomegaly
- Aplastic crisis
- Gallstone
Bleeding disorders
- 59 -
Haemophilia
Categorised into A and B, and then into mild, moderate, and severe dependent on the severity
of the condition.
Haemophilia A
Sex linked disorder with reduced or absent factor VIII activity
Clinical Features:
- Abnormal bleeding
Haemophilia B (Christmas disease)
Haemophilia B (Christmas disease) with reduced or absent IX activity
Von Willebrand’s disease (vWD)
The factor Von Willebrand factor has two main roles in the coagulation pathway:
- It assists the adhesion of platelets to injured endothelium
- It is a carrier protein for factor VIII:C
Clinical features:
- Mucocutaneous bleeding – e.g. Nose and gums
- Menorrhagia in the older female patients
Coagulation Inhibitors
For maintenance of the blood system and activation of coagulation at injured sites there must
be a balance between coagulation and anti-coagulation. The major inhibitors of coagulation
occurring in the human body are: Antithrombin and activated protein C (APC). Abnormalities in
these factors can cause imbalance in this process.
Protein C and S deficiencies
Thrombocytopenia
Literally a “penia” lack of, “thrombocytes”, platelets. Occurs with a reduction in platelts.
Purpura occurs as a result of reduced platelets around 20x10 9/L level.
Immune-Mediate Thrombocytopenic Purpura
The most common cause of thrombocytopenia in kids. This is a purpura (cutaneous
manifestation) of reduced platelets, as a result of immune-mediate destruction. This mainly
occurs in the spleen. As a result of the above there is an increase in megakaryocytes with in
the bone marrow as a compensatory mechanism. Possible suggested cause is viral.
Clinical features:
- Skin bleeding
- No systemic features – No fever
- No hepatomegaly or splenomegaly
- Fundal haemorrhage
Treatment
Supportive, nothing, or platelts. Can also use immunoglobulins which can ‘mop up’ the
antigens (give a broad immunoglobulin) or use steroids
Disseminated Intravascular Coagulation (DIC)
Where physiological generation of thrombin is not under control. Triggering release of
thrombin into the circulatory system leafs to deposition of fibrin in the microcirculation. Cause:
Endothelial damage (e.g. burns or trauma), or pathological release of tissue factor (e.g.
cancers, amniotic fluid embolism).
Clinical features:
- Haemorrhage
- Thrombosis
- Dysfunction of the major systems
- 60 -
Respiratory system
Infections
Upper Respiratory Tract Infections
Common colds, pharyngitis, tonsillitis, acute otitis media, and sinusitis.
Larygneal/Tracheal infection
Mucosal inflammation causes swelling
- Stridor, inspiratory rasping sound
Viral Croup
Viral croup is caused by Parainfluenza viruses, and influenzae and RSV. The mucosal
inflammation and laryngeal oedema, along with increased secretion in the trachea and airway
can cause stridor, and barking cough.
Clinical Features:
- Stridor
- Barking Cough
- Hoarse Voice
- Pink colour
- Febrile
Spasmodic or recurrent croup
In those children who develop stridor and characteristic cough without any other respiratory
systems, due to a hyperreactivity of the airways.
Bacterial tracheitis (pseudomembranous croup)
Cause – Staphylococcus aureus, or H. Influenzae
Clinical Features:
- Febrile
- Appears toxic
- Rapid progression
- Laryngoscopy shows thick secretions
Acute epiglottis
A medical emergency
Caused by H.Influenzae B, and therefore reducing in this country with HiB immunisation
Clinical features:
- Febrile, look-toxic
- Painful throat. So painful, child refrains from swallowing and talking, saliva builds up in
mouth
- Soft inspiratory stridor
- Rapidly progressing airway obstruction
Do not examine, and avoiding moving the throat as this can exacerbate inflammation
Lower Respiratory Tract Infections
Bronchitis
Inflammation of the bronchi
e.g. Bordetella Pertussis (Whooping cough)
Clinical features:
- 2-3/7 Hx of cold, with onset of paroxysmal or spasmodic cough, followed by distinctive
inspiratory whoop.
- Cough may cause vomiting
- 61 -
Bronchiolitis
The most common serious respiratory infection, especially seen in the winter!
Cause - Resp Syncitial virus
Clinical features:
- Dry cough
- Tachypnoea
- Subcostal and intercostals recession
- Sternal retraction in the very young child
- Hyperinflation of chest
- Fine end-inspiration crackles
- Wheeze
- Tachycardia
- Cyanosis or pallor
Pneumonia
Cause – Group B Beta-Haemolytic Streptococcus. E-Coli and Gram-negative bacilli.
Clinical Features:
- Febrile
- Cough
- SOB
- Lethargy
Asthma
Common
10-15% of children diagnosed as having asthma at some point in childhood.
Approx 20-30 children die of acute asthma each year in the UK
Airways
Variable airflow obstruction (like breathing through a straw)
Airways are normally large. In asthma the airways are restricted (use your hand to
demonstrate)
Cause: Bronchial inflammation with a prominent eosinophilic infiltrate
-
Airway inflammation
Airway muscle spasm
Mucosal oedema
Trapping of air in alveoli on CXR
Airway narrowing
Increased airway secretion
Airway twitchiness (bronchial hyperactivity – hyperactivity to allergens)
Triggers
- Infection – usually viral
- Allergens
- Other e.g. emotional, cold air
Clinical Features:
- Cough – Generally worse at night
- Wheeze – Expiratory (Polyphonic noise (multiple areas of narrowing) as opposed to
monophonic in upper airway obstruction (where there is one area of narrowing)
- SOB
- Chest tightness
- Look for barrel chest
- Nasal flaring
- Harrison’s Sulcus – Permanent indentation of the chest wall along the costal margins at
the level of diaphragm insertion as a result of chronic asthma (or rickets)
- 62 -
-
Reduced peak flow
What
-
to look for:
Resp rate
Colour
Work of breathing
o Suprasternal recession – tracheal tug
o Intercostal recession
o Subcostal recession
o Ability to talk in sentences
- Conscious state
- Peak flow – Should be taken
o Standing up
o Biggest breath possible
o Blow out
o Seal lips around the tube
o Three times – Make it competitive
Peak flow per height in men
Differential Diagnosis
- Hyperventilation – In anxiety
- Viral induced wheeze ~0-1 years
- Vocal cord dysfunction – often related to anxiety, attention deficit
- Croup
- Foreign body – If sudden onset
What
-
to do:
Any change in conscious level get an ambulance
Reassure, realx – talk to them
Try bronchodilator
o Salbutamol, bricanyl pmdi spacer
Reassess
o RR
o Work of breathing
o Colour
o Conscious state
Change in last 10 years:
NO LONGER USED
- Cromogylacte/Intal
- Syrups e.g. Salbutamol
- High dose INH steroids
- Old devices/rotahalers
- 63 -
NOW USED:
- New INH steroids – Orange INH Flixotide
- Long acting B2 stimulants (agonists)
- Combination
o Purple (Serotide) = Flixotide + Long Acting B2 agonist (serovent)
- Leukotriene receptor antagonists (inflammatory mediator inhibitor)
- High doses bronchodilator via PMDI and spacer
Side effects
- Bronchodilator
o Tremor
o Nausea
o Tachypnoea
o Headache
- Steroids in standard doses very safe, and a lot safer than not using them
o Local
Hoarse voice
Sore throat
Thrush ( wash out mouth)
o Systemic
?bones
?growth
?cataracts
adrenal suppression (high doses)
Treatment
Acute:
ABC
1 puff into spacer of Salbutamol, with 5 breaths (x10 times)
If no better nebs and oral prednisolone
Chronic
INH steroids (Brown) PREVENTER
B2 agonists (Blue) RELIEVER
Others:
PURPLE (serotide) = Flixotide + LA B2 agonists
ORANGE (INH Flixotide)
Pre-School Children
Should all have inhalers via a volumatic (for use with Ventolin, Becotide, Becloforte, ventide)
or aerochamber (GSK), a universal port spacer
Types of Inhalers
- Easy breathe
- Normal inhaler
o Deep breathe in, out, then breathe in push down inhaler, hold breath for 10secs
- Turbohaler
o As above, with twist to activate
- Accuhaler
o As above, with twist to activate
Pharmacology – Drug names/Spacers
-
Becotide® Steroid
o beclometasone dipropionate 50, 100 or 200 micrograms/metered inhalation
Ventolin® B2 agonist
- 64 -
Salbutamol, in accuhaler Accuhaler® (dry powder for inhalation), disk
containing 60 blisters of salbutamol (as sulphate) 200 micrograms/blister with
Accuhaler® device
Seretide® Steroid + B2 agonist
o 100, 250, or 500 Accuhaler® (dry powder for inhalation), disk containing 60
blisters of fluticasone propionate 100 micrograms, salmeterol (as xinafoate)
50 micrograms/blister with Accuhaler® device
Flixotide® Steroid
o Accuhaler® (dry powder for inhalation), disk containing 60 blisters of
fluticasone propionate 50 micrograms/blister
Serovent® B2 agonist
o Accuhaler® (dry powder for inhalation), disk containing 60 blisters of
salmeterol (as xinafoate) 50 micrograms/blister with Accuhaler® device
Atrovent®
o Bronchodilator
o Aerocaps® (dry powder for inhalation; for use with Atrovent Aerohaler®),
green, ipratropium bromide 40 micrograms
AeroChamber® Plus(GSK)
o Spacer device, medium-volume device. For use with Airomir®, Alvesco®,
Atrovent ®,Atrovent ® Forte, Combivent®, Duovent®, Salbulin®, and Qvar®
inhalers
Leukotriene receptor antagonists
o Montelukast: 10 mg daily in the evening; CHILD 6 months–5 years 4 mg daily in
the evening, 6–14 years 5 mg daily in the evening
o Zafirlukast: 20 mg twice daily; CHILD under 12 years, not recommended
Long acting B-2 agonist
o Theophylline
o Aminophylline
o
-
-
-
-
-
-
-
Asthma History
- Good structure
- SOB, Cough, when?
- Peers?
- What do they mean by wheeze? Demonstrate
- Current Rx
- No. of previous admissions
- Smoking
- Atopy hayfever, eczema, Family history
- Pets
Chronic Lung Infection – See Neonates
Recurrent Cough
Cystic Fibrosis
Autosomal recessive disorder. The most common AR disorder of Europeans.
Characterised by chronic suppurative lung disease, and chronic exocrine pancreatic
insufficiency. A gene located on chromosome 7 codes for the protein called cystic fibrosis
transmembrane regulator (CFTR), which is defective in CF. CFTR is a cyclic AMP-dependent
chloride channel blocker. Over 800 different mutations have been discovered in CF, but the
F508 mutation is found in 78% of cases.
Some of the Clinical features:
- Newborn
o Prolonged neonatal jaundice
o Meconium ileus in newborn 10%
o Malabsorption and possible subsequent failure to thrive
- 65 -
Respiratory
o Chest infections (recurrent)
o Bronchiectasis
o Nasal polyposis
o Pulmonary fibrosis
- Gastrointestinal
o Rectal prolapse
o Biliary stricture
o Gynaecomastia
o Intussusception
- Cardiac
o Cor pulmonale
- Others
o Infertility in affected males
o Affected females subfertile
o Diabetes Mellitus
o Vasculitis
o CF athropathy
Investigations:
- Sweat test – Abnormal function of the sweat glands results in excessive concentrations
of sodium and chloride in the sweat (80-125mmol/L in cystic fibrosis, 10-14mmol/L in
normal children). This forms the basisi of the essential diagnostic procedure, the sweat
test, in which sweating is stimulated by pilocarpine iontophoresis.
-
Ondine’s Curse
A rare breathing disorder where patients who fall asleep stop breathing and die. (Based on Ondine,
a Mermaid, who cursed a mortal with apnoea on sleep)
- 66 -
Cardiovascular
Normal Cardiology
-
-
Foetus
o Umbilical cord: Two arteries (deoxygenated) and one vein (oxygenated)
o Left atrial pressure is low, as relatively little blood returns from the lungs
o The right atrium is higher than in than in the left, as it receives all the systemic
venous return including blood from the placenta
o The flap valve of the foramen ovale is held open, blood flows across the atrial
septum into the left atrium and then into the left ventricle, which in turn pumps
it to the upper body
Within the first few breathes
o Resistance to pulmonary blood flow falls and the volume of blood flowing
through the lungs increases sixfold
o This results in a rise in the left atrial pressure
o Meanwhile the volume of blood returning to the right atrium falls as the placenta
is excluded from the circulation
o The change in the pressure differences causes the flap valve of the foramen
ovale to be closed.
o The ductus arteriosus, which connects the pulmonary artery to the aorta in
foetal life, will normally close within the first few hours or days.
o Some babies with congenital heart lesions rely on blood flow through the duct
(duct-dependent circulation). Their clinical condition will deteriorate dramatically
when the duct closes.
- 67 -
-
Postnatally
o Murmurs are common, especially PDA. The non-pathological murmurs are:
Systolic
Soft
S……..
o Innocent Murmurs
Still’s murmur (parasternal vibratory ejection systolic murmur) – which
has a very characteristuc low-frequency ‘twanging’ or musical quality. It
is made to disappear on hyperextension of the back and neck (Scott’s
manoeuvre)
Venous Hum – Is a superficial continous murmur heard beneath the
clavicles and in the neck which can be abolished by head movements, by
compression of the Ipsilateral jugular vein or by lying the child spine
The innocent right ventricular outflow tract murmur (pulmonary flow
murmur) is a soft early to mid-systolic ejection murmur heard at the
eight upper sternal border butdoes not radiate to the back. In the prem
and newborn infant an innocent pulmonary flow murmur may be audible
radiating to the axillae and to both lungs at the back
Innocent carotid bruits are common in normal children.
- 68 -
Heart Failure
Causes:
- Neonates – obstructed systemic circulation
o Hypoplastic left heart syndrome
o Critical aortic valve stenosis
o Severe coarctation of the aorta
o Interruption of the aortic arch
- Infants
o VSD
o AVSD
- 69 -
o
Large PDA
Symptoms:
- Breathlessness (particularly on feeding or exertion)
- Sweating
- Poor feeding
- Recurrent chest infections
Signs:
- Poor weight gain or ‘FTT’
- Tachypnoea
- Tachycardia
- Heart murmur, gallop rhythm
- Enlarged heart
- Hepatomegaly
- Cool peripheries
Chromosomal Abnormalities
- Down’s Syndrome Trisomy 21
o AVSD, VSD, ASD, Tetralogy of fallot
- Edwards Syndrome Trisomy 18
o Complex
- Patau’s syndrome Trisomy 13
o Complex
- Turner’s Syndrome 45XO
o Aortic valve stenosis, Coarctation of the aorta
- Chromosome microdeletion
o Aortic arch anomalies
Common Congenital Heart lesions
-
-
Non-Cyanotic
o VSD 32%
o PDA 12%
o PS 8%
o ASD 6%
o Coarctation of the aorta 6%
o AS
Cyanotic
o Tetralogy of fallot 6%
o Transposition of the great arteries 5%
Non-Cyanotic congenital heart disorders
Left to
-
right shunt
VSD
ASD
PDA
Aortic stenosis
Pulmonic stenosis
Coarctation of the aorta
Ventricular septal defect
Some children are born with a hole in the wall between the
two lower chambers of the heart, called the ventricles. This
hole is called a ventricular septal defect. Because the hole can
vary in location and size, the cardiologist may observe the
child to see whether the hole will close on its own.
Clinical Features:
- Asymptomatic
- 70 -
-
-
Heart failure with SOB and FTT
Recurrent chest infections
Cyanosis
Endocarditis (Late)
Parasternal thrill
Heart murmur at lower left sternal edge
o Small defect: Loud pansystolic murmur
o Large defect: Unimpressive ejection murmur from flow across pulmonary valve
Variable pulmonary component of second sound
Tachypnoea, tachycardia, and hepatomegaly
Patent ductus arteriosus
The ductus arteriosus is a blood vessel that connects the aorta
and the pulmonary artery in the fetus. That blood vessel is
supposed to close when the baby is born. If it does not close,
the congenital heart defect is called a patent ductus arteriosus.
Cardiac surgery may be required to correct the problem.
Clinical Features:
- In preterm infants, a PDA may be suspected by detecting
a bounding pulse (due to increased pulse pressure) and a systolic murmur at the left
sternal edge
- When severe – heart failure
- Most older children present with a continuous murmur beneath the left clavicle
- The murmur continues into diastole because the pressure in the pulm artery is lower
than that in the aorta throughout the cardiac cycle.
- Pulse pressure is increasing causing a collapsing pulse.
Pulmonary Valve Stenosis
Clinical Features:
- Most children with pulm valve stenosis are asymptomatic
- It is diagnosed clinically
- A small number of neonates with critical pulmonary stenosis have a duct-dependent
pulmonary circulation and present in the first few days of life
Signs:
- An ejection systolic murmur best heard in the second and third left intercostal spaces,
radiating into the back
- An ejection click best heard in the second and third left intercostal spaces
- When severe: A prolonged right ventricular impulse, with delayed pulmonary valve
closure on auscultation.
Atrial septal defect
Two main types:
- Ostium Secundum
o A deficiency of the foramen ovale
and surrounding atrial septum (see
diagram)
- Ostium primum
o A deficiency of the AV septum and is
characterised by :
An inter-atrial communication
between the bottom end of
- 71 -
the atrial spetum and the AV valves
An abnormal AV junction
Abnormal AV valves (a trileaflet left AV valve is its hallmark)
Clinical features:
- None (commonly)
- Recurrent chest infections/wheeze
- Heart failure
- Arrhythmias
- A fixed and widely split second hearty sound – due to the right ventricular stroke
volume being equal in both inspiration and expiration
- An ejection systolic murmur best heard in the third intercostal space – due to increased
flow across the right ventricular outflow tract
because of the left-to-right shunt
- A rumbling mid-diastolic murmur best heard at
the lower left sternal edge – due to increased
flow across the tricuspid valve, because of the
left-to-right shunt at the atrial level.
Coarctation of the aorta
Often associated with other lesions, the most common
being a bicuspid aortic valve and VSD
Clinical Features:
- Neonate
o Severe coarctation presents with a duct-dependent systemic circulation, and
circulatory collapse occurs on closure of the duct
o Less severe may present with symptoms of heart failure, or with a heart murmur
between the shoulder blades.
- Older children
o Hypertension
Aortic Valve stenosis
May not be an isolated lesion. It is often associated with mitral valve stenosis and coarctation
of the aorta, and their presence should always be excluded.
Clinical features:
- Neonate
o There may be a duct-dependent systemic circulation or severe heart failure
- Later life
o Mist children with mild or moderate stenosis present with an asymptomatic
murmur
o Severe stenosis – May present with reduced exercise tolerance, chest pain on
exertion or syncope.
- Signs
o Small volume, low rising, plateau-type pulses
o Carotid thrill
o Ejection systolic murmur maximal in the aortic area and radiating to the neck
o Delayed and soft aortic second sound
o Apical ejection click
o May have no murmur where critical aortic stenosis, has caused stenosis so to
the extent of no increased pressure after contracture.
Cyanotic congenital heart disorders
Right to left shunt
-
Tetralogy of fallot
Transposition of the great arteries
Total anomalous pulmonary venous return
- 72 -
-
Hypoplastic left heart sundrome
Pulmonary atresia
Truncus arteriosus
Tricuspid atresia
Single ventricle with pulmonary stenosis
Persistent foetal circulation
Elbstein malformation of the tricuspid valve
Tetralogy of fallot
- A large outlet VSD
- Overriding of the aorta with respect to ventricular septum
- Right ventricular outflow obstruction
- Right ventricular hypertrophy
Clinical Features:
Symptoms
- A few children present with:
o Severe cyanosis in the first few days of life with a duct-dependent pulmonary
circulation,
- Most are diagnosed in the first month of two of life following identification of a murmur
- Late infancy – Classical but somewhat rare presentation
o Severe cyanosis
o Hypercyanotic spells
o Squatting on exercise
- Signs
o Clubbing
o A long, loud ejection systolic murmur is best heard in the third left intercostal
space, usually with a single second heart sound
o With increasing right ventricular outflow tract obstruction, which is
predominantly muscular and below the pulmonary valve, the murmur will
shorten and cyanosis will increase.
o During a hypercyanotic spell, the murmur will be very short or inaudible
If tetralogy of fallot, there is a VSD, The upper part of the septum swings in front of the
pulmonary artery causing a stenois. There is hypertrophy of the right ventricle to deal with the
increased pressure and an overriding aorta. If a patient presents acutely, with cyanosis,
distress, resus, it may due to the upper septum causing a contracture over the pulmonary
stenosis. This may relieve itself spontaneously.
- 73 -
Transposition of the great arteries
-
-
Aorta and pulmonary arteries are swapped. Two parallel Circulations occur: SVC and
IVC Aorta, which goes round body and back to right atrium. Left atrium, to
pulmonary artery, to lungs, oxygenated and back to left atrium.
Maintenance of life can be maintained via a patent ductus arterious.
Clinical features:
Symptoms:
- Cyanosis is predominant symptom
- These children usually present on day 1 or 2 when
spontaneous closure of the ductus arteriosus leads
to a marked reduction in mixing of the desaturated
and saturated blood.
Signs
- Cyanosis
- There may be finger clubbing
- Remainder of examination is dependent on presence
associated abnormalities.
Arrhythmias
SV re-entry tachycardia
The most common childhood arrhythmia.
Heart rate – 250 – 300 beats/min
It can cause poor cardiac output and pulmonary oedema.
The term re-entry is used because a circuit of conduction is setup, with premature
activation of the atrium via an accessory pathway.
Presentation:
- Symptoms of heart failure in the neonate or young infant
- It is a cause of hydrops fetalis (defined as the presence of fetal subcutaneous tissue
edema accompanied by serous effusion(s) in 1 or more body cavities) and intrauterine
death.
Congenital complete heart block
Usually related to presence of anti-Ro antibodies in maternal serum (connective tissue
disorder).
- 74 -
Rheumatic Fever
Two major, or one major and two minors, criteria plus evidence of preceding group A
streptococcal infection (markedly raised ASO titre or other strep antibodies, or group A
streptococcus on throat culture)
-
-
Major manifestations
o Pancarditis (50%)
Endocarditis
Myocarditis
Pericarditis
o Syndenham’s Chorea (10%)
2-6 months after Strep infection
o Polyarthritis (80%)
o Erythema marginatum (<5%)
Rash on trunk and limbs, pink macules spread outwards, causing pink
border with fading centre
o Subcutaneous nodules (rare)
Minor manifestations
o Fever
o Polyarthralgia
o History of rheumatic fever
o Raised acute-phase reactants e.g. CRP, ESR
o Prolonged PR interval on ECG
Infective endocarditis
Cause: Most common alpha-haemolytic streptococcus (strep viridans)
At risk:
All children of any age with CHD, including neonates
VSD
Coarctation of the aorta
PDA
Prosthetic valve
Clinical signs:
- Fever
- Anaemia and pallor
- Splinter haemorrhages
- Clubbing (Late)
- Necrotic skin lesions
- Changing cardiac signs
- Splenomegaly
- Neuro signs from cerebral infarct
- Retinal infarcts
- Arthritis/Arthralgia
- Microscopic haematuria
Myocarditis/cardiomyopathy
Treatment is symptomatic with diuretics
Gastrointestinal tract
What’s your concern?
- Dehydration
- In who?
o Very young
- 75 -
o
o
o
Immunosuppressed
Diabetes
Cyanotic heart disease
Vomiting
Posseting:
Non-forceful return of milk, describes the small amounts of milk which often accompany the
return of swallowed air (‘wind’)
Occurs in nearly all babies from time to time
Regurgitation:
Non-forceful return of milk, larger, more frequent losses
Regurgitation usually indicates the presence of gastro-oesophageal reflux.
Vomiting:
Forceful ejection of gastric contents. It is a common problem in infancy and childhood. Usually
benign and is often caused by feeding disorders or mild gastro-oesophageal reflux or
gastroenteritis.
Causes of regurgitation/vomiting:
Medical
Gastroenteritis
GORD
Feeding problems
Infection:
- Resp tract
- Urinary tract
- Meningitis
Dietary protein
intolerance
Peptic ulceration
H. Pylori infection
Migraine
Surgical
Intestinal obstruction
- Pyloric stenosis
- Duodenal
stenosis/atresia
- Intussusception
- Malrotation
- Volvulus
- Duplication
- Hirschsprung’s
disease
- Foreign body
(bezoar)
Rare
Raised intracranial
pressure
Inborn error’s of
metabolism
Congenital Adrenal
Hyperplasia
Coeliac disease
Renal failure
Cyclical vomiting
Bulimia/anorexia nervosa
Diagnostic Clues in a vomiting infant
-
Bile staining vomit
o Intestinal obstruction must be excluded
Blood in vomit
o Suggests Oesophagitis or peptic ulceration or oral/nasal bleeding
Coffee ground
o Digested Haemoglobin
Redcurrant jelly stool
o Intussessception
Projectile vomiting in first few weeks of life
o Pyloric stenosis?
Symptoms of UTI, CNS, or GI infection?
Dehydration
Abdominal distension
o Is there lower intestinal obstruction
Gastro-Oesophageal Reflux
Reflux is common:
- In children with cerebral palsy, when energetic management, surgically if necessary,
may transform the child’s quality of life
- 76 -
-
In newborn infants with bronchopulmonary dysplasia (chronic lung disease of
prematurity)
Following surgery for oesophageal atresia or diaphragmatic hernia
Complications of Gastro- Oesophageal Reflux
- Failure to thrive
- Feeding problems
- Oesophagitis – pain, bleeding, iron deficiency
- Pulmonary aspiration leading to bronchitis or pneumonia
- Peptic stricture – associated with Oesophagitis
- Dystonic movements of head and neck – Sandifer’s syndrome
- Apnoea in preterm infants
- Apparent life-threatening events (ALTEs) or sudden infant death syndrome (SIDS) –
controversial
Pyloric Stenosis
In Pyloric stenosis, there is hypertrophy of the pylorus causing gastric outlet obstruction. It
presents at between 2 and 7 weeks of age, irerespective of gestational age. Common in boys
(4:1), particularly first-borns, there may be a family Hx.
Clinical features:
- Projectile vomiting (not bile-stained), which increases in frequency and severity with
time
- Constant hunger even after vomiting; only when markedly dehydrated do they refuse to
feed
- A hypochloreamic alkalosis with a low plasma potassium from vomiting acid stomach
contents
- Weight loss or poor weight gain if presentation is delayed.
Diagnosis:
- Test feed performed
- Gastric peristalsis may be seen as a wave moving from left to right across the abdomen
- Pyloric mass or ‘olive’ is usually palpable in the right upper quadrant
- If the stomach is overdistended with air, it will need to be emptied by a nastrogastric
tube to allow palpation
- USS
- Barium meal is only performed when the diagnosis remains in doubt.
Treatment:
Ramstedt’s Pylorotomy
- The operation is preceded by gastric lavage and carried out under continuous
nasogastric aspiration
- A longitudinal incision is made through the hypertrophied muscle as far as the mucosa;
the mucosa is left intact and is seen bulging into the incision
- The cut edges are separated
Crying
Crying of sudden onset may be due to:
- Urinary tract
- Meningeal or middle ear infection
- To pain from an unrecognised fracture
- Oesophagitis
- Torsion of the testis
Miserable crying infant:
- Severe nappy rash
- Constipation
- Coeliac disease
‘The baby is always crying’:
- May be a pointer to potential or actual non-accidental injury
- 77 -
Infant ‘colic’
A common symptom complex which occurs during the first few months of life. Paroxysmal,
inconsolable crying or screaming accompanied by drawing up of the knees takes place, several
times a day, particularly in the evening.
The condition usually resolves by 4 months of age
Occasionally cows milk protein intolerance or gastro-oesophageal reflux may be responsible
Acute abdo pain
Intra-abdominal
Surgical
Medical
Acute appendicitis
Mesenteric adenitis
Intestinal obstruct inc
Gastroenteritis
intussussception
Urinary tract:
Inguinal hernia
- UTI
Peritonitis
- Acute
Inflamed Meckel’s
pyelonephritis
diverticulum
- Hydronephritis
Pancreatitis
- Renal calculus
Trauma
Henoch-Schonlein
purpura
DKA
Sickle cell disease
Hepatitis
IBD
Constipation
Recurrent abdo pain in
childhood
Gynae in pubertal
females
Psychological
Lead poisoning
Acute porphyria (rare)
Unknown
Extra-abdominal
URTI
Lower lobe pneumonia
Testicular torsion
Hip and spine
Acute Appendicitis
Commonest cause of abdo pain in childhood requiring surgical intervention
Uncommon in children <3 years
Symptoms:
- Anorexia
- Vomiting
- Abdo pain, initially central and colicky (appendicular midgut colic) but then localising to
the right iliac fossa (from localised peritoneal inflammation)
Signs:
- Flushed face with oral foetor
- Low-grade fever 37.2-38ºC
- Abdo pain aggravated by movement
- Persistent tenderness with guarding in the right iliac fossa (McBurney’s Point)
In pre-school children:
- The diagnosis is more difficult, particularly early in the disease
- Faecoliths are more common and can be seen on a plain abdo X-ray
- Perforation is more common, as the omentum is less well developed and fails to
surround the appendix
Non-Specific Abdominal Pain (NSAP)
Abdo pain, which resolves in 24-48 hour
- 78 -
Intussusception
Invagination of proximal bowel into a distal segment. Coomonly involves the ileum passing into
the caecum and colon through the ileocaecal valve.
How? Viral Inflammation of peyer’s patches, forms lead point with peristaltic contraction
Commonest causes of intestinal obstruction in infants after the neonatal period.
Usually between 2 months and 2 years
Presentation:
- Paroxysmal, severe colicky pain and pallor – during episodes of pain, the child becomes
pale, especially around the mouth, and draws up his legs
- Sausage-shape mass – often palpable in the abdomen
- Passage of a characteristic redcurrant helly stool comprising blood-stained mucus – this
is a characteristic sign but tends to occur later in the illness and may be first seen after
PR
- Abdominal distension and shock
Management:
- Rectal air insufflation – 75% efficiency
- Surgery
Meckel’s Diverticulum
- 2% of population – ileal remnant of vitellointestinal duct, contains ectopic gastric
mucosa or pancreatic tissue
- 2% of people with Meckel’s have clinical manifestation
- 2 inches long
- 2 feet from ileocaecal junction
- First 2 years
Treatment – Surgical resection
Malrotation
Malrotation is an abnormal arrangement or twisting of the
intestine inside the abdomen, which is also known as volvulus,
that may result in loss of blood flow to the intestine. This
abnormal arrangement or twisting of the intestine can also cause
blockages of the intestines by causing a kinking of the intestine
without loss of blood flow. A delay in recognizing these conditions
can result in damage to the intestine as well as danger to the life
of the child.
Recurrent abdo pain
Gastroenteritis
Causes:
- Commonest cause
o 60% Rotavirus infection
- Other viruses
o Adenovirus
o Calicivirus
o Corona and astroviruses
- Bacterial causes
o Campylobacter jejuni (NB. Guillain Barre)
o Shigella and Salmonella – Dysenteric type
o Cholera
- 79 -
o
Enterotoxigenic E.Coli
Major Concern – Dehydration
Clinical Features of Dehydration in an infant:
- Sunken fontanelle
- Reduced level of consciousness
- Dry mucous membranes
- Reduced skin turgor
- Eyes sunken and tearless
- Sudden weight loss
- Oliguria
- Tachypnoea
- Tachycardia and Hypotension
- Peripheral vasoconstriction
- Reduced cap refill time
Isonatraemic and Hyponatraemic Dehydration
When total body deficit or sodum and water is proportional and plasma sodium remains within
the normal range (isonatraemic) or when sodium losses exceed those of water, plasma sodium
falls (hyponatraemic) which is associated with a shift of water from extravascular to
intravascular compartments.
The increase in intracellular volume leads to an increase in brain volum, sometimes resulting in
convulsions, whereas the marked extravascular depletion leads to a greater degree of shock
per unit of water loss.
Hypernatraemic Dehydration
Where, water loss exceeds the relative sodium loss and plasma sodium concentration
increases. Usually as a result of high insensible water losses.
The extravascular fluid becomes hypertonic with respect to the intracellular fluid and there is a
shift of water into the extravascular space from the intracellular compartment.
Signs of extravascular fluid depletion are therefore less per unit of fluid loss, and depression of
fontanelle, reduced tissue elasticity and sunken eyes are less obvious.
Particularly dangerous as water is drawn out of the brain and cerebral shrinkage within a rigid
skull may lead to multiple, small cerebral haemorrhages and convulsions.
Management
Mild dehydration (<5% body weight loss)
Moderate dehydration (6-10% body weight loss)
Severe dehydration (>10% body weight loss)
IV rehydration, treat shock + maintenance + continuing losses
Hypernatraemic
Management is particularly difficult. Once circulation has been restored, a too rapid reduction
in plasma sodium concentration and osmolality will lead to a shift of water into cerebral cells,
resulting in cerebral oedema and possible convulsions. The reduction in plasma sodium should
therefore be slow, over 48 Hours, in order to not exceed a reduction in plasma sodium of
10mmol/L per 24hours
Post-Gastroenteritis Syndrome
- 80 -
Dietary intolerance, for example lactose, with watery diarrhoea, following an episode of
gastroenteritis.
Malabsorption
Present with:
- Abnormal stools
- Failure to thrive
- Specific nutrient deficiencies
Ceoliac disease
Enteropathy in which the gliadin fraction of gluten provokes a damaging immunological
response in the proximal small intestinal mucosa. As a result, the rate of migration of
absorptive cells moving up the villi, from the crypts is massively increased but is insufficient to
compensate for increased cell loss from the villous tips. Villi become progressively shorter, and
then absent, leaving a flat mucosa.
Transient Dietary Protein Intolerance
Transient rather than life-long as in ceoliac. Clinical manifestations include:
- Diarrhoea and/or vomiting with FTT
- Eczema
- Acute colitis
- Migraine
- Occasionally an acute anaphylactic reaction with urticaria, stridor, bronchospasm and
shock. .
Bile Salt deficiency
From cholestatic disorders or following resection of the distal ileum, leads to a substantial
malabsorption of fat and fat-soluble vitamins.
Transport Defects (Rare)
- Glucose-Galactose malabsorption
o Severe, life threatening diarrhoea from the introduction of milk feeds and
affected children are only able to tolerate fructose as their dietary carb.
- Acrodermatitis enteropathica
o Congenital defect in zinc transport in the S.intestine. Affected children present in
infancy, with a symmetrical erythematous rash mainly affecting mucocutaneous
junctions around the mouth and anus. Plasma zinc is very low, as are the
activities of zinc-dependent enzymes such as AlkPhos in plasma.
Toddler Diarrhoea
Chronic non-specific diarrhoea. Unknown aetiology
IBD
Crohn’s
- Transmural, focal, sub-acute or chronic IBD. It affects any parts of GIT, from mouth to
anus.
- Affected intestine is thickened and adhesions between affected loops are common
- Perianal skin tags, fissures, and fistulae are common.
- Histologically – Non-caseating epitheloid cell granulomata
- Clinical features:
o Abdo pain
o Diarrhoea
o Growth failure with pubertal delay
o Oral and perianal ulcers
- Extraintestinal symptoms
- 81 -
o
o
o
o
o
Growth failure
Intermittent fever
Arthritis
Uveitis
Erythema nodosum
Ulcerative Colitis
- Recurrent, inflammatory and ulcerating disease involving the mucous membrane of the
colon.
- Presentation – Classically, rectal bleeding, diarrhoea, colicky pain and weight loss
- Extraintestinal symptoms
o Erythema nodusum
o Pyoderma gangrenosum
o Arthritis
o Spondylitis
Constipation
Hirschsprung’s disease
Absence of ganglion cells from the mysenteric and submucosal plexuses of part of the large
bowel results in a narrow, contracted segment. The abnormal bowel extends from the rectum
for a variable distance proximally, ending in a normally innervated, dilated colon.
Presentation:
- Usually in neonate period with intestinal obstruction heralded by failure to pass
meconium within the first 24h of life
- Abdo distension
- Later bile-stained vomiting develop
- Rectal examination – May reveal a narrowed segment and withdrawal of the finger
often releases a gush of liquid stools or flatus.
Liver
Neonatal liver disease
Clinical Features of Liver disease:
- Encephalopathy
- Jaundice
- Epistaxis
- Spider naevi
- Varices with portal hypertension
- Muscle wasting from malnutrition
- Bruising and petechiae
- Splenomegaly with portal hypertension
- Hypersplenism
- Hepatorenal failure
- Ascites
- Hypotonia
- Peripheral neuropathy
- Rickets secondary to Vit D deficiency
- Clubbing
- Liver palms
- Loss of fat stores secondary to malnutrition
Causes:
- Unconjugated
o Breast milk jaundice
o Infection (particularly urinary tract)
o Haemolytic anaemia e.g. G6PD deficiency
- 82 -
-
o Hypothyroidism
o High GI obstruction
o Crigler-Najjar syndrome - severe unconjugated hyperbilirubinaemia
Conjugated (>20% of total bilirubin)
o Bile duct obstruction
Biliary atresia
Choledochal cyst
o Neonatal hepatitis
Congenital infection
Inborn errors of metabolism
Alpha-1-antitrypsin deficiency
Galactosaemia
Tyrosinaemia
CF
Total parenteral nutrition (TPN) cholestasis
o Intrahepatic biliary hypoplasia
Alagille’s syndrome
Alagille’s Syndrome
Alagille syndrome is defined clinically as the combination of biliary hypoplasia:
- Cardiovascular abnormalities
- Vertebral abnormalities
- Characteristic facies
- Ocular abnormalities
Jaundice
Physiological – Due to immaturity of liver – Can’t conjugate bilirubin
- >48 hours age
- Commoner if breast fed or low birth weight
- Baby physically well
- Worsened by excessive bruising or cephalohaemotoma
- Typically gone with in a week
Pathological
- Excess bilirubin production e.g. Haemolysis (Common in Rhesus, Mother –ve, Baby
+ve), Bruising
- Delayed conjugation e.g. Hepatitis, metabolic
- Delayed enterohepatic excretion
- E.g. atresia – stenosis or absent of biliary tree, would be a conjugated jaundice
- Treatment – Light box
When
-
to Check a bilirubin level
< 48 hrs old, suggests pathological
Symptomatic e.g. XS wt loss, poor feeding
When the midwife says so!
Check Conjugated bilirubin
- Prolonged >2 weeks bottle fed, 3 weeks if breast fed
Symptomatic, e.g. Pale stools, dark urine
Bile duct obstruction
Biliary Atresia
1 in 14 000 live births
A progressive disease in which there is destruction or absence of the extrahepatic biliary tree
and intrahepatic biliary ducts.
Leads to chronic liver failure and death unless surgical intervention is performed.
- 83 -
Babies with biliary atresia have a normal birthweight but fail to thrive as the disease
progresses.
They are jaundiced and from the second day their stools are pale and their urine dark,
although both the jaundice and stool colour may fluctuate. Hepatomegaly is present and
splenomegaly will develop secondary to portal hypertension.
Treatment:
Consists of surgical bypass of fibrotic ducts, hepatoportoenterostomy (Kasai procedure), in
which the jejunum is anastamosed to patient ducts in the cut surface of the porta hepatitis. If
surgery achieved before the age of 60 days, 80% achieve bile drainage
Choledochal cysts
Cystic dilatations of the extrahepatic biliary system. About 25% present in infancy with
choleostasis.
In the older age group, choledochal cysts present with abdo pain, a palpable mass, and
jaundice or cholangitis.
Investigation: USS, or radionucleide scanning
Neonatal Hepatitis
Alpha-1-antitrypsin deficiency
Deficiency of protease Alpha-1-antitrypsin. Autosomal recessive with an incidence of 1 in
2000-4000 in the UK.
Presentation:
- Persistent neonatal jaundice
- Some develop bleeding
- Including cranial haemorrhage from Vit K def, particularly if breast fed
- Hepatomegaly
- Splenomegaly develops with cirrhosis and portal hypertension
Galactosaemia
Very rare – 1 in 40 000 incidence
Clinical features when fed milk:
- Poor feeding
- Vomiting
- Jaundice
- Hepatomegaly
- Untreated
o Chronic Liver failure
o Cataracts
o Developmental delay
- If Gram-negative Sepsis occurs
o Shock
o Haemorrhage
o DIC
Viral Hepatitis
Commonly caused by hepatitis viruses
Faecal-oral transmission - self-limited infections: A, E
Blood-borne - potentially cause chronic infection: B, C, D (G)
Hepatitis A
Virology
- picornavirus (enterovirus - RNA)
Incubation Period- 2-4 weeks
Epidemiology
- faecal-oral spread - poor hygiene or poor sanitation
- virus is shed in faeces in large amounts during the prodrome and initial illness
so only minor defects in hygiene are needed to spread the disease.
- blood (would need to have blood to blood contact in viraemic phase)
- 84 -
Clinical Course
- may be subclinical, especially in children
- prodrome with fever, chills, myalgia, nausea, vomiting, diarrhoea, abdominal
pain
- then jaundice, dark urine, pale faeces, hepatomegaly
- LFTs - transaminases usually in 1000s, alkaline phosphatase slightly raised,
hyperbilirubinaemia and bilrubinuria
Complications
- prolonged cholestasis (jaundice and raised alkaline phosphatase) after the acute
phase can occur (responsive to steroids)
- fulminant hepatic failure rarely
Diagnosis
Treatment
Prevention
- serology - positive Hepatitis A IgM
- supportive
- good hygiene
- active immunisation - killed vaccine – 2 doses produce long-lasting
immunity
- passive immunisation - human normal immunoglobulin – lasts about 3 months
Hepatitis A is most infectious before the symptoms begin
Hepatitis E
Virology
- calicivirus (RNA)
Incubation Period
Epidemiology - similar to hepatitis A
SE Asia, Central America, Africa, Russia
Clinical Course - similar to hepatitis A
Complications
- fulminant hepatic failure in pregnant females
Diagnosis
- serology – positive Hepatitis E IgM
Treatment
- supportive
Prevention
- hygiene (no immunisation available)
- 2-10 weeks
Hepatitis A and E produce very similar clinical pictures. Both can be associated with travel abroad but
hepatitis A vaccine is well tolerated, of high efficacy and has a good uptake meaning that travel related
acute hepatitis is more likely to be E. Hepatitis E is rarely tramsmitted in the UK.
Hepatitis A and E are diseases of childhood in endemic areas with lifelong immunity after
infection. Hepatitis E produces high morbidity and mortality in pregnant women - not usually a problem
to those in endemic areas who are likely to be immune but pregnant travellers are at risk.
Hepatitis B
Virology
- hepadnavirus (DNA)
Incubation Period
- 4-20 weeks
Epidemiology
- blood-borne - injecting drug users, needlestick
- sexual
- vertical - common in high prevalence areas
Clinical Course
acute infection - prodrome with fever, chills, myalgia, nausea, vomiting,
diarrhoea, abdominal pain, arthritis, urticarial rash, then jaundice, dark urine,
pale faeces, hepatomegaly
Chronic infection – often asymptomatic unless complications ensue
LFTs - acute infection - transaminases in 1000s, hyperbilrubinaemia, slightly
raised alkaline phosphatase; chronic infection - slight to moderate increase in
transaminases
Complications
- acute - fulminant hepatic failure
- chronic carriage in 10% following acute acquired infection
- 'high risk' chronic infection - cirrhosis, hepatocellular carcinoma,
association with polyarteritis nodosa
Diagnosis
- serology (see below), liver biopsy in chronic hepatitis
- 85 -
Treatment
- supportive during acute illness
- chronic, severe - interferon-alfa, adefovir, lamivudine, liver transplant
Prevention
yeast
- active immunisation - HBsAg produced by recombinant DNA technique in
- passive immunisation - human hyperimmune immunoglobulin (HBIg) (shortlived protection
- care with sharps, blood and body fluids
- safer sex
- screening or pregnant women and immunisation of infants at risk
Serological markers for hepatitis B infection
HBsAg
surface antigen
HBsAb
surface antibody
HBcIgG
core IgG antibody
HBcIgM
core IgM antibody (= acute infection)
HBeAg
e antigen
HBeAb
e antibody
HBV-DNA
hep B DNA measured by PCR (quantitative)
N.B. core antigen does not appear in the blood (only in the liver)
- 86 -
HBsAg
HBsAb
HBcIgG HBcIgM HBeAg
HBeAb
HBV-DNA
acute hepatitis B
+
-
+
+
+
-
++
'high risk' carrier
+
-
+
-
+
-
+++
'high risk' carrier (pre-core
mutant strain)
+
-
+
-
-
+
+++
'low risk' carrier
+
-
+
-
-
+
+/-
previous infection - now immune
-
+
+
-
-
+/-
-
Immunised
-
+
-
-
-
-
-
The only marker which proves acute infection is HBcIgM
The level of HBV-DNA is a better marker of high risk of infectivity and chronic liver disease than HBeAg
Hepatitis C
Virology
Incubation Period
Epidemiology
-
flavivirus (RNA)
- 2-26 weeks
blood-borne - injecting drug users, blood products in past
(sexual -rare)
(vertical - uncommon)
Clinical Course
- the acute infection is milder than hepatits A or B - often subclinical
- but about 90% go on to be chronic carriers
- LFTs - moderate to high transaminases in acute infection, can be normal in
chronic infection and no help as a guide to need for treatment
Complications
- chronic infection
- chronic hepatitis, cirrhosis, hepatocellular carcinoma,
- association with cryoglobulinaemia, glomerulonephritis
Diagnosis
- serology – HCV Ab (N.B. antibodies only positive 3 months after infection),
HCV-RNA by PCR, liver biopsy
Initial screen is by HCV Antibodies, if positive then chronic hepatitis C can be
confirmed by finding positive HCV-RNA. Liver biopsy is needed to stage the
severity of the chronic hepatitis
As antibodies can take 3 months to develop after acute infection they are of no
help in diagnosis at this stage. Presence of HCV-RNA but negative antibodies
would confirm acute infection.
Treatment
- chronic, severe - interferon-alfa +/- ribavirin,
- liver transplant for end-stage disease
Prevention
- care with sharps, blood and body fluids
- 87 -
- needle exchange schemes for injecting drug users
- safer sex
Hepatitis D
Virology
Epidemiology
Clinical Course
Diagnosis
Treatment
Prevention
-
defective RNA virus, needs HBsAg for protein coat
uncommon, blood-borne in HBV carrier
severe hepatitis, can be chronic
serology, PCR
supportive
prevent HBV
Hepatitis G
Virology
- flavivirus (RNA), related to HCV
Epidemiology
- blood-borne - injecting drug users, blood products approx 1% blood donors
infected
Clinical Course
-mild transient liver function abnormalities
Complications
- none known
Diagnosis
- serology, PCR (not routinely available)
Treatment
- supportive
Prevention
- none
Other infective causes of hepatitis
Viral causes
EBV (may be with glandular fever but can occur alone)
CMV
Yellow fever (very rare as vaccination to high risk areas compulsory)
Non-viral causes
Leptospirosis (from infected rat urine - clues from the history)
Q fever (animal exposure)
Toxoplasmosis (rare)
Syphilis (rare)
So if not hepatitis A to E then check serology for CMV, EBV, leptospirosis, Q fever
- 88 -
Almost any infection can cause mild liver function test abnormalities
Acute Liver failure
Causes:
Infection
Poisons/Drugs
Metabolic
Autoimmune hepatitis
Reye’ Syndrome
Viral Hep
Paracetamol, isoniazid, halothane, Amanita Phalloides
Wilson’s disease, tyrinosaemia
Reye’s Syndrome is an acute non-inflammatory Encephalopathy with microvesicular fatty
infiltration of the liver. Although the aetiology is unknown, there is a close association with
aspirin therapy. Since stopping giving aspirin to children aged less than 12 years, Reye’s
syndrome has virtually disappeared.
Chronic Liver failure
Causes:
- Chronic hepatitis
o Postviral hep
o Autoimmune hep
o Drugs
o IBD
o Primary sclerosing cholangitis +/- UC
- Wilson’s disease >3 years
- Alpha-1-antitrypsin deficiency
- CD
- Secondary to:
o Neonatal liver disease
o Bile duct lesions
Autoimmune Hepatitis
Mean age 7-10 years. Girls > Boys. May present as:
- An acute hepatitis
- A Fulminant hepatitic failure
- Chronic liver disease with autoimmune features such as skin rash, lupus
erythematosus, arthritis, haemolytic anaemia or nephritis.
Diagnosis based on:
- Hypergammaglobinaemia (IgG >20g/L)
- Positive autoantibodies e.g. Smooth muscle antibodies, ANA’s
- A low serum complement (C4)
- And typical histology
Treatment:
90% respond to Prednisolone and azathioprine
Cystic Fibrosis
Abnormal bile acid concentration and biliary disease is seen in CF as the CFTR is found in
biliary epithelial cells.
Wilson’s Disease
Autosomal recessive disorder with incidence of 1 in 200 000. Many mutations now identified on
chromosome 13.
Defect is:
- A combination of reduced synthesis of caeruloplasmin (the copper-binding protein) and
- 89 -
Defective excretion of copper in the bile, which leads to accumulation of copper, in the
liver, brain, kidney and cornea.
Rarely presents in children under 3 years
Clinical Features:
- Any form of liver disease
- Neurological features common in second decade
o Deterioration in school performance
o Mood and behaviour change, and extrapyramidal signs such as incoordination,
tremor and dysarthria.
- Renal tubular dysfunction, with Vit D-resistant rickets
- Heamolytic anaemia
- Copper accumulation in cornea (Kayser-Fleischer rings) are not seen before age of 7.
-
Congenital Hepatic Fibrosis
Presents in children over 2 years old with hepatosplenomegaly, abdo distension, and portal
hypertension.
Cirrhosis and portal hypertension
Cirrhosis is the end stage of many forms of liver disease. It is defined pathologically as
extensive fibrosis with regenerative nodules. It may be secondary to hepatocellular disease or
to chronic bile duct obstruction. The main pathophysiological effects of cirrhosis are diminished
hepatic function and portal hypertension with splenomegaly, varices and ascites.
Hepatocellular carcinoma may develop.
Management of children with liver disease
Mainly supportive
Nutrition
Malnutrition may be due to protein malnutrition, fat malabsorption, anorexia, and fat soluble
vitamin deficiencies (Vit A, D, E, and K)
Rx – Provide a high-proten, high-carbohydrate diet with 50% more calories than the
recommended dietary allowance. In children with cholestasis, medium-chain triglycerides,
which are absorbed by the portal circulation, will provide fat, but 20-40% long chain
triglycerides are required to prevent essential fatty acid deficiency. Many children will require
NG tube
Fat-soluble vitamins
- Vitamin K Deficiency
o In liver disease, may be due to malabsorption or diminished synthesis. Watersoluble forms of vitamin K are available.
- Vitamin A Deficiency
o Causes night blindness in adults and retinal changes in infants. It is easily
prevented with oral Vitamin A
- Vitamin E Deficiency
o Causes peripheral neuropathy, haemolysis and ataxia. It is very poorly absorbed
in cholestatic conditions and high oral doses are required.
- Vitamin D Deficiency
o Causes rickets and pathological fractures. It is prevented by using a watersoluble form of Vitamin D. Vitamin D-resistant rickets indicates renal tubular
acidosis.
Pruritis
Many children with cholestasis have severe pruritis. It is alleviated by phenobaritone to
stimulate bile flow, cholestyramine, which is a bile salt resin, ursodeoxycholic acid, an oral bile
acid, or evening primrose oil (arachniadonic acid) applied to the skin.
Encephalopathy
- 90 -
In children, encephalopathy is managed by treating the precipitating factor (sepsis, GI
haemorrhage), by protein restriction or by using oral lactulose to reduce ammonia
reabsorption by lowering colonic pH and increasing colonic transit.
Liver transplantation
Indications for transplantation in chronic liver failure are:
- Sever malnutrition unresponsive to intensive nutritional therapy
- Recurrent complications (bleeding varices, resistant ascites)
- Failure of growth and development
- Poor quality of life
Complications post-transplantation include:
- Primary non-function of the liver (5%)
- Hepatic artery thrombosis (10-20%)
- Biliary leaks and strictures (20%)
- Rejection (30-60%)
- Sepsis, the main cause of death
Endocrine
Diabetes Mellitus
Classification:
- Type 1 Insulin-dependent
o Most childhood diabetes
- Type 2 Non-insulin-dependent
o Usually older children, obesity-related, positive family history, not prone to
ketosis
- Type 3 Other specific types
o Genetic defects in β-Cell function
o Genetic defects in insulin action
o Infections e.g. Congenital rubella
o Drugs e.g. Corticosteroids
o Pancreatic exocrine insufficiency e.g. CF
o Endocrine diseases e.g. Cushing’s Syndrome
o Genetic/Chromosomal syndromes e.g. Down’s and Turner’s
- Type 4 Gestational Diabetes (GDM)
All most all children with diabetes mellitus are insulin-dependent (type I).
Symptoms and Signs of diabetes:
- Early
o Most common – the ‘classic triad’
Excessive drinking (polydipsia)
Polyuria
Weight loss
o Less Common:
Enuresis (secondary)
Skin sepsis
Candida and other infection
- Late – DKA
o Smell of acetone on breath (‘Pear-drop’ halitosis)
o Vomiting
o Dehydration
o Abdominal pain
- 91 -
o
o
o
o
Hyperventilation due to acidosis (Kussmaul breathing)
Hypovolaemic shock
Drowsiness
Coma
Diagnosis:
Usually confirmed in a symptomatic child finding a markedly raised random blood glucose
(>11.1mmol/L WHO), glycosuria and ketonuria. Where there is any doubt, a fasting blood
glucose (>7.8mmol/L) or a raised glycosylated Hb (HbA1c) are helpful.
Insulin
Made chemically identical to human insulin by recombinant DNA technology or chemical
medification of pork insuline.
Available in:
- Short-acting
o E.g. Actrapid and Humulin S
- Medium-acting
o E.g. Insulatard and Humulin I
- Long-acting
o E.g. Ultratard
- Mixed – Short and intermediate-acting insulins
o E.g. Mixtard 30/70 and Humulin M3
Most available insulin is human and in concentrations of 100 U/ml (U-100)
Insulin may be injected into the S/C tissue to the upper arm, the anterior and lateral aspects
of the thigh, buttocks and the abdomen. Rotation of the injection sites is essential to prevent
lipohypertophy or, more rarely, lipoatrophy. Skin pinched up, and injected at 45 degree angle.
In young children, insulin is usually given twice a day, before breakfast and evening meals as a
mixture of short-acting (approx 30%) and medium or long-acting (approx 70%). In general
about 2/3 is given before breakfast, and 1/3 before evening meal.
Older children and teenagers are increasingly using a 3 or 4 injections a day regimen (‘basalbolus’) in which a short-acting insulin (often Lispro) is given before each meal and some longacting insulin late evening to provide insulin overnight. This allows greater flexibility by relating
the insulin more closely to food intake and exercise.
Shortly after presentation, when some pancreatic function is preserved, insulin requirements
often become minimal, the so-called ‘honeymoon period’. Requirements subsequently increase
to 0.5-1 or even up to 2 units/kg per day.
Diet
The diet and insulin regimen need to be matched, to avoid hypoglycaemia.
Factors affecting blood glucose levels
Increase
Omission of insulin
Food (especially carbs)
Illness
Menstruation
Growth hormone
Corticosteroids
Sex hormones at puberty
Stress of an operation
Decrease
Insulin
Exercise
Alcohol
Some drugs
Marked anxiety/excitement
- 92 -
Hypoglycaemia in Diabetes
Most children develop well defined symptoms when their blood glucose falls below about
4mmol/L
Symptoms are highly individual and change with age but most complain of:
- Hunger
- Sweatiness
- Feeling faint or dizzy or of a ‘wobbly feeling’ in their legs
- If unrecognised or untreated, hypoglycaemia may progress to seizures and coma.
- Parents can often detect hypoglycaemia in their children by their pallor and irritability,
sometimes presenting as unreasonable behaviour.
Diagnosis:
- Check Blood Glucose Measure
Treatment:
- Administration of an easily absorbed glucose in the form of sweets (glucose tablets e.g.
Dextrosol or similar)
- Oral Glucose gels (e.g. Hypostop) are easily and quickly absorbed from the buccal
mucosa and so are helpful if the child is unwilling or unable to cooperate to eat.
- Glucagon injection if severe hypoglycaemia IM
Diabetic Ketoacidosis
Essential early investigations
- Blood glucose >15mmol/L
- U & E’s, creatinine (dehydration)
- Blood gas analysis (Severe metabolic acidosis)
- Urinary glucose and ketones (both are present)
- Evidence of a precipitating cause e.g. infection (blood and urine cultures performed)
- Cardiac monitor for T-wave changes of hypokalaemia
- Considered salicylate level
- Weight
Management
1. Fluids:
Dehydration should be corrected gradually over 48-72 H. Rapid rehydration should be
avoided as it can lead to cerebral oedema,
2. Insulin:
Insulin infusion (0.05-0.1 units/kg per h) is started. Aim for gradual reduction
3. Potassium:
Hypokalaemia will occur with rehydration and insulin so give as prevention as soon as
urine is passed
4. Acidosis:
Acidosis should self-correct
5. Re-establish oral fluids, S/C insulin and diet:
Do not stop IV insulin infusion until after S/C insulin has been given
6. Identification and treatment of an underlying cause:
DKA may be precipitated by an underlying infection. Antibiotics may be indicated.
Hypoglycaemia
Hypoglycaemia is a common problem in neonates but is seen much less often beyond this
period. It is often defined as a plasma glucose less than 2.6mmol/L, although the
development of clinical features will depend on whether other energy substrates can be
utilised. Clinical features include:
- Sweating
- Pallor
- Central nervous system signs of irritability, headache, seizures and coma
- 93 -
The neurological sequelae may be permanent if hypoglycaemia persists and include epilepsy,
severe learning difficulties and microcephaly. This risk is greatest in early childhood during the
period of most rapid brain growth.
Infants have high energy requirements and relatively poor reserves of glucose from
gluconeogenesis and glycogenesis. They are at risk of hypoglycaemia with fasting. Infants
should never be starved for more than 4 h duration e.g. preoperatively. A blood glucose should
be checked in any child who:
- Becomes septicaemic or appears seriously ill
- Has a prolonged seizure
- Develops an altered state of consciousness
This is often done at the bedside using glucose sensitive strips, whose accuracy is improved by
use of a reflectance meter. However, the strips only indicate that the glucose is within a low
range of values and any low reading must always be confirmed by laboratory measurement.
If the cause of the hypoglycaemia is unknown, it is vital that blood is collected at the time of
the hypoglycaemia and the first available urine sent for analysis so that a valuable opportunity
for making the diagnosis is not missed.
Tests:
- Blood
o Confirm hypoglycaemia with lab blood glucose
o Growth hormone, cortisol, insulin, C-peptide, fatty acids, acetoacetate
- First urine after hypoglycaemia
o Dicarboxylic acids, glycine conjugates, carnitine derivatives, Consider saving
blood and urine for toxicology, e.g. salicylate, sulphonylurea
Causes of hypoglycaemia beyond the neonatal period:
- Fasting
o Insulin excess
Excess exogenous insulin e.g. in diabetes mellitus
Beta-cells tumours/disorders
Drug-induced
Autoimmune
Beckwith’s Syndrome
o Without hyperinsulinaemia
Liver disease
Ketotic hypoglycaemia of childhood
Inborn errors of metabolism
Hormonal deficiency
- Reactive/non-fasting
Galactosaemia
Leucine sensitivity
Fructose intolerance
Maternal diabetes
Hormonal deficiency
Aspirin/Alcohol poisoning
Treatment
Hypoglycaemia can usually be corrected with an IV infusion of glucose (2-4ml/kg of 10%
dextrose)
Hypothyroidism
There is minimal thyroxine transfer from mother to the foetus, although severe maternal
hypothyroidism can affect the developing brain. The foetal thyroid predominantly produces
‘reverse T3’, a derivative of T3 which is largely inactive. After birth there is a surge in the level
of thyroid-stimulating hormone (TSH) which is accompanied by a marked rise in T4 and T3
levels. The TSH declines to the normal adult range within a week. Preterm infants may have
- 94 -
very low levels of T4 for the first few weeks of life whilst the TSH is within the normal range;
under these circumstances, additional thyroxine is not required. An example of a patient with
hypothyroidism may have a low but normal range Free T4, and total T4, but high Total TSH
(above 0.5-4.0). There is a hypothyroid with compensation
Clinical features:
- Congenital
o FTT
o Feeding problems
o Prolonged jaundice
o Constipation
o Pale, cold, mottled dry skin
o Coarse facies
o Large tongue
o Hoarse cry
o Goitre
o Umbilical hernia
o Delayed development
- Acquired
o Short stature/growth failure
o Cold intolerance
o Dry skin
o Cold peripheries
o Bradycardia
o Thin, dry hair
o Pale, puffy eyes with loss of eyebrows
o Goitre
o Slow-relaxing reflexes
o Constipation
o Growth/short stature
o Delayed puberty
o Obesity
o Slipped upper femoral epiphysis
o Deterioration in school work
o Learning difficulties
Congenital Hypothyoridism
Detection of congenital hypothyroidism is important as it is:
- Relatively common, occurring in 1 in 4000 births
- One of the few preventable causes of learning difficulties
Causes of congenital hypothyroidism:
- Maldescent of the thyroid and athyrosis
- Dyshormonogenesis, an inborn error of thyroid hormone synthesis, in about 5% of
cases, although commoner in some ethnic groups with consanguineous marriage
- Iodine deficiency, the commonest cause of congenital hypothyroidism worldwide but
rare in the UK
- Hypothyroidism due to TSH deficiency
Juvenile Hypothyroidism
This is usually caused by autoimmune thyroiditis. Other autoimmune disorders e.g. Diabetes
mellitus, may develop, particularly in children with Down’s or Turner’s Syndrome. In some
families, Addison’s disease may also occur.
Hyperthyroidism
This usually results from Grave’s disease (autoimmune thyroiditis secondary to the production
of thyroid stimulating immunoglobulins (TSIs))
Clinical features:
- Systemic
o Anxiety, restlessness
o Increased appetite
- 95 -
-
o Sweating
o Diarrhoea
o Weight loss
o Rapid growth in height
o Advanced bone maturity
o Tremor
o Tachycardia, wide pulse pressure
o Warm, vasodilated peripheries
o Goitre (bruit)
o Learning difficulties/behaviour problems
o Psychosis
Eye signs (uncommon in children)
o Exophthalmos
o Ophthalmoplegia
o Lid retraction
o Lid Lag
Parathyroid disorders
Hypoparathyroidism is rare in childhood. Parathormone (PTH) plays a key role in mobilisation
of calcium by osteoclasts and the excretion of phosphate in the urine. In addition to a low
serum calcium, there is a raised serum phosphate and a normal alkaline phosphatase. The PTH
level is very low.
Hypoparathyroidism in infants is usually due to a congenital deficiency (DiGeorge’s syndrome),
associated with thymic aplasia, defective immunity, cardiac defects and facial abnormalities. In
older children, hypoparathyroidism is usually an autoimmune disorder associated with
Addison’s disease.
In pseudohypoparathyroidism there is end-organ resistance to the action of PTH. Serum
calcium and phosphate levels are abnormal but the PTH levels are normal or high. Other
abnormalities are short stature, obesity, s/c nodules, short fourth metacarpals and mild
learning difficulties. There may be teeth enamel hypoplasia and calcification of the basal
ganglia. A related state, in which there are the physical characteristics of
pseudohypoparathyroidism but the calcium, phosphate and PTH are all normal, is called
pseudopseudohypoparathyroidism. There may be a positive family Hx of both disorders in the
same kindred.
Treatment of acute symptomatic hypocalcaemia is with an IV infusion of calcium gluconate.
The 10% solution of calcium gluconate must be diluted as extravasation of the infusion will
result in severe skin damage. Chronic hypocalcaemia is treated with oral calcium and high
doses of Vit D analogues, adjusting the dose to maintain the plasma calcium concentration jus
below the normal range. Hypercalcuria is to be avoided as it may cause nephrocalcinosis and
so the urinary calcium excretion should be monitored.
Adrenal cortical insufficiency
CAH is the commonest non-iatrogenic cause of insufficient cortisol and mineralocorticoid
secretion.
Primary adrenal cortisol insufficiency (Addison’s disease) is rare in children. It may result from:
- An autoimmune process, sometimes in association with other autoimmune endocrine
disorders, e.g. diabetes mellitus, hypothyroidism
- Haemorrhage/infarction – Neonatal, meningococcal septicaemia (usually fatal)
- Adrenoleucodystrophy, a rare neurodegenerative disorder
- TB, now rare
Features of adrenal cortical insufficiency:
- 96 -
Acute
o Hyponatraemia
o Hyperkalaemia
o Hypoglycaemia
o Dehydration
o Hypotension
o Circulatory collapse
- Chronic
o Vomiting
o Lethargy
o Brown pigmentation (gums, scars, skin creases)
o Growth failure
Diagnosis:
- This is made by finding hyponatraemia and hyperkalaemia, often associated with a
metabolic acidosis and hypoglycaemia
- The plasma cortisol is low or normal and the plasma ACTH concentration high (except in
hypopituitarism). With an ACTH (synacthen) test, plasma cortisol concentrations remain
low in both adrenal failure and in long-standing pituitary/hypothalamic Addison’s
disease. A normal response excludes adrenal cortical insufficiency.
Management:
- An adrenal crisis required urgent treatment with IV saline, glucose and hydrocortisone.
Long term treatment is with glucocorticoid and mineralocorticoid replacement
-
Cushing’s Syndrome
Glucocorticoid excess in children is usually a side-effect of long-term glucocorticoid treatment
(IV, oral, or, more rarely, inhaled, nasal or tropical) for conditions such as nephritic syndrome,
asthma or bronchopulmonary dysplasia. Corticosteroids are potent growth suppressors and
prolonged use in high dosage will lead to reduced adult height.
This unwanted side-effect of systemic corticosteroids is markedly reduced by taking
corticosteroid medication in the morning on alternate days.
Clinical features:
- Growth failure/short stature
- Face and trunk obesity
- Red cheeks
- Hirsutism
- Striae
- Hypertension
- Bruising
- Carbohydrate intolerance
- Muscle wasting
- Osteoporosis
- Psychological problems
Inborn Errors Of Metabolism
Presentation:
- Poor feeding and FTT with persistent or recurrent vomiting
- Jaundice or hepatomegaly
- Lethargy, convulsions or coma
- Unusual smell of the body or urine
There may be a severe metabolic acidosis, ketosis or raised plasma ammonia.
In older children, inborn errors of metabolism need to be considered as a cause of:
- Unusual odour of the body or urine
- 97 -
-
-
Intermittent, unexplained vomiting or coma with acidosis, ketosis or raised ammonia
Children developing coarse facies, dislocated lens, abnormal hair, renal calculi and
hypopigmentation
Unexplained learning difficulties, developmental delay or convulsions
Disorders of amino acid metabolism
o Phenylketonuria
Occurs in 10 000 – 20 00 live births.
It is either due to a deficiency of the enzyme phenylalanine hydroxylase
or in the synthesis or recycling of the bioptern cofactor for this enzyme
If untreated, it usually presents with infantile spasms and developmental
delay at 6-12 months of age
o Homocystinuria
Due to cystathionine synthetase deficiency
Presentation is with FTT and developmental delay and subluxation of the
ocular lens
There is progressive learning difficulty, psychiatric disorders and
convulsions. Skeletal manifestations resemble Marfan’s syndrome.
The ciplexion is usually fair with brittle hair
Thromboembolic episodes may occur at any age
Almost half respond to large doses of the coenzyme pyridoxine.
o Albinism
Due to a defect in biosynthesis and distribution of melanin.
o Tyrosinaemia
Type 1 is a rare autosomal recessive disorder of fumarylacetoacetase.
Accumulation of toxic metabolites results in damage to the liver and renal
tubules.
The organic acidaemias
Urea enzyme defects
o Enzyme defects have been identified for all stages of the urea cycle.
Disorders of carbohydrate metabolism
o Galactosaemia
o Glycogen storage disorders
Hyperlipidaemia
One of the main risk factors for coronary heart disease. Identification and treatment of
hyperlipidaemia in childhood may delay the onset of CV disease in later life.
Familial hypercholesterolaemia (FH)
Autosomal dominant disorder of lipoprotein metabolism is due to a defect in the LDL receptor.
About 1 in 500 of the population are affected. The serum LDL cholesterol concentration is
markedly raised (>3.3mmol/L). The condition is associated with premature coronary heart
disease, which occurs in half by 50 years of age in males and by 60 years in females.
Neurology
Headache
- Tension – Symmetrical, gradual onset
- Migraine – More severe, visual disturbance, possible nausea and vomiting, abdo pain
- Raised intracranial pressure – Worse lying down, with morning vomiting.
Seizures
A disturbance of the function of the brain which may present with abnormal behaviour,
sensation, level of consciousness, or abnormality in motor activity.
- 98 -
Febrile Seizure/convulsion
Seziure associated with fever, benign in nature, and occur in about 3% of children. The
seizure, is usually brief, tonic-clonic or generalised clonic in manifestation and has developed
from a extracranial viral infection.
Epilepsy
-
-
-
Generalised epilepsies
o Clinical features: Loss of consciousness, no warning, symmetrical and detectable
on EEG with bilateral synchronous seizure
o Types:
Absence – Acute loss of consciousness
Myoclonic – Brief, with jerky movements of limbs
May be normal, consider, falling asleep!!
Tonic – Increase in tone across whole body
Tonic-Clonic – More severe, contraction of muscles following an increase
in tone. Movements are often rhythmical in nature
Atonic – Myoclonic jerk with transient loss of muscle tone. Usually causes
patient to fall.
Localisation-related epilepsies
o Partial seizure with “aura” before onset, and possible loss of consciousness
o Types:
Simple partial
Complex partial
Partial seizure with secondary generalisation
Epilepsy Syndromes
o For example
Infantile spasms
Lennon-Gestaut syndrome
Management:
Anticonvulsants : Calproate, Carbamezipine, Lamotrigine, Vigabatrin, Gabapentin
Cerebral palsy
Prevalence 1.7-3.0/1000 live births (UK)
Due to non-progressive damage to the brain, showing persistent disorder of movement and
posture. It is a permanent impairment of voluntary movement or posture,due to damage to
the immature brain, that has occurred at any time during brain development. It is a
heterogeneous group of conditions. Manifestations change over time, but the underlying lesion
does not progress or regress.
Clinical Manifestations:
- Altered tone
o Tone is the resistance of a muscle to passive stretch
- Spasticity
o Abnormal increase in tone with passive stretch-velocity dependent
- Ataxia
o Loss of smooth approach to object; poorly coordinated actions
- Athetosis
o Slow writhing movements of distal limb during voluntary movement
- Chorea
o Rapid, sudden, involuntary movements
- Dystonia
o Abnormal tone (high or low) usually resulting in abnormal posture due to
sustained abnormal contractions of combination of agonist and antagonist
muscles
Cause:
- 99 -
-
Antenatal – Cerebral malformation, Genetic, infection (CMV, rubella, chorioamnionitis),
toxins e.g. Cocaine, trauma/vascular accidents in utero, placental insufficiency
Intrapartum – Asphyxia (Only 10% attributed to this cause), prematurity,
hypoglycaemia, infection, toxins e.g. Bilirubin
Postnatal – e.g. Head trauma, hydrocephalus, vascular accidents, anoxic event,
encephalopathy
Types:
- Spastic (70%)
o
Monoplegic, hemiplegic, diplegic, quadraplegic, unilateral or bilateral??
Little’s disease – spastic, weak and clumsy limbs. Extension and
adduction. Scissoring gait.
Double hemiplegia – Bilateral spastic weakness of face, arm and leg
Infantile hemiplegia – Baby’s arms and legs show spastic weakness.
Associated with epilepsy.
- Ataxic – Cerebellar ataxia (Cerebellum – Coordination), Loss of power, hypotonia, and
tremor
- Choreoathetoid – Involuntary movements, usually chorea, athetosis, and dystonia of
limbs, face, trunk and the bulbar muscles.
Associated Problems
- Feeding difficulties/GI dysmotility
- Bowel and bladder problems
- Impaired vision, squints
- Impaired hearing
- Learning difficulties
- Epilepsy
- Orthopaedic problems
- Resp infection, pressure sores
- Psychological and behavioural problems
Management:
- Good assessment of physical impairment(s) and the resulting disability
- Appropriate use of therapists
o Physiotherapists
o Occupational therapists
o Speech and language therapists
- Family support and education
- Education and social services input
- Medical
o Orthotics
o Mobility aids
o Surgery
o Botulinum Toxin A: ‘Botox’
Chemcially denervates muscle by cleaving synaptosomal associated
proteins and preventing acetylcholine release from presynaptic
membrane
Inject into specific muscles for desired effect, which lasts 10-14 weeks
o Baclofen
GABA analogue which impedes excitatory neurotransmission at spinal
level – interrupts spinal cord reflexes
Can be given orally
Can be used intrathecally via a continous infusion using a baclofen pump
o Anti-Convulsants
Ataxia
Clinical features of cerebellar disease:
Dysdiadochokinesis
- 100 -
Ataxia
Nystagmus
Intention Tremor
Scanning Dysarthria
Heel-shin test positivity
-
Cerebellar ataxia – unsteady gait, difficulty standing on one leg, dysdiadochokinesis,
intention tremor
o Ataxia telangiectasia
Autosomal recessive disorder
Clinical features: Trunkal ataxia, on development of walking, Slurred
speech, oculomotor abnormalities, learning difficulties, wheel-chair bound
by round 10 years olds, and telangiectasia
o Friedreich’s ataxia
Autosomal recessive disorder
Progressive gait and limb ataxia, dysarthria, a loss or proprioception and
pyramidal weakness
Cerebral haemorrhage
Extradural Haemorrhage
Usually follows trauma
Haematoma will act as a space-occupying lesion, causing neurological disturbance, and
possible focal neurological signs
Subdural haematoma
Tearing of veins as they cross subdrual space
Consider Non-Accidental Injury
Retinal haemorrhages may be seen
Subarachnoid haemorrhage
Acute onset of head ache, neck stiffness and sometimes fever.
Neural tube defects and hydrocephalus
Anecephaly
Failure of development of most of cranium and brain. Majority still born.
Encephalocele
Extrusion of brain and the meninges through a defect in the skull on the midline.
Spina Bifida Occulta
Vertebral arch in spine fails to fuse. Usually an incidental finding, but may have overlying hair
growth of lipoma
Meningocele
Extrusion of meninges, pia, arachnoid, dura and CSF from vertebral arch, protruding from
back.
Good prognosis with surgery
Myelomeningocele
Extrusion of meninges, pia, arachnoid, dura, CSF, and spinal cord from vertebral arch,
protruding from back.
Clinical features:
- Variable Paralysis of lower limbs
- Sensory losses
- Neuropathic bladder/bowel
- Scoliosis
- 101 -
-
Hydrocephalus
Hydrocephalus
Increased pressure in ventricular system as a result of:
Causes:
- Non-communicating (obstruction)
o Congenital e.g. Aqueduct stenosis, Dandy-Walker malformation
o Post-intracranial infection
o Malignancy
- Communicating (Failure to observe circulating CSF)
o Subarachnoid haemorrhage
o TB meningitis
o Arnold-Chari malformation
- Important points to consider
o Head circumference
o Signs of raised intracranial pressure
o Rx - Insertion of a ventricular shunt, look and feel for shunt
Neuromuscular
Disorders of:
- Muscle
o P/C:
-
-
-
Duchenne/Becker’s/Congenital muscular dystrophy
Progressive muscle degeneration with inheritance
Polymyositis/dermatomyositis
Dystrophia myotonia
Dominant inheritance
Profound hypotonia, with resp difficulty.
Metabolic myopathies
Congenital myopathies
Anterior horn cell
o P/C: Weakness, wasting, absent reflexes
o E.g.
Spinal muscular atrophy
Poliomyelitis
Peripheral nerve
o P/C:
Hereditary motor sensory neuropathies (Type 1 = Charcot- Marie-Tooth
disease)
Hypertrophic affected nerves, due to demyelination an subsequent
attempts at remyelination
Symmetrical gradually progressive distal (rather than proximal)
muscle wasting
Guillane-Barré Syndrome (post-infectious polyneuropathy)
2-3 weeks after an URTI or Campylobacter Gastroenteritis (jejuni)
Sensory disturbance in legs. Symmetrical weakness.
Bell’s Palsy
Facial weakness due to paresis of facial nerve, with unknown viral
aetiology.
Neuromuscular transmission
o P/C:
Myasthenia Gravis
Abnormal Muscle fatigue
Neurocutaneous disorders
Neurofibromatosis
Tuberous Sclerosis
- 102 -
Autosomal dominance inheritance. Majority however are new mutations
Clinical features sometimes seen:
- Adenoma sebaceum (angiofibromata), on face, butterfly distribution
- Depigmented “ash-leaf”-shaped macules, which flouresce under Wood’s light (UV)
- Shagreen’s patches – Cutaneous roughened macules usually over lumbar spine region
- Mental retardation
- Epilepsy
- Subungual fibromata
- Retinal phakomata – White areas due to local degeneration
- Cardiac Rhabdomyoma
- Polycystic kidneys
Sturge-Weber Syndrome
Port-wine stain in the trigeminal distribution, usually V1 and V2, with underlying angiomas of
the leptomeninges and choroids.
Clinical features:
- PWS, Trigeminal region, unilateral
- Epilepsy
- Learning difficulties
- Hemiplegia
Neurodegenerative disorders
Shows development regression and abnormal neurological signs. Some have identified enzyme
defect, some have unknown aetiology.
Lipid Storage disorders
- Tay-Sachs disease
o Cause – Hexoseaminidase A defect
o Autosomal recessive inheritance
o Majority Ashkenazi Jews
o Development regression, Cherry red spot at the macula, 0% 5 year survival
from birth.
- Gaucher’s disease
o Cause - Beta-glucosidase defect
o 1 in 500 Ashkenazia Jews
o Childhood chronic presentation - Splenomegaly, bone marrow suppression,
normal IQ, bone involved
o Infantile Acute – Splenomegaly, neurological degeneration
- Neimann-Pick Syndrome
o Cause - Sphingomyelinase defect
o Neonatal, feeding difficulties, failure to thrive
o Hepatomegaly, splenomegaly, hypotonia, degeneration of hearing and vision
o 0% 5 year survival from birth
Subacute Sclerosing Panencephalitis
SSPE is believed to be the result of persistence of the measles antigen in the CNS. About half
of cases have had a natural measles infection in their first two years of life.
High titres of measles antibody are usually present in the blood and CSF.
Death is inevitable but long periods of survival have been reported.
Clinical features:
Features develop between 4 and 10 years after the original infection, and are attributed to
progressive, sclerosing demyelination with severe neurological impairment and include:
- Mental deterioration - regression of abilities with dementia
- Strange behaviour
- Convulsions
- Myoclonic jerks, which are generalised, repetative and occur at five to ten second
intervals
- Pyramidal signs, with cerebellar ataxia
- 103 -
Investigations:
Investigations in this condition included
- Raised immunoglobin levels to measles
- The EEG appearance may be diagnostic
- There may be oligoclonal bands in the CSF
Schizencephaly
Schizencephaly is a brain malformation characterized by infolding of cortical gray matter along
a hemispheric cleft near the primary cerebral fissures. The malformation was described by
Yakovlev and Wadsworth (1946) who coined the term schizencephaly. Schizencephaly is
thought to represent a defect in neuronal migration, i.e., a true malformation, in contrast to
the porencephalies which may be encephaloclastic. Early understanding of schizencephaly was
derived from pathologic specimens; subsequently, pneumoencephalography, CT, and MRI
demonstrated the defects in life.
Renal/Urinary
Assessment of the kidneys and urinary tract
-
Plasma creatinine concentration
Glomerular filtration rate (GFR)
Creatinine clearance
Urine dipstick
o RBC
o WCC
o Protein
o Ketones – ?Diabetes, UTI
o Nitrites – ?UTI
o PH
o Glucose
o Specific Gravity Osmolality
o Epithelial cells – Shows contamination
Antenatal Diagnosis of Urinary Tract anomalies
-
-
-
Ultrasound
o Non-invasive anatomical assessment
Functional scanning
o Static nuclear medicine
Isotope-labelled substance e.g. Dimercaptosuccinic acid (DMSA), injected
IV. Good for detection of renal scars
o Dynamic nuclear medicine scanning
Isoptope-labelled substance e.g. MAG 3, STPA, that is excreted by
glomerular filtration, and gives information on blood flow, renal function
and drainage. Particularly useful in detecting urinary obstruction
Micturating cystourethrograthy (MCUG)
o Filling the bladder with contrast via a urethral catheter outlines the bladder and
is used to identify the vesicoureteric reflux.
Intavenous urography (IVU)
o Rarely indicated in children unless detailed anatomy of the calyces or ureter is
required.
- 104 -
Anomalies detectable on antenatal US screening p254
Potter’s Syndrome – Absence of both kidneys (renal agenesis)
Multicystic Kidney – Failure of union of the ureteric bud (which forms the ureter, pelvis,
calyces, and collecting ducts) with the nephrogenic mesenchyme.
Horsehoe or Pelvic Kidney – Abnormal caudal migration, may predispose to infection or
obstruction to urinary drainage.
Duplex System – Premature division of the ureteric bud, which can vary from simply a bifid
renal pelvis to complete division with two ureters.
Prune Belly Syndrome – Failure of fusion of the infraumbilicial midline structure results in
exposed bladder mucosa (bladder extrophy) or absent abdominal musculature with a large
bladder and dilated ureters (megacystis-megaureters) and cryptorchidism.
Obstruction to Urine Flow – May occur at the pelviureteric or vesicoureteric junction
Neuropathic Bladder – Disruption of nerve supply
Urinary Tract Infection
Symptomatic UTI before the age of 11 years
Girls – 3%
Boys - 1%
50% recurrence within a year
-
Up to half have a structural abnormality of their urinary tract
A UTI may damage the growing kidney by forming a scar, predisposing to hypertension
and to chronic renal failure if the scarring is bilateral
Clinical Features:
- Newborn
o Symptoms are non-specific and septicaemia may develop rapidly
o Fever
o Lethargy or irritability
o D and V
o FTT
o Prolonged neonatal jaundice
o Febrile convulsions
- Beyond infancy
o Dysuria, loin pain, frequency
o Dysuria without a fever is often due to vulvitis in girls or balanitis in boys rather
than a UTI
o Symptoms of UTI may also occur following sexual abuse
Urine Sample:
- Children in nappies
o A ‘clean catch’ sampling into a waiting sterile pot when the nappy is removed.
o Adhesive plastic bag applied to the perineum after careful washing
o Suprapubic Aspiration (SPA), in the severely ill child is method of choice
- Older child
o MSU
Predisposing factors:
- Infecting organism
- 105 -
Usually a result of bowel flora entering the urinary tract via the urethra.
Most Common first:
E-Coli
Proteus (More common in boys and predisposes to Phosphate
stones)
Pseudomonas (usually indicates a structural abnormality in urinary
tract affecting drainages)>
Incomplete bladder emptying
o Infrequent voiding, resulting in bladder enlargement
o Vulvitis
o Hurried micturation
o Obstruction by a loaded rectum from constipation
o Neuropathic bladder
Vesicoureteric reflux (VR)
o VR is a development anomaly of the vesicoureteric junctions.
Urine returning to the bladder from the ureters after voiding results in
incomplete bladder emptying
Kidney infection (pyelonephritis)
Bladder voiding pressure is transmitted to the renal papillae; this may
contribute to renal damage if voiding pressures are high
o
-
-
All children need investigation after a first UTI
Investigation tools:
- Ultrasound Scan
- Abdominal Xray
- DMSA (Dimerceptosuccinic acid)
- MCUG (Micturating Cystourethrogram)
- Indirect cystogram
Under
-
1 year:
Renal USS
DMSA (after 3/12)
MCUG (After 8/52) –
o Wait for cystitis to resolve before MCUG
o 50% of patients at this age probably get vesicoureteric reflux
Between 1-5 years
- Renal USS
- DMSA
o No MCUG as catheter uncomfortable
5 years
- Renal USS
o At 5 years olnly around 10% will get veicoureteric reflux, so chance of scarring is
reduced
MCUG
- Can demonstrate vesicoureteric reflux
- Obstruction in boys – Can show posterior urethral valve obstruction which may foetally
present with dilated kidneys, or in life when UTI causes gross obstruction retention.
Treatment:
Oral Trimethoprim for 5 days, with prophylactic Trimethoprim to cover until further
investigation. Or IV augmentum/gentamicin (Broad spectrum)
Asymptomatic Bacteriuria
Bacteria in urine, which may be treated with antibiotics but will reoccur, without symptoms.
Does not cause renal damage.
- 106 -
Enuresis
Nocturnal Enuresis
Bed wetting at night
Reasons:
- Psychological
- Sphincter control
- UTI
- Faecal retention severe enough to reduce bladder volume and cause bladder neck
dysfunction
- Polyuria from diabetes or chronic renal failure.
Management:
1. Explanation – Explain to both parent and child
2. Star chart – Child earns praise and a star each morning if his bed is dry. Wet beds don’t
have attached blame
3. Enuresis alarm – A sensor placed in child’s pants, which sound if becomes wet. This
works, if child wakes, gets up, changes sheets
4. Desmopressin – Short term relief from bedwetting, can be achieved by use of the
synthetic analogue of antiduiretic hormone, desmopressin, taken as tablets or nasal
spray.
Daytime Enuresis
Lack of bladder control during the day in a chid old enough to be continent. May be caused by:
- Lack of attention to bladder sensation, a manifestation of a developmental or
psychogenic problem which may be secondary to stress of part of a general behavioural
problem
- Detrusor instability
- Bladder neck weakness
- Neurogenic bladder
- UTI
- Constipation
- An ectopic ureter
Secondary Enuresis
Loss of previously achieved urinary continence may be due to:
- Emotional upset, the commonest cause
- UTI
- Polyuria from an osmotic diuresis in diabetes mellitus, or a renal concentrating disorder
e.g. Sickle cell or Chronic Renal failure
Investigations:
- Urine sample for infection, glycosuria, proteinuria
- Assessment of urinary concentrating ability by measuring osmolality of an early
morning urine sample
- US of renal tract
Maple Syrup Urine Disease
The decarboxylation of leucine, isoleucine and valine is achieved by a complex system using
thiamine pyrophosphate as a coenzyme. A deficiency of this enzyme system results in maple
syrup urine disease. This condition is so named because of the sweet odour of maple syrup
found in body fluids, especially in urine.
There are different forms of this disease:
- Classic maple syrup urine disease: This form of the condition has the most severe
clinical manifestations.
o Development of vomiting and poor feeding during the first week of life, after
being normal at birth.
- 107 -
The child then becomes lethargic and falls into a coma over the next few days.
There may be convulsions
Hypoglycamia may develop
On examination there is:
Increased tone, rigid muscles and severe opisthotonus, which may
alternate to periods of flaccidity.
o Immediate treatment is aimed at removing the branched chain amino acids and
their metabolites from the tissues and body fluids, which is best effected with
peritoneal dialysis. Treatment of hydration or hypoglycaemia does not resolve
the problem
o Long term treatment is with a low branched-chain amino acid diet.
Intermittent maple syrup urine disease: Children affected by this condition are
seemingly normal. They subsequently develop vomiting, ataxia, odour of maple syrup,
lethargy, and coma during stress such as infection or surgery.
o Investigations during attacks reveal:
Raised plasma levels of leucine, isoleucine and valine. Alloisoleucine, a
stereoisomer of isoleucine not normally found in the blood, is present.
Urine analysis - raised levels of leucine, isoleucine, and valine; also there
are raised levels of the ketoacids of these amino acids.
Mild maple syrup urine disease: Children affected by this condition develop milder
disease after the neonatal period
o Features include:
Mildly to moderately mentally retarded.
Increased levels of leucine, isoleucine and valine in the blood
Excretion of ketoacids of these amino acids in the urine
Diagnosis occurs during an intercurrent illness when clinical features such
as vomiting, hypertonicity, muscular rigidity, odour of maple syrup in
urine.
o
o
o
o
-
-
Proteinuria
Transient proteinuria may occur during febrile illness or after exercise.
Persistent proteinuria can be quantified by:
- A 24H urine
- A timed collection (Protein excretion should not exceed 4mg/h per m 2) or,
- More usefully in younger children, by measuring urine protein/creatinine ratio in an early
morning sample (protein should not exceed 20mg/mmol of creatinine)
Common cause:
- Orthostatic proteinuria – Where proteinuria is found only when the child is upright e.g.
During the daytime
- Glomerular abnormalities
o Minimal change disease
o Glomerulonephritis
o Abnormal glomerular basement membrane
- Increased glomerular perfusion pressure
- Reduced renal mass
- Hypertension
- Tubular Proteinuria
Nephrotic Syndrome
Proteinuria
Oedema
Hypoalbuminaemia
(Ref Range
Neonate 25-35g/L
- 108 -
Child 35-55g/L)
Heavy proteinuria results in a low plasma albumin and oedema. The cause of the condition is
unkown, but a few cases are secondary to systemic diseases such as:
- Henoch-Schonlein Purpura
- Vasculitis (e.g. SLE)
- Infections (e.g. Malaria)
- Allergens (e.g. Bee stings)
Clinical Signs:
- Periorbital oedema (particularly on waking)
- Scrotal, leg and ankle oedema
- Ascites
- SOB due to pleural effusions and abdominal distension
Investigations at presentation of nephrotic syndrome:
- Urine protein
- FBC and ESR
- U & E’s, creatinine, albumin
- Complement levels – C3, C4
- Antistreptolysin O titre and throat swab
- Urine microscopy and culture
- Urinary sodium concentration
- Hepatitis B antigen – Membranous nephropathy (steroid-resistant) is associated with
Hep B
Types:
- Steroid-sensitive nephrotic syndrome
o In over 90% of children, it resolves with corticosteroid therapy
o These children do not progress to renal failure
o Features suggesting steroid sensitive are:
Age between 1 and 10 years
No macroscopic haematuria
Normal blood Pressure
Normal complement levels
Normal renal function
- Steroid-resistant nephritic syndrome
o These children should be managed by a paediatric nephrologists
o Management of oedema is by diuretic therapy, salt resitriction, and Captopril
(ACEI)
- Congenital Nephrotic Syndrome
o Presents in first 3 months of life
o It is rare
o It is associated with early end stage renal failure and high mortality
o When renal failure develops, nephrectomy and dialysis are instituted until the
child is large enough for transplantation.
An oedematous child – Test for proteinuria to diagnose nephrotic syndrome
Complications:
- Hypovolaemia
- Infection – pneumococcal peritonitis – Low Ig’s
- Intravascular thrombosis – DVT, renal vein thrombosis (hypovolaemic, low antithrombin
III)
- Hypercholesterolaemia
Haematuria
Urine red in colour, or positive on dipstick
- 109 -
Brown Urine – Suggess Glomerular haematuria
Most common cause:
- UTI
Other causes:
- Non-Glomerular
o Infection (viral, TB, schistosomiasis, bacterial)
o Trauma to genitalia, urinary tract or kidney
o Stones
o Tumour
o Sickle cell
o Bleeding disorders
o Renal vein thrombosis
o Hypercalciuria
- Glomerular
o Acute glomerulonephritis
o Chronic glomerulonephritis
o IgA nephropathy
o Familial nephritis
o The basement membrane disease
Acute Nephritis
Usually in childhood this follows a streptococcal sore throat or skin infection.
Causes:
- Post infectious (inc. Strep)
o Diagnosed by a raised ASO titre and lower complement C3 levels that return to
normal after 3-4 weeks
o Prognosis is good
- Vasculitis
o Common findings are: fever, malaise, weight loss, skin rash and arthropathy
o Henoch-Schönlein Purpura
Is the combination of:
Characteristic Skin Rash
Arthralgia
Periarticular oedema
Abdominal pain
Glomerulonephritis
Usually occurs between the ages of 3 and 10 years
Male, Female 2:1
Peaks during winter months
Often preceded by an URTI
IgA and IgG interact to produce complexes that activate complement and
are deposited in the affected organs, precipitating an inflammatory
response with vasculitis.
o SLE
o Wegner’s granulomatosis
o PAN
o Polyarteritis
- IgA nephropathy
o May present with episodes of macroscopic haematuria, commonly in association
with URTI.
o Histological findings and management as for HSP
- Mesangiocapillary glomerulonephritis
- Anti-glomerular basement membrane disease (Goodpasture’s syndrome)
In acute nephritis, increased glomerular cellularity restricts glomerular blood flow and
therefore filtration is decreased. This leads to:
- 110 -
-
Decreased urine output and volume overload
Hypertension, which may cause seizures
Oedema, characteristically around the eyes
Haematuria and proteinuria
Renal masses
Most frequently – Bilateral enlarge kidneys due to autosomal recessive polycystic kidney
disease
Renal calculi
Uncommon in childhood. When they occur, predisposing causes must be sought:
- UTI
- Structural anomalies of the Urinary tract
- Metabolic abnormalities
Most common cause:
- Phosphate stones associated with Proteus Infection
Other Causes:
- Calcium containing stones occur in idiopathic hypercalciuria
- Deposition of calcium in the parenchyma (nephrocalcinosis) may occur with
hypercalciuria, hyperoaluria, and distal renal tubular acidosis.
o Nephrocalcinosis may be a complication of frusemide therapy in the neonate
- Cystine and Xanthine stones are rare
Clinical Features:
- Haematuria
- Loin or abdo Pain
- UTI
- Passage of a stone
Management:
- Removal of stone,
- Lithotripsy
- High fluid intake
- If metabolic abnormality then specific treatment
Renal tubular disorders
Abnormalities of renal tubular function may occur at any point along the nephron
Generalised proximal tubular dysfunction – Fanconi Syndrome
Causes:
- Idiopathic
- Secondary to inborn errors of metabolism
- Cystinosis (autosomal recessive disorder causes intracellular accumulation of cystine
- Glycogen storage disorders
- Lower’s Syndrome (oculocerebrorenal dystrophy)
- Galactosaemia
- Fructose intolerance
- Tyrosinaemia
- Wilson’s disease
Cardinal features:
- 111 -
-
Excessive urinary loss of amino acids, glucose, phosphate, bicard, sodium, calcium,
potassium and urate
Polydypsia and polyuria
Salt depletion and dehydration
Hyperchloraemia metabolic acidosis
Rickets and osteoporosis
FTT/Poor growth
Specific Transport Defects
-
-
-
-
-
-
-
-
-
-
Glycosuria
Reduced reabsorption of Glucose
o Asymptomatic
Cystinuria
Reduced reabsorption of Cystine and dibasic aminoacids
o Renal calculi
Vit D Resistant rickets
Reduced reabsorption of Phosphate
o Rickets
Pseudohypoparathyroidism
Increased reabsorption of Phosphate
o Obesity
o Depressed nasal bridge
o Short fingers (2nd, 4th and 5th)
Hyperuricosuria
Increased secretion and reduced reabsorption of Uric acid
o Renal calculi
Renal tubular acidosis type II
Reduced reabsorption of Bicarbonate
o Metabolic acidosis
o Alkaline urine
o Growth failure
Hypercalciuria
Reduced reabsorption of Calcium
o Nephrocalcinosis or renal stone
Nephrogenic diabetes insipidus
Reduced reabsorption of water in distal tubule
o Polydypsia and polyuria
o Fever
o FTT/Poor growth
Renal tubular acidosis type I
Reduced secretion Hydrogen Ions
o As for type II RTA and nephrocalcinosis
Bartter’s (Rare)
Reduced reabsorption of Chloride
o Hypokalaemic
o Metabolic alkalosis
o Normal blood pressure with increased renin
o Polydipsia and polyuria
o Growth failure
Acute Renal Failure
A sudden reduction in renal function. Oliguria (<0.5 ml/kg per h) is usually present. Types:
-
Prerenal
o The commonest cause in children
Renal
- 112 -
Salt and water retention; blood, protein and casts are often present in the urine;
and there may be symptoms specific to an accompanying disease (e.g. HenochSchonlein Purpura)
Postrenal
o From urinary obstruction
o
-
Causes:
Prerenal
Hypovolaemia
Gastroenteritis
Burns
Sepsis
Haemorrhage
Nephrotic Syndrome
Circulatory failure
Renal
Vascular
HUS
Vasculitis
Embolus
Renal vein thrombosis
Tubular
Acute tubular necrosis
Ischaemic
Toxic
Obstructive
Glomerular
Glomerulonephritis
Interstitial
Interstitial nephritis
Pyelonephritis
Acute-on-chronic renal
failure
Postrenal
Obstruction
Congenital
Acquired
Haemolytic Uraemic Syndrome
A triad of:
- Acute renal failure
- Microangiopathic haemolytic anaemia
- Thrombocytopenia
The most common renal cause of ARF in childhood. Thought to be due to activation of
neutrophils which damage vascular endothelium.
Typical HUS is secondary to GI infection with verocytotoxin-producing E.Coli O157:H7 or, less
often, Shigella. It follows a prodrome of bloody diarrhoea. Although the platelet count is
reduced, the clotting is normal (unlike in DIC). Other organs such as brain, pancreas and heart
also be involved.
Clinical features:
- < 5 years
- Blood diarrhoea – 5-10 days later oliguria, pallor, lethargy, and petechiae
- Other organ damage – CNS (fits, coma), colitis, pancreatitis
- Haemolysis Anaemia, Jaundice
Investigations:
- Bloods, U&E’s, Calcium (reduced), Phosphate (increased), FBC, Film, Coagulation
screen (normal)
- Urine – mild haematuria, proteinuria
- Stool MC&S – Can send for E-Coli verotoxin
Treatment:
- Supportive
- Fluids
- Hyperkalaemia – Insulin/dextrose therapy, Salbutamol, Potassium restriction, Dialysis
- Transfusion – Platelets/Bloods
- 113 -
Hypertension
Causes of hypertension:
-
-
-
-
Renin-dependent
o Renal parenchymal disease
o Renovascular e.g. Renal artery stenosis
o Renal tumours
Coarctation of the aorta
Catecholamine excess
o Phaeochromocytoma
o Neuroblastoma
Endocrine causes
o Congenital adrenal hyperplasia
o Cushing’s syndrome or corticosteroid therapy
Essential hypertension
Chronic Renal Failure
Causes:
Structural malformations
Glomerulonephritis
Hereditary nephropathies
Systemic diseases
Miscellaneous/unknown
40%
25%
20%
10%
5%
Clinical Features: (Presents with)
- Anorexia and lethargy
- Polydypsia and polyuria
- FTT/Growth failure
- Bony deformities from renal osteodystrophy (renal rickets)
- Hypertension
- Acute-on-chronic renal failure (precipitated by infection or dehydration)
- Incidental finding of proteinuria
Dermatology
Vascular naevi
- Salmon patch
- Port-Wine Stain
o PWS
Capillary malformation of ecstatic vessels
o Sturge-Weber Syndrome
Port-wine stain in the trigeminal distribution, usually V1 and V2, with
underlying angiomas of the leptomeninges and choroids.
- Strawberry naevus
- Cavernous haemangioma
Napkin Dermatitis
An irritant dermatitis, sparing the skin folds
Infantile seborrhoeic dermatitis
Starts in first few weeks of life, and tends to affect the body folds, including axillae, groin and
neck
- 114 -
Candida
Often occurring in patients with dermatitis.
Juvenile Plantar dermatosis
Clinical Features:
- Presents with red, dry, fissured and glazed skin, principally over the forefoot, but
sometimes the whole sole
Cause:
Synthetic materials in shoes
Urticaria Pigmentosa
Accumulations of Mast cells cause brown-red macules on skin. Worse when rubbed
Hystiocytosis X
Infiltration of Langerhans cells which act in malignant fashion causing skin and major organ
complications.
Orthopaedic
Variations of normal posture:
-
-
-
Bow Legs (Genu varum)
o Bowing of the tibiae causing the knees to be wide apart while standing with their
feet together.
o Another cause of bow legs is rickets
o Marked bow legs may also occur in Blount’s disease (infantile tibia vara) an
uncommon condition predominantly seen in Afro-Caribbean children. There is
beaking of the proximal medial tibial epiphysis on X-ray. Orthoses (splints and
special footwear) and surgical correction may be required.
Knock-knees (Genu valgum)
o In this condition, the feet are wide apart when standing with the knees held
together. It is seen in many children between 2 and 7 years of age and usually
resolves.
Flat feet (Pes Planus)
o Flatness of the medial longitudinal arch and the presence of a fat pad which
subsequently disappears.
o Marked flat feet can be the presentation of a collagen disorder such as ElhersDanlos syndrome.
In-toeing
Causes:
- Metatarsus varus – an adduction deformity of a highly mobile forefoot
o Clinical features:
Occurs in infants
Passively correctable
Heel is held in the normal position
No treatment required unless it persists beyond 5 years if age is
symptomatic
- Medial tibial torsion – at the lower leg, when the tibia is laterally rotated less than
normal in relation to the femur
o Clinical features:
Occurs in toddlers
May be associated with bowing of the tibiae
Self-corrects within about 5 years
- Persistent anteversion of the femoral neck – At the hip, when the femoral neck is
twisted forward more than normal
o Clinical features:
- 115 -
Presents in childhood
Usually self-corrects by 8 years of age
May be associated with hypermobility of the joints
Children sit between their feet with the his fully internally rotated (‘W’
sitting)
Most do not require treatment but femoral osteotomy may be required
for persistent anteversion
Out-Toeing
This is ucommon but may occur in infants between 6 and 12 months of age. When bilateral, it
is due to lateral rotation of the hips and resolves spontaneously
Toe-Walking
This is common in 1 to 3 year old children. It may become persistent, usually from habit, but
may be due to mild cerebral palsy. It may also be due to isolated tightness of the Achilles
tendons. In older boys, Duchenne’s Muscular dystrophy should be excluded.
Disorders of the hip, knee and feet
Causes:
Age
1-3 years
3-10 years
11-16 years
Painful limp
Septic
arthritis/oestomyelitis
Transient Synovitis
Trauma – accidental/NA
Transient synovitis
Septic
arthritis/oestomyelitis
Trauma
Juvenile idiopathic
arthritis
Perthe’s Disease (acute)
Malignancy e.g.
Leukaemia
Slipped upper femoral
epiphysis
Juvenile idiopathic
arthritis
Trauma
Septic
arthritis/oestomyelitis
Bone tumours
Painless limp
DDH
Neuromuscular e.g.
cerebral palsy
Unequal leg length
Juvenile idiopathic
arthritis
Perthe’s disease (chronic)
DDH
Neuromuscular disorders
Juvenile idiopathic
arthritis
Slipped upper femoral
epiphysis
Juvenile idiopathic
arthritis
Dysplastic hip
Developmental Dysplasia of the Hip (DDH)
Spectrum of disorders ranging from dysplasia to subluxation through to frank dislocation of the
hip.
- Ortolani
o Relocation back into the acetabulum on abduction
- Barlow
o Check id hip can be dislocated posteriorly out of the acetabulum
Pavlik harness
- 116 -
Transient Synovitis (TS, irritable hip)
Most common cause of acute hip pain in children. It occurs in children of 2-12 years old. It
often follows or is accompanied by a viral infection. Presentation is with sudden onset of pain
in the hip or a limp. There is no pain at rest, but there is decreased ROM, particularly external
rotation.
Perthe’s Disease
This is due to ischaemia of the femoral epiphysis, resulting in avascular necrosis, followed by
revascularisation and reossification over 18-36 months. It mainly affects boys (Male: female
ratio 5:1) of 5-10 years of age.
Presentation:
- Insidious, with the onset of a limp or hip pain
- May be initially be mistaken for transient synovitis
- Bilateral in 10-20%
- X-rays show increased density in the femoral head, which subsequently becomes
fragmented and irregular.
- Even if the initial X-ray is normal, a repeat may be required if clinical symptoms persist.
- A bone scan and MRI scan can be helpful in making the diagnosis.
Slipped Upper Femoral Epiphysis
Displacement of the epiphysis of the femoral head postero-inferiorly. It is most common at 1015 years of age during the adolescent growth spurt, particularly in obese boys
Skeletal maturation may be found to be delayed.
Presentation:
- With a limp or hip pain, which may be referred to the knee
- There is restricted abduction and internal rotation of the hip
- The onset may be acute, following minor trauma
- In 20% bilateral
- Diagnosis confirmed on X-ray, although a frog lateral view is sometimes required.
- Management is surgical, usually with pin fixation in situ
- Severe slips may require subsequent corrective realignment osteotomy once the
epiphysis has fused or, rarely, open reduction of the hip, but this carries a risk of
avascular necrosis.
Painful Knee
-
Osgood-schlatter disease
o Overuse syndrome commonly occurring in physically active males around
puberty, resuling in detachment of cartilage fragments from the tibial tuberosity
(traction apophysitis).
o There is localised tenderness and swelling over the tibial tubercle
o Disease is bilateral in 25-50%
o Most resolve with reduced physical activity, trying to avoid over-restriction for a
disorder which is self-limiting. A knee immobiliser splint may be helpful.
o In some patients the disorder fails to resolve over several months; a period of
immobilisation or, rarely, excision of the ossicle may then be required.
- 117 -
-
-
-
-
Chondromalacia patellae
o In this condition there is softening of the articular cartilage of the patella. It
most often affects adolescent females, causing pain when the patella is tightly
apposed to the femoral condyles, as in standing up from sitting or on walking up
stairs. Treatment is with rest and physiotherapy for quadriceps muscle
strengthening
Osteochondritis dissecans
o Pain is caused by separation of bone and cartilage from the medial femoral
condyle following avascular necrosis. Complete separation of articular fragments
may result in loose body formation. Treatment is initially with rest and
quadriceps exercises; sometimes arthroscopic surgery is required.
Subluxation and dislocation of patella
o Subluxation produces the feeling of instability or giving way of the knee.
Treatment is with quadriceps exercises; surgery to realign the pull of the quads
on the patellar tendon is occasionally required.
o Dislocation of the patella laterally occurs suddenly. Reduction occurs
spontaneously or on gentle extension of the knee. An x-ray is required to
differentiate loose bodies from bone fracture. Immobilisation, and sometimes
surgery, is required.
Injuries
Talipes Equinovarus (clubfoot)
- Positional talipes
o From intrauterine compression is common.
o The foot is of normal size and the deformity is mild and can be corrected to the
neutral size with passive manipulation. Otfen the baby’s intrauterine posture can
be recreated
- Talipes Equinovarus
o Is a complex abnormality
Entire foot inverted and supinated
Short foot
Thin calf muscles
Forefoot adducted
Heel rotated inwards (varus) and in plantar flexion (equinus)
Talipes Calcaneovalgus
The foot is dorsiflexed and everted. It usually results from intrauterine moulding and selfcorrects. Passive foot exercises are sometimes advised. There is an association with DDH.
Pes Cavus
In Pes davus, there is a high arched foot. When it presents in older children, it is often
associated with neuromuscular disorders e.g. Friedreich’s ataxia and type I hereditary motor
sensory neuropathy (peroneal muscular atrophy). Treatment is required if the foot becomes
still or painful.
Disorders of the back, spine and neck
-
Back
o
o
o
Pain
Muscle spasm
Poor posture
Scheuermann’s disease – an osteochondritis of the thoracic vertebrae in
adolescents resulting in a fixed kyphosis; diagnosed in x-ray
o Spondylolysis/Spondylolisthesis – Stress fracture of the pars interarticularis of
the vertebrae, typically lower lumbar (spondlylolysis)
o Vertebral osteomyelitis/discitis
o Tumours
- 118 -
Spinal cord/Root compression
Idiopathic pain syndrome – Diagnosed when no physical cause is found; may be
exacerbated by psychological stress
Scoliosis
o Lateral curvature in the frontal plane of the spine.
o Causes:
Idiopathic
Congenital e.g. hemivertebrae, spina bifida, VACTERL association
Secondary
Torticollis
o Most common cause of torticollis (wry neck) in infants is a sternomastoid tumour
(congenital muscular torticollis). It occurs in the first few weeks of life and
presents with a mobile, non-tender nodule, which can be felt within the body of
the SCM muscle. There may be restriction of head turning and tilting of the
head. The condition usually resolves in 2-6 months. Passive stretching is
advised, but its efficacy is unproven.
o
o
-
-
Painful limb
Common Presentation – ‘Growing pains’ or Nocturnal Idiopathic pain
Episodes of generalised pain in the lower limbs, in preschool children. The pain often wakes the
child from sleep and settles with massage or comforting. It occurs less often during the day,
the child is otherwise healthy and there is no evidence of musculoskeletal disease.
Hypermobolitity
Thumbs and little fingers can be extended onto the forearms; elbows and knees can be
extended beyond 10°; hands can be placed flat on floor with legs straight; often complain of
generalised limb pain and may also wake at night with pain.
Osteomyelitis
Infection of the metaphysic of the long bones. The most common sites are the distal femur and
proximal tibia, but any bone may be affected. It is usually due to hgaematogenous spread of
the pathogen, but may arise by direct spread from an infected wound. The skin is swollen
directly over the affected site. Where the joint capsule is inserted distal to the epiphyseal
plate, as in the hip, osteomyelitis may spread to septic arthritis.
Cause:
- Most commonly caused by Staph Aureus
- Other pathogens include Streptococcus and Haemophilus Influenzae.
In sickle cell anaemia, there is an increased risk of Staphlococcal and Salmonella
Osteomyleitis
- Chronic infection can cause a localised abscess in the bone (Brodie’s abscess) but it is
uncommon
- TB infection can occur, but is uncommon in UK
Presentation:
- Usually with a markedly painful, immobile limb (pseudoparesis) in a child with febrile
illness
- Directly over the infected site there is swelling and exquisite tenderness, and it may be
erythematous and warm
- Moving the limb causes severe pain
- There may be a sterile effusion of an adjacent joint
- Presentation may be insidious in infants, in whom swelling or reduced limb movement is
the initial sign
- Beyond infancy, presentation may be with back pain in the vertebral infection or with a
limp or groin pain in infection of the pelvis.
- Occasionally, there are multiple foci (e.g. Disseminated Staphlococcal or H.Influenzae
infection)
Investigation:
- Blood cultures are usually positive and the WBC and acute-phase reactants (CRP) are
raised
- 119 -
X-rays are initially normal, other than showing soft tissue swelling; it takes 7-10 days
for subperiosteal new bone formation and localised bone rarefaction to become visible
- The presence and site of infection can usually be identified on a radionuclide bone scane
- USS may show periosteal elevation at presentation
Treatment:
- Prompt treatment with parental antibiotics is required for several weeks to prevent
bone necrosis, chronic infection with a discharging sinus, limb deformity and
amyloidosis
- Antibiotics are given IV until here is clinical recovery and the acute-phase reactants
have returned to normal, followed by oral therapy for several weeks
- Aspiration or surgical decompression of the subperiosteal spac may be performed if the
presentation is atypical or in immunocomprised children.
- Surgical drainage is performed if the condition does not respond rapidly to antibiotic
therapy
- The affected limb is initially rested in a splint and subsequently mobilised.
-
Bone Tumours
- Osteogenic sarcoma and Ewing’s tumour – are rare
o They present with pain or swelling or occasionally a pathological fracture
- Osteoid osteoma is a benign tumour
o Affects adolescents, especially boys, usually involving the femur or tibia
o The pain is more severe at night an improves with salicylate therapy
o There may be some localised tenderness
o X-ray is usually diagnostic, with a sharply demarcated radiolucent nidus or
osteoid tissue surrounded by sclerotic bone.
o If x-ray is normal, a CT or MRI scan is required
o Rx – Surgical removal
Arthritis
Presentation:
- May be acute when there is combination of pain, swelling, redness, heat and ROM
- It must be distinguished from joint pain (arthralgia)
- In Chronic arthritis, there may be insidious onset of early morning stiffness of the
joints, ‘gelling’ after inactivity, the development of a limb or slowness on walking
Causes of polyarthritis:
Infection
IBD
Vasculitis
Haematological disorders
Malignant disorders
Connective tissue disorders
Other
Bacterial – septicaemia/septic arthritis,
TB
Viral – rubella, mumps, adenovirus,
Coxsackie B, Herpes, Hepatitis,
parvocirus
Other – Mycoplasma, Lyme disease,
rickettsia
Reactive – GI infection, Strep infection
Rheumatic fever
Crohn’s disease, UC
Henoch-Schonlein purpura, Kawasaki’s
disease
Haemophilia, sickle cell disease
Leukaemia, Neuroblastoma
Juvenile idiopathic arthritis, juvenile
ankylosing spondylitis, systemic lupus
erythematosus (DLE), dermatomyositis,
missed connective tissue disorder
(MCTD), polyarteritis nodosa (PAN)
CF
- 120 -
Septic Arthritis
- Serious bone infection of the joint space as it can lead to bone destruction.
- It is most common in children less than 2 years old
- Usually results from haematogenous spread, but may also occur following a puncture
wound or infected skin lesions, e.g. Chickenpox
- In young children, it may result from spread from adjacent osteomyelitis into joints
where the capsule inserts below the epiphyseal growth plate.
- Beyond the neonatal period, the most common organism is Staphylococcus aureus, and
usually only one joint is affected.
- H.Influenzae was an important cause in young children prior to Hib immunisation and
often affected multiple sites.
Presentation:
- Usually with an erythematous, warm, acutely tender joint, with a reduced ROM, in an
acutely unwell, febrile child.
- Infants often hold the limb still (pseudoparesis, pseudoparalysis) and cry if it is moved
- A joint effusion may detectable in peripheral joints
- Although a sympathetic joint effusion may present in osteomyelitis, the tenderness is
over the bone.
- Diagnosis of septic arthritis of the hip can particularly difficult in toddlers, as the joint is
well covered by S/C fat
- Initial presentation may be with a limp or pain referred to the knee
Investigation:
- Increased WBC, and Acute-phase reactants
- USS of deep joints, such as the hip, is helpful to identify an effusion
- X-rays are used to exclude trauma and other bony lesions
- In septic arthritis, the x-rays are initially normal, apart from widening of the joint space
and soft tissue swelling.
- A bone scan may be helpful
- Aspiration of the joint space under USS guidance may reveal organisms and a positive
culture in some but not all instances.
Treatment:
- A prolonged course of antibiotics, initially IV (e.g. flucloxacillin, which in young children
is combined with a third generation cephalosporin to cover H.Influenzae), should be
given.
- Washing out of the joint or surgical drainage may be required if resolution does not
occur rapidly or if the joint is deep-seated such as the hip
- The joint is initially immobilised in a functional position, but subsequently must be
mobilised to prevent further deformity.
Juvenile Idiopathic Arthritis (Juvenile Chronic Arthritis)
A chronic arthritis lasting more than 6 weeks, presenting before 16 years of age.
Revised classification:
- Systemic (9%)
- Oligoarthritis
o Persistent (49%)
o Extended (8%)
- Polyarthritis (Rheumatoid factor (RF)-negative) (16%)
- Polyarthritis (Rheumatoid factor (RF)-positive) (3%)
- Psoriatic arthritis (7%)
- Enthesitis-related arthritis (7%)
- Other arthritis (1%)
Systemic arthritis (Still’s disease)
Usually affects young children.
Clinical features:
- Acute illness, marked malaise
- 121 -
-
High, spiking fever
Anorexia, weight loss
Salmon-pink rash at height of the fever
Aches and pains in the joints and muscles (arthralgia, myalgia), but there is often no
arthritis at presentation
Lymphadenopathy, hepatosplenomegaly and occasionally pericarditis
Anaemia, raised neutrophil and platelet count, and markedly raised acute-phase
reactants
Polyarticular
Occurs at all ages, in girls more than boys. Any joint may be affected, but most often there is
a symmetrical involvement of the wrists and hands, knees and ankles. The cervical spine and
temporomandibular joint may also be affected.
A few children, usually females in the second decade, can be classified as having juvenile
rheumatoid arthritis (JRA). They have a symmetrical arthritis, remain rheumatoid factor
positive (seropositive polyarticular) and have a pattern of disease similar to adult R/A. They
may have or develop S/C rheumatoid nodules and over half develop chronic arthritis.
Pauciarticular/oligoarthritis
Usually occurs in young children, affecting the knees, and, less often, the ankles and wrists.
There is an increased risk of developing eye disease, a chronic anterior uveitis, especially in
females who are ANA-positive. In some children, furher large joints become involved during
the first 6 months; they are categorised as having ‘extended oligoarthritis’
Enthesitis-related arthritis (juvenile spondyloarthropathy, B27-associated arthritis)
Predominantly affects older boys, who present with a large joint arthritis, usually of the lower
limbs, or a swollen digit (sausage finger). They may have the HLA-B27 tissue type and a
positive family Hx. In addition, there may be inflammation of the insertion of tendons into
bone e.g. Achilles tendon, and of the plantar fascia (enthesitis). Subsequently there may be
sacroiliac and spinal involvement. Acute symptomatic iritis may occur in these children and
requires ophthalmological referral, but other complications are rare.
Juvenile Psoriatic Arthritis
This often involves the interphalangeal joints and may present with a sausage-shaped swelling
of a digit. It may occur before the onset of skin lesions or nail pitting. For inclusion in this
category there needs to be either arthritis and psoriasis or arthritis plus two of dactylitis, nail
abnormalities or a family history of psoriasis in a first-degree relative.
Other arthritis
This is used when the child’s disease does not fit into any of the main defined categories or
overlaps more than one category.
Genetic Skeletal Dysplasia
These are generalised developmental disorders of bone, of which there are several hundred
types. They usually result in reduced growth and abnormality of bone shape rather than
impaired strength, except for osteogenesis imperfecta. The bones of the limbs and spine are
often affected, resulting in short stature. Intelligence is usually normal. Improved knowledge
of the molecular basis of collagen and its disorders is allowing better understanding and
delineation of some of these disorders.
Achondroplasia
- Autosomal dominant inheritance
- But there are 50% new mutations
Clinical features:
- Short stature from marked shortening of the limbs, a large head, frontal bossing and
depression of the nasal bridge.
- The hands are short and broad
- 122 -
-
A marked lumbar lordosis develops
Hydrocephalus sometimes occurs
Thanatophoric Dysplasia
- This results in stillbirth
- Infants have a large head, extremely short limbs and a short chest
- The appearance of the bones on x-ray are characteristic
- The importance of the correct diagnosis of this order is that, in contrast to
achondroplasia, its inheritance is sporadic. It may be identified on antenatal USS
Cleidocranial Dysostosis
- Autosomal dominant inheritance
- Absence of part or all of the clavicles and delay in closure of the anterior fontanelle and
of ossification of the skull.
- The child is often able to bring the shoulders together in front of the chest to touch
each other as a ‘party trick’
- Short stature is usually present
Arthrogryposis
- Heterogeneous group of congenital disorders in which there is stiffness and contracture
of joints.
- The cause is usually unknown, but there may be an association with oligohydramnios,
widespread congenital anomalies, or chromosomal disorders
- It is usually sporadic
- Marked flexion contractures of the knees, elbows and wrists, dislocation of the hips and
other joints, talipes equinovarus and scoliosis are common, but the disorder may be
localised to the upper of lower limbs
- The skin is thin, S/C tissue is reduced and there is marked muscle atrophy around the
affected joints.
- Intelligence is usually unaffected
- Management is with physio and correction of deformities
Osteogenesis Imperfecta (Brittle bone disease)
- A group of disorders of collagen metabolism causing bone fragility, with bowing and
frequent fractures.
- Type I (Most common type)
o Autosomal dominant
o Fractures occur during childhood, which require splinting to minimise joint
deformity
o Affected children also have a blue appearance to the sclerae and may develop
hearing loss
o Prognosis is variable
- Type II (Severe, lethal type)
o Inheritance variable, but mostly autosomal dominant or due to new mutations
o Multiple fractures present at birth. Many infants are stillborn.
Osteopetrosis (Marble Bone Disease)
Osteopetrosis is also known as marble bone disease to denote the main features of the
disease:
- Overgrowth and sclerosis of bone
- Thickening of the cortex
- Narrowing or even obliteration of the medullary cavity
Despite such a quantity of dense bone the skeleton is brittle and liable to fracture. Prognosis is
poor, but bone marrow transplantation may be curative. A less severe autosomal dominant
form may present during childhood with fractures.
Marfan’s Syndrome
- Autosomal dominant disorder of connective tissue associated with:
- 123 -
o
o
o
o
o
o
-
o
Major
o
o
Tall stature
Long thin digits (arachnodactyly)
Hyperextensible joints
A High arched palate
Dislocation (usually upwards) of the lenses of the eyes and severe myopia.
Body proportions are altered, with long, thin limbs resulting a grater distance
between the pubis and soles (lower segment) than from the crown top pubis
(upper segment)
Arm span is greater than height
problems are:
Cardiovascular, due to degeneration of the media of vessel walls, resulting in
dilated, incompetent arotic root with valvular incompetence and mitral valve
prolapse and regurgitation.
Aortic aneurysms of the aorta may dissect or rupture.
Malignancy
Cancer presentation:
- A localised mass
- The consequences of disseminated disease e.g. Bone marrow infiltration, causing
systemic ill-health
- The consequences of pressure from a mass on local structures or tissue e.g. airway
obstruction secondary to enlarged lymph nodes
Leukaemia
Types:
- Acute Lymphoblastic Leukaemia (ALL) accounts for 80%
- Acute myeloid/acute non-lymphocytic (AML/ANLL) leukaemia
- Chronic myeloid leukaemia and other myeloproliferative disorders are rare
Clinical presentation
Clinical signs and symptoms result from infiltration of the bone marrow or other organs with
leukaemic blast cells. In most children, leukaemia presents insidiously over several weeks with
some or all of:
- Malaise
- Infections
- Pallor
- Abnormal bruising
- Hepatosplenomeglay
- Lymphadenopathy
- Bone pain
- In some children, but not all:
o Blood count is abnormal, with low haemoglobin and thrombocytopenia and
evidence of circulating blast cells
Prognosis of ALL:
- Related to age
- Tumour load (measured by WBC)
- Speed of response to initial chemotherapy
- Presence or absence of specific cytogenetic/molecular genetic abnormalities in tumour
cells
- High WBC (>50x109/L, age <1 year or > 10 years and persistence of leukaemic blasts
in the bone marrow at day 28 of treatment are all important variables in defining
treatment intensity.
- 124 -
Lymphomas
Lymphomas are malignancies of the cells of the immune system. Lymphomas can be divided
into Hodgkin’s, seen more frequently in adolescence and Non-Hodgkin’s (NHL), more common
in childhood.
Non-hodgkin’s Lymphoma
In most cases of NHL, the clinical features reflect the pattern of migration of normal lymphoid
cells, with lymph nodes being the predominant site of disease.
Presentation will depend on the site of disease:
- T cell malignancies may present as ALL or NHL, with both being characterised by a
mediastinal mass with varying degrees of bone marrow infiltration.
- B cell malignancies present more commonly as NHL, localised lymph node disease
usually in the head and neck or abdomen. Abdominal disease presents with pain, a
palpable mass or even intussuception in cases with involvement of the ileum.
Staging must include radiological assessment of all nodal sites (CT or MRI) and examination of
the bone marrow and CSF. Treatment is multi-agent chemotherapy, a more intensive course
being employed for advanced B cell disease.
Principal presentation, treatment and prognosis in NHL
Site
Type
Treatment
Localised
Usually of B-cell
Short, moderately
origin
intensive mutliagent chemo
Intra-thoracic –
Typical T-cell
As for ALL
anterior
disease
Prognosis
Good
Approaching that
for ALL
- 125 -
mediastinal mass,
pleural effusion
Intra-abdominal
disease – Bulky
gut or lymph node
masses
Typical advanced
B-cell disease
Very intensive
multi-agent
chemotherapy
Previously very
poor; now much
improved
Hodgkin’s disease
Relatively uncommon in prepubertal children. It usually presents as painless
lymphadenopathy, most frequently in the neck. Lymph nodes are much larger and firmer than
the benign lymphadenopathy commonly seen in children. The clinical history is often long, and
systemic symptoms (sweating, pruritis, weight loss and fever – the so-called ‘B’ symptoms)
are uncommon, even in more advanced disease
After diagnostic biopsy, the disease is staged to determine treatment. Intra-abdominal disease
is generally assessed radiologically, and staging laparotomy with biopsies and splenectomy, is
no longer performed. Lymphangiography is a technically difficult examination in small children
and is rarely required.
Combination chemotherapy (possibly with RadioRx to sites of bulky disease) is the treatment
for all except those with localised disease who receive radiotherapy alone. Overall, about 80%
of patients can be cured; even for those with disseminated disease, about 60% can be cured.
Brain Tumours
In contrast to adults, brain tumours in children are almost always primary and 60% are
infratentorial . Signs and symptoms are usually of raised intracranial pressure:
- headache (classically worse lying down)
- Vomiting (especially on waking in morning)
- Squint secondary to VIth nerve palsy
- Nystagmus
- Ataxia
- Personality or behavioural change
Location:
- Astrocytoma (40%)
o Juvenile cerebellar astrocytoma is cystic, often slowly growing, and the results of
treatment with surgery are excellent
o Non-juvenile astrocytoma occurs at all sites but more frequently in the cerebral
hemispheres. They vary from relatively benign to highly malignant (glioblastoma
multiforme), which have poor prognosis
- Medulloblastoma (20%)
o Nearly always arise in the midline of the posterior fossa
o Presentation is with ataxia as well as headache and vomiting. The tumour may
seed through the CNS via the CSF and up to 20% have spinal mets on diagnosis.
o Treatment with whole CNS radiation after maximal surgical resection has
produced 5 year survival of 50%
- Ependymoma (8%)
o Mostly occur in the posterior fossa where it behaves like a medulloblastoma, but
can also arise in the ventricles or spinal cord.
- Brain stem glioma (6%)
o Peak incidence is in early childhood
o Presents with cranial nerve defects, ataxia and pyramidal tract signs, but
frequently without raised intracranial pressure.
o Diagnosis is often based on clinical findings and CT/MRI scan, as biopsy can be
hazardous.
- 126 -
-
o Prognosis is poor (<20% survival)
Craniopharyngioma (4%)
o A developmental tumour arising from the squamous remnant of Rathke’s pouch.
o It is not truly malignant but is locally invasive and grows slowly in the
suprasellar region.
o It presents with raised intracranial pressure, visual field loss, and pituitary
dysfunction, typically as growth failure.
o Surgical excision with or without subsequent radiation is required.
Neuroblastoma
Arises from neural crest tissue in the adrenal medulla and sympathetic nervous system. It is
an unusual tumour in that spontaneous regression sometimes occurs in very young infants.
There is a disease spectrum from benign ganglioneuroma to malignant neuroblastoma.
It is most common before the age of 5 years.
Presentation:
Common
Less Common
Pallor
Paraplegia
Weight loss
Cervical Lymphadenopathy
Abdominal mass- classically adrenal in
Proptosis
origin
Hepatomegaly
Periorbital bruising
Bone pain
Skin nodules
Limp
Diagnosis:
- Clinically
- Radiologically
- Raised urinary catecholamine (VMA, HVA) levels
- Confirmatory biopsy is usually obtained and evidence of metastatic disease detected
with bone marrow sampling, bone scan and MIBG (Metaiodobenzyl Guanidine) scan.
MIBG is a radiolabelled tumour-specific agent, with provides a sensitive radioisotope
scan to measure disease extent and monitor response to treatment.
Prognosis:
- Based on age and stage of disease at diagnosis
- Majority of children over 1 year present with advanced siease and have a poor
prognosis
- Associated with poorer prognosis: Overexpression of N-myc oncogene and evidence of
deletion of material on chromosome 1 (del1p) in tumour cells.
Treatment
- Surgery in localised primaries without mets can be curative
- Chemo is used in advanced disease
Wilm’s Tumour
Originates from embryonal renal tissue. There is a Wilm’s tumour susceptibility gene which has
identified from the rare association between Wilm’s tumour and sporadic aniridia which was
known to be associated with loss of genetic material from chromosome 11.
Over 80% present before the age of 5 years. It is very rarely seen after 10 years of age.
Presentation:
Common
Uncommon
Abdominal mass
Abdominal pain
Anorexia
Haematuria
Hypertension
Diagnosis:
- 127 -
Radiological diagnosis from USS or CT is usually characteristic, showing an intrinsic
renal mass distorting the normal structure.
Treatment:
- Current UK protocol immediate nephrectomy, for patients with respectable tumours, vs.
with initial chemo followed by delayed nephrectomy
- All children require chemo
- Radiotherapy restricted to those with advanced disease
Prognosis:
- Good, 80% cure rate
- For those 15% with metastasic disease is 60%, but relapse carries a poor prognosis.
-
Rhabdomyosarcoma
Originates from primitive mesechymal tissue. There is a wide variety of primary sites, resulting
in varying presentation and prognosis.
- Head and neck
o Most common, causing proptosis, nasal obstruction and blood stained nasal
discharge
- Genitourinary tumours
o Next most common, and cause dysuria, and urinary obstruction or bloodstained
vaginal discharge
- Metastatic disease (Lung, liver, bone or bone marrow)
o Approx 15% of patients at diagnosis
o Treatment depends on the site, size and extent of disease.
Bone tumours
Malignant bone tumours are uncommon before puberty.
Osteogenic sarcoma is more common than Ewing’s sarcoma, but Ewing’s sarcoma is seen more
often in young children. Both have a male predominance.
Limbs are most common site. Persistent localised bone pain is a characteristic symptom,
usually preceding the detection of a mass. At diagnosis most patients are well and even
metastatic disease (most commonly lungs) is asymptomatic.
A bone x-ray shows destruction and variable periosteal new bone formation. In Ewing’s
sarcoma there is often a substantial soft tissue mass.
Rentinoblastoma
Very rare, but accounts for about 5% of severe visual impairment in children. It may be
unilateral or bilateral. All bilateral tumours are thought to be hereditary, as are about 20% of
unilateral cases.
Retinoblastoma susceptibility gene on chromosome 13.
The pattern of inheritance is dominant but with incomplete penetrance.
Most cases present within the first 3 years of life. Children from families with the hereditary
form of the disease should be screened regularly from birth.
Presentation:
- A white papillary reflex replaces the normal red reflex or
- Squint
Treatment:
- Cure but preserve vision
- Enucleation of the eye may be necessary for more advanced disease.
- Treatment with chemo to shrink the tumour followed by local laser treatment to the
retina is being used successfully.
- 128 -
Liver tumours
Are rare. Primary liver tumours in the newborn are more likely to be benign (haemangioma).
Primary malignant liver tumours are mostly hepatoblastoma (65%) or hepatocellular
carcinoma (25%). Hepatocellular carcinoma may arise in children with pre-existing liver
disease.
Initial Presentation:
- Abdominal distension or with a mass
- Pain and jaundice are rare
Investigations:
- USS or CT scan confirms a large intrinsic liver mass, occasionally with calcification
- Elevated serum AFP is detected in nearly all cases of hepatoblastoma and in some cases
of hepatocellular carcinoma.
Germ cell tumours
Are rare but may be benign or malignant. They arise from the primitive germ cells which
migrate from yolk sac endoderm to form gonads in the embryo. Benign tumours are most
common in the sacrococcygeal region and most malignant germ cell tumours are found in the
gonads. Serum markers (AFP and B-HCG) are invaluable in confirming the diagnosis and in
monitoring response to treatment.
Malignant germ cell tumours are very sensitive to chemo, and a good outcome can be
expected for disease at sites other than the brain.
Langerhans cell histocytosis (histocytosis X)
A rare disorder characterised by an abnormal proliferation of histiocytes. It is no longer
believed to be a truly malignant condition. However, it’s sometimes aggressive behaviour and
its response to chemo place it within the practice of the oncologists. There is a spectrum of the
disorder from localised systemic forms.
-
-
-
Solitary lesions of bone (eosinophilic granuloma)
o These may present at any age with pain, swelling or fracture. X-ray reveals a
characteristic lytic lesion with a well-defined border. Biopsy is usually necessary
and full skeletal survey is required to identify multiple lesions. Curretage,
intracavity steroid injection and low-dose radiotherapy are all successful forms of
treatment. Asymptomatic lesions may not require any treatment
Mutliple bone lesions
o These can occur at any sire and frequently involve the skull
Diabetes insipidus
o The association of skull disease with proptosis and hypothalamic infiltration
causing diabetes insipid was previously known as Hand-Schuller-Christian
disease. Diabetes insipidus can occur with other patterns of presentation, and
once established, is not usually reversed by successful treatment of the
underlying disease; long-term treatment with desmopressin is usually required.
Systemic LCH
o The most aggressive from of LCH (previously known as Letterer-Siwe disease)
tends to present in infancy with seborrhoeic rash and soft tissue involvement of
the gums, ears, lungs, liver, spleen, lymph nodes, and bone marrow
o Clinical presentation may be characteristic but the diagnosis should be confirmed
by biopsy, usually from skin or lymph node.
- 129 -
Extras
Sudden Infant Death Syndrome (SIDS)
Sudden infant death syndrome is the commonest cause of death in the first year of life,
causing one to two deaths per thousand livebirths. There were 1487 deaths attributed to this
condition in 1987 in England and Wales. (re: CAP (child accident prevention) survey).
Recently much focus has been placed on the unknown subset of infants perishing from what
has been described as sudden infant death syndrome, but who have in fact been the victims of
more definable illnesses, including inborn errors of metabolism and abuse. It is for these
potentially preventable reasons that sudden infant death syndrome should be regarded as a
diagnosis of exclusion, and careful examination and investigation be carried out in each and
every case. This is never more true than when more than one child in a family has died.
Aetiology
The aetiology of true sudden infant death syndrome is unexplained. Theories abound, including
those to do with brainstem abnormalities leading to poor cardiorespiratory regulation.
A number of risk factors have been noted.
Diagnosis
Obvious differentials include causes of death such as overwhelming sepsis and undetected
congenital malformation.
A differential diagnosis includes:
- Inborn errors of metabolism, particulary medium chain acyl-CoA dehydrogenase
deficiency which is said to account for up to 5% of sudden infant death in some
populations
- Infanticide, whether as part of neglect, physical abuse or as part of Munchausen
syndrome by proxy
Prevention
- Babies should sleep supine rather than prone
- Babies should be placed with the feet near the end of the cot
- More cot deaths could be prevented by reducing maternal smoking
- Keep the room temperature as close to 18 degrees C as possible
- Do not smoke while pregnant
- One of the factors attributed to the fall in number of cot deaths (comparing 1992 and
1993) is the reduced use of duvets and an increase in use of blankets; also there is an
increased tendency for babies to sleep in the parents' bedroom or bed - both of these
changes are now recommended as activities to reduce the risk of SIDS
- Medical advice should be sought early if the baby is unwell
Note that cardiac and respiratory monitoring equipment may be recommended to parents of an
infant, with a history of a previous child dying of SIDS
Child Psychiatry
Emotional or behavioural problems:
Presentation may be very different dependent on the age.
6 years: School, ADHD, Male vs. female
11 years: School refusal, Rebellion, eating disorder
15 years: Mood swings, behaviour, drugs/smoking, truancy, self harm/suicide
- 130 -
Attachment Theory
Attachment behaviour results in the individual attaining or retaining proximity to some other
differentiated and preferred individual.
Bowlby stated that infants are predisposed virtually from birth to interact socially with
caregivers as part of a survival-enhancing motivational system. There is a process of
integration of early relationship experiences into an overall organisation of attachment through
internal working models i.e. dynamic internal representations. These models organise our
perception of, and interpretation of, our experience with others.
Children’s internal working models guide their behaviour with others. If they have experienced
rejection or abuse they tend to develop negative thoughts about themselves. If children
expect their attachment figure to be unavailable they won’t turn to them when upset. This can
lead to the parent feeling rejected and set up a vicious cycle.
Features of attachment behaviour
Proximity-seeking to attachment figure, especially when threatened.
Use of attachment figure as a "secure base" from which to explore environment.
Separation from attachment figure leads to "separation protest" by the infant. Permanent
separation leads to impaired capacity to feel secure and explore environment.
This behaviour is seen between 6 and 36 months.
Classification of attachment in children
Attachment styles in infants can be classified according to a classification derived from an
experimental situation, where infant and parent are subject to a series of separations and
reunions, (Strange Situation Procedure).
Ainsworth Strange Situation Procedure
1.
2.
3.
4.
5.
6.
7.
Caregiver (CG) sits with infant on floor for 3 minutes.
Stranger enters room & engages infant for 3 minutes.
Caregiver leaves for up to 3 minutes.
CG returns and spends 3 minutes with infant.
CG leaves and infant left alone for 3 minutes.
Stranger returns for 3 minutes.
CG returns.
The attachment and exploratory behaviour of the infant, especially on reunion, is observed and
rated, to give a classification:
Type
Type
Type
Type
A - Insecure Avoidant.
B - Secure.
C - Insecure Ambivalent.
D - Disorganised or unclassifiable.
Insecure avoidant infants, (type A), have caregivers who fail to respond to their distress. The
infant explores with little reference to caregiver. Infant learns to suppress emotional distress,
may show little behavioural reaction to separations and reunions with caregiver, but is
physiologically aroused and stressed.
Infants classified as securely attached, (type B), have caregivers who respond predictably and
sensitively to their needs. The infant uses caregiver as a secure base from which to explore,
becomes distressed in caregiver’s absence, greets her positively on return, and then continues
to explore.
Insecure ambivalent, (type C), infants have caregivers who respond inconsistently. Infant is
distressed on separation, difficult to settle and angry on reunion. Infant cannot learn
- 131 -
contingencies about situations (e.g. when punished, when comforted) and copes by splitting
off conflicting emotions.
Infants classified as disorganised, (type D), often show a mixture of types A and C. The
caregiver is often depressed, alcohol dependent or abusive. It is thought that the child is
either afraid of or for the caregiver. It is a strong predictor of aggression in school and
behaviour problems generally.
Percentages of each style vary according the population studied.
Typical figures in North European cultures are:
A 20%
B 65%
C 15%
D up to 4%
It is important to remember:
Attachment styles at this age refer to a pattern of relationship i.e. strictly infants are securely
or insecurely attached to a particular caregiver. Children may show different attachment styles
with different caregivers, (e.g. mother and father), but children show a hierarchy of
attachment figures and the style shown with the adult that does the majority of caregiving is
usually dominant and predictive of later functioning.
Secure or insecure attachment does not refer to the intensity of feeling the infant has for the
parent but the behavioural interaction. Although insecure attachment may be associated with
abuse, most insecurely attached children are strongly attached to their parents in the sense of
loving them and being loved by them.
Why is attachment important?
Attachment relations, that is, the emotions or expectations, (secure or insecure), arising from
the parent child relationship, form the basis for rudimentary conceptual models of self and
others. These can be updated by later experience but are fairly stable over time and may form
one way in which caregiving styles are transmitted across generations.
These conceptual "working models" influence later adult narrative capacity- the way people tell
stories about their experiences of being parented, (as assessed by the Adult Attachment
Interview). This may link to the way people see themselves, their ability to reflect on
themselves and think about others.
Securely attached children tend to be more sociable and co-operative at school entry, more
able to generate happy endings on a picture completion task at 10-11 years. Insecurely
attached children, when followed up, have more problems at school Avoidant children tend to
be clingy and relatively passive. Ambivalent children tend to isolate themselves and show
unprovoked outbursts of aggression.
Can attachment styles change?
Attachment styles can change if the caregiving environment changes for the child. In general,
early attachment status is less predictive of later functioning the more the environment, (and
hence the child-caregiver relationship), changes. About 75% of children retain the same
attachment classification throughout childhood.
Cognitive Development
Piaget’s Theory
1. Sensori-motor
Birth to ~24 months.
Thinking dominated by sensations and motor activity.
2. Preoperational
2 to 7 years.
Object permanence.
Egocentrism.
- 132 -
Social perspective taking is limited.
3. Concrete operational
7 to 11 years.
Concepts begin to override percepts.
4. Formal operational
~11 years to adulthood.
Thought is no longer structured by concept of actual object.
Can think about what might be.
Can think about their own thinking, and internal states.
Possibly a basis for adolescent self-consciousness.
Piaget’s theories are not the only theories of cognitive development, but they are the
most influential. Experimental evidence seems to support them, though children
may pass through the stages earlier or less uniformly than originally thought, (not
all reach formal operational stage).
Social Cognition
Child begins to understand feelings of others in 2nd year.
Realises can hurt and comfort others.
Begins to understand goals of others in 2nd to 3rd years.
Pretend games, questions about other people.
Begins to understand social rules from 21/2- 3 years.
Rules may apply differently to family members.
Initially reward-punishment based morality, which is internalised into moral values
over childhood.
Understanding other minds
Undifferentiated, egocentric perspectives
3 to 6 years.
Confusion between acts and feelings and intended and unintended behaviour. Attributes own
perspective to the other person.
Differentiated and subjective perspectives
5 to 9 years.
“Same situation equals same viewpoint”.
Relationship is one-way rather than mutual.
Difficulty understanding: “it’s the thought that counts”.
Self-reflective/second person and reciprocal perspectives
6 to 12 years.
Can distinguish actions from intentions.
Begins to understand other people have different and distinct inner life.
Realises may have to influence other’s mental state to get what s/he wants.
Third-person and mutual perspectives
10 to 15 years.
Can take observer position in social interactions.
Might suggest mutually satisfying compromises.
Reflects on self as actor & object.
In-depth and societal-symbolic perspectives
12 years to adulthood.
Idea of a personal unconscious develops.
- 133 -
Memory development
Freud described infantile amnesia, and suggested active repression of memories took place.
Early implicit memory, “memory for how”, may still influence behaviour. The average age of
earliest retrieved memory is 3.5 years. Earliest form of long-term memory is that for generic
events. Autobiographical memory is interlinked with narrative capacity - adults help children
construct stories about their experiences, all early memories are co-constructed.
Family Life Cycle
Consideration of life cycle stage of a family may help predict the type of problems they are
likely to face. Family lifecycle stages can be defined in a number of ways and are culturally
dependent. With stepfamilies, there are added complications of resolving separations and
forming new boundaries with ex-spouses' families of origin, together with forming of new
relationships with new spouses and their families of origin.
One possible framework is:
1. Couple or family formation.
2. Birth of (first) child.
3. Individuation of the young child; going to school.
4. Individuation of the adolescent; puberty and sexual development.
5. Leaving home (separation from family of origin by the children).
6. Older adulthood; for the parents, the "empty nest" syndrome.
Older
Adulthood
Couple
formation
Leaving
home
Birth of
(first) child
Individuation
of the
adolescent
Individuation
of young
child
This represents the family life cycle
from the perspective of childrearing.
For each stage, the family has a
number of issues to be resolved, and
which set the family and its members,
certain tasks. The concept gives a
standard against which to evaluate
individual families.
Life is never quite so simple! For
reconstituted families the concept
needs to be reformulated. The
complexities of resolving separations
and forming new boundaries with exspouses’ families of origin, forming new
relationships with new spouses and
their family of origin make the situation
problematic.
1. Couple or family formation
Major tasks for family
Both partners move away from families of origin and towards each other to form a new family
unit. This involves balancing loyalties to partner, friends, families of origin and own needs.
Acceptance of some loss of freedom: replaced by a sense of belonging.
Renegotiate boundaries with previous social worlds so that the new social world is acceptable
to both partners, (avoiding too much loss).
Adjust to a variety of unexpected differences and unmet expectations, caused by differences
between the families of origin.
Tasks for couple
- 134 -
Establish sufficient intimacy to meet each other’s emotional needs without smothering the
partner.
Resolve fundamental issues of power and dominance.
Accept and tolerate different emotional responses and style of partner.
Ensure that personal space is available for communication between the couple.
Divide responsibilities in a mutually acceptable way.
Establish a workable means of conflict resolution.
COMMON PROBLEMS
Loyalty conflicts- e.g. “He’s not good enough for you”.
Rejection scenes.
Adults who do not like the role.
Formation problems - too close or too distant.
Power Conflicts - overt, (chronic bitching) or covert, (tense atmosphere).
Need for affection unmet.
2. Birth of (first) child
Tasks for family
Adjust to new arrival, and balance loyalties to self, partner and child.
Reorganise patterns of family functioning.
Renegotiate boundaries with social world.
Tasks for parents
Maintain intimacy in marriage while meeting needs of the child.
Adjust to new role as parent with implications for career, ‘freedom’ etc.
To love the infant; accepting his or her biological dependency.
Cope with tiredness and learn new skills.
Cope with extra responsibility and divide labour with partner.
Resolve differences in parenting styles.
Tasks for child
Formation of primary bond with parents.
Implicit learning of family rules, especially social and emotional behaviour.
N.B. Subsequent children will require older children to adjust to change in sibling structure
and cope with jealousy and responsibilities.
COMMON PROBLEMS
Loyalty conflicts – overt or covert.
Coping with responsibility – parental mental illness, perhaps overwhelmed by parenthood.
Grandparents may take over.
Neglect of infant’s needs, e.g. husband goes out every night.
3. Individuation of young child
Tasks for family
Balancing the increasing autonomy of the child with a sense of belonging.
Maintaining reasonable control – not over or under burdening the child.
Allowing the child to experience and express a range of emotions so that the child recognises
the needs of self and others.
Encouraging the child to develop independent behaviour outside the home.
Tasks for parents
Facing separation from the child – loss of intimacy or fears of welfare.
Establish consistent, age-appropriate rules to reward & regulate behaviour.
Encourage the child to recognise the needs of others, but also to develop self-esteem so that
the child does not sacrifice itself for others.
Recognise and encourage individual strengths.
Encourage play in and out of home at an age-appropriate level.
Encourage acceptance of gender identity.
- 135 -
Tasks for children
Facing fears of new situations and increasing separations from parents.
Adjusting to different rules and expectations from peers and adults.
Balancing own needs with those of others.
Learning to contribute and take responsibility.
Develop relationships outside family.
Accepting gender identity.
Resolving jealousy and rivalries.
COMMON PROBLEMS
Separation problems – may be present in child or parent.
Power problems – child may be presented as stubborn etc, especially where parents disagree.
Conduct problems – low expectations and inconsistent control may lead to behaviour that
ignores the needs of others.
4. Individuation of the adolescent
Tasks for family
Maintaining a degree of mutual loyalty, trust and respect between adolescent and parents,
despite increasing distances in relationships. Compensated by developing greater intimacy in
peer group relationships.
Redefining issues of autonomy, control and responsibility.
Acceptance and facilitation of the changes in emotional behaviour needed in transition to
adulthood.
Encourage greater freedom and privacy for adolescent.
Acceptance of puberty and impending sexual maturity.
Tasks for parents
Acceptance of loss of the ‘child’.
Accept loss of control – may include overt rebellion.
Tolerating differences which adolescent uses to differentiate from family and parent identities.
Tasks for adolescent
Accept loss of parents as ‘protectors’ – cope with fears of loneliness, responsibility, sexuality,
identity and autonomy.
Learning to assertiveness with peers and adults and take responsibility for important decisions.
Learn to express emotions and needs in adult way – ‘new rules’.
Completion of tests and ‘rites of passage’ in adolescence.
Develop confiding relationships with peers.
COMMON PROBLEMS
Struggles for autonomy - overt or covert:
Overt: Commonly a rebellious teenager is presented by parents with a request to ‘fix him’.
The adolescent may resist or sullenly comply – but is usually fearful or ambiguous about
growing up. He may be labelled as a ‘changed personality’.
Covert: The teenager is tied to the family by loyalty or mutual consent. The problem may
present as ‘psychiatric’, psychosomatic, school refusal, eating disorder etc. The symptom is
best seen as a way of expressing frustration while the teenager remains superficially loyal.
Family legacies and missions – expectations may exist about careers, responsibilities, and
‘substitution’ effects commonly create problems.
5 & 6. Stage of departure of the children
Tasks for family
To separate without breaking relationships.
Acceptance of greater equality between parents and offspring.
Mutual understanding of expectations between parents and departed offspring.
Reorganisation of intra-family boundaries.
- 136 -
Tasks for parents
Facing the pain of loss of offspring and the ‘empty nest’.
Acceptance of greater equality with offspring.
Re-examination of needs and limitations of self and marriage. Development of new purpose
may be needed.
Learning to accept children’s choice of partners as potential relatives.
Acceptance of possible new roles as grandparents.
Tasks for adult children
Letting go of family of origin.
Coping with responsibilities of housing, work and social world.
Putting one’s own development and needs before family loyalty.
May involve adjustment to new rules if living with others.
Coping with mature social and sexual relationships.
Developing identity and priorities; choosing a lifestyle that suits them.
COMMON PROBLEMS
‘Binding’ – the young adult may present with vague neurotic or social problems sometimes
may return home repeatedly or simply unable to leave.
‘Expelling’ – the young adult is thrown out and attempts are made to sever family
relationships.
‘Delegating’ – the offspring is allowed to leave on condition they fulfil a ‘mission’.
The parent most involved with parenting may present with anxiety or depression.
Longstanding covert marital problems may surface.
- 137 -
Child Protection
Categories:
- Physical Injury
- Sexual Abuse
- Emotional Abuse
- Neglect
- Not mutually exclusive, often overlap
Non-accidental injury
- 7 ‘pointers none ‘diagnostic’
- delay in seeking medical help or not seeking help at all
- Vague inconsistent history, lacking in detail
- Account of the history not consistent with the injury observed
- Abnormal parental affect - lack of concern, preoccupation with own problems
- Parents behaviour gives cause for concern - hostility, paranoia, failure to wait for senior
advice
- Child’s appearance and interaction with parents abnormal - sad, withdrawn, ‘frozen
watchfulness’
- Disclosure by child
Sexual abuse
- Disclosure/statement by the child
- Symptoms due to local trauma or infection
- Symptoms attributable to emotional effects
- Self-Harm
- Sexualised Conduct or inappropriate sexual knowledge in young children
- Sexually transmitted diseases
- Pregnancy
Symptoms Attributable to Emotional Effects
- poor concentration
- low mood
- self-harm
- drug/alcohol abuse
- anorexia
- enuresis or encopresis
Emotional abuse(Gabarion)
- Rejecting the child - The adult refuses to acknowledge the child’s worth and legitimacy
of the child’s needs
- Isolating the child - Adult cuts the child off from normal social experiences and contacts
and prevents the child from making friendships - makes the child believe she is alone in
the world
- Terrorising the child - Adult verbally assaults the child - creates a climate of fear, bullies
and frightens the child - makes the child feel the world is hostile
- Ignoring - The adult deprives the child of essential stimulation and responsiveness,
stifling emotional growth and intellectual development
- Corrupting - The adult mis-socialises the child- stimulates the child to engage in
destructive anti-social behaviour - reinforces that deviance and makes the child unfit for
normal social experiences
infants
- Sleep/feeding problems
- Irritability, anxious
- Disordered attachment
- Withdrawal, apathy
- Developmental delay
- Self-stimulation - head banging, rocking
- Lack of social responsiveness
- 138 -
pre-school children
- Delay in language acquisition
- Behavioural problems - poor attention
- Growth retardation
- Aggressive
- Withdrawn, fearful, anxious
- Lack of selective attachment - e.g indiscriminate affection to strangers
school
-
age
Learning difficulties - poor concentration and overactivity
Inappropriate attachment to carers
School - disruptive in school, failure in school, truancy
Behavioural problems - running away, stealing bullying
Enuresis or encopresis
low self-esteem, poor social interaction, rejection by peers
Other behaviour patterns - repetitive rocking, self-mutilation, masturbation
adolescents
- As above
- Depression
- Escalating aggression
- Anxiety
- DSH – overdosing
- Psychosomatic illnesses
- Drug and alcohol abuse
- Criminal activities
Neglect
child’s needs
- Food – appropriate
- Shelter
- Clothing
- Protection - suitability of substitute carers, safety
- Optimum Health - preventative services, curative services
- Hygiene
- Comfort - warmth etc
- Physical Affection
- Interaction
- Stimulation
- Control
Neglect is the persistent or severe neglect of a child, or the failure to protect a child from
exposure to any kind of danger, including cold and starvation
- Or extreme failure to carry out important aspects of care resulting in significant
impairment of the child’s health or development, including non-organic failure to thrive
- Difficult to diagnose because by definition has to be present for a period of time
- All age groups can be affected, but the pre-school child is the most vulnerable
- A child is neglected if the child’s basic needs are unmet
- Manifestations of this are when the child is:
- Malnourished, ill-clad dirty, without proper shelter or sleeping arrangements
- without supervision, unattended
- ill and lacking essential medical care
- denied normal experiences that produce feelings of being loved, wanted, secure and
worthy (emotional neglect)
- failing to attend school regularly
The neglected child is likely to have difficulties with
- basic trust
- ability to govern his own/behaviour
- 139 -
-
social interaction
problem solving capacity
resourcefulness
medical indicators
- Developmental delay
- Poor attention and emotional immaturity
- Eating disorders - stealing and/or hoarding food
- failure to attend for routine medical examinations, immunisations, hearing and vision
assessments or refusal of appropriate medical treatment
- Frequent A&E attendance for accidents caused by inappropriate or inadequate
supervision
What to do if you suspect abuse
- Pass on and share information with: ‘your manager’:
- Medical students = Senior Nursing or Medical staff
- Child Protection Unit
- Social Services
- Police
Role of Child Psychiatry
- Priority is the child’s safety and meeting emotional and developmental needs in a
nurturing and adaptive environment. Therapy cannot provide a substitute for this
- Possible interventions:
o Individual work
o group work
o parent training
o family therapy
Behavioural Management
Underlying principles
Behaviour is modified according to its outcome - change environmental contingencies in
order to change child's behaviour cf. changing the child "from within".
Draws on learning theories of operant conditioning.
Reinforcement
Behavioural outcome, (reward), that increases the probability or frequency of occurrence of
the behaviour that immediately precedes it.
Positive reinforcers act as rewards.
Negative reinforcers act by the removal of something unpleasant (e.g. avoidance).
What behaviours might you not want to reinforce?
What types of reinforcers can you think of?
What would be suitable reinforcers for children of different ages?
Subtypes of positive reinforcement
Reinforcement of incompatible behaviours.
Shaping.
Differential reinforcement.
How to make it clear that the behaviour and the outcome are linked…
Reinforce with a CICC:
CICC-
clear
immediate
consistent
contingent
What to do with unwanted behaviours…
What does punishment do?
- 140 -
Some punishments can be reinforcing!
Is physical punishment ever justified?
Extinction
Withholding reward after inappropriate behaviour.
In the absence of the reinforcer, the behaviour disappears over time BUT it may
increase in frequency before it disappears.
One type of extinction is time out from reinforcement.
Reinforcement is withdrawn for a particular length of time e.g. "naughty chair". As a
rule of thumb, duration of "time-out" in minutes = child's age in years.
How to do this in practice
2 preliminary questions to consider:
1. Are there practical barriers to the parents managing their children?
(Think about practical support or nursery placements).
2. Are there emotional barriers to the parents managing their children?
(This approach requires parents to acknowledge the importance of their responses in
maintaining the child's behaviour and requires effort on their part).
Get as much information as possible:
Detailed account of 1 or 2 episodes in the history - keep questioning until you are
happy you could reconstruct the event fully.
Diary keeping.
Use A-B-C format:
Antecedent
What happened before
the behaviour
Behaviour
What happened during
the behaviour
Consequence
What happened
afterwards
The chart may suggest solutions e.g. modifying antecedents or consequences.
Work with the family.
Choose a single behaviour to target.
Target the behaviour that is most likely to meet with success and which parents are
motivated to change.
Make goals specific and achievable.
Make sure all the family understand the "rules" - often helpful to given written
instructions.
Monitor the response before and after treatment.
Persist: things may get worse before they get better.
Star Charts
Star charts act as a positive reinforcer.
Can be useful in bedwetting or tantrums from ~4-5 years old.
Important points:
Define behaviour as clearly as possible, preferably in positive rather than negative
terms.
- 141 -
Do not pick the most difficult behaviour first; pick one that is easily treatable.
Stars are won for the defined behaviour and only for the defined behaviour, (not for
‘being good’), even if performed ‘with bad grace’.
You cannot lose stars for bad behaviour.
Decide the value of stars beforehand. In younger children, sticking a colourful sticker on
the chart may be enough. In older children, consider cashing in every 5 (or whatever)
stars for a small treat e.g. an extra 10 minutes up at night, a small toy or rewarding
the child when they reach (say) the 5th 12th and 25th stars.
Put stars on the chart promptly.
Attention Deficit Hyperactivity Disorder
(ADHD)
Key Features:
- Inattention.
- Over-activity.
- Impulsivity.
Inattention
- Poor attention to tasks.
- Poor attention to detail.
- Apparently not listening.
- Poor self-organisation
- Avoids tasks requiring sustained mental effort.
- Easily distracted.
- Doesn’t finish tasks
- Loses homework etc.
- Seems forgetful, but memory OK on testing.
Overactivity
- Fidgets and squirms.
- Leaves seat in class, at meals etc.
- Runs, climbs rather than walks.
- Noisy, cannot play or work quietly.
- Persistent overactivity not moderated by social demands.
Impulsivity
- Blurts out answers.
- Fails to wait turn.
- Interrupts and intrudes on others activities.
- Talks excessively without response to social constraints.
Diagnosis
Need to have problems in all areas which are:
- Excessive compared with norm for child of that age or developmental ability.
- Present from early age.
- Pervasive i.e. present in more than one social setting.
- In US laxer diagnostic criteria, hence quoted rates in US>>UK.
o (6-7% vs 0.5- 1%).
What else could it be?
- Age or developmentally appropriate boisterousness, disobedience or cheekiness.
- Conduct disorder.
- Disinhibited attachment disorder - lack of parental care leads to failure to form secure
emotional attachments.
- Anxious inattentiveness caused by stress.
What else could it be?
- Overactivity caused by prescribed drugs.
- Overactivity associated with pervasive developmental disorder e.g. autism.
- Rarely behavioural disturbance secondary to depression, psychosis or closed head
injury.
Assessment
- 142 -
Therefore need to consider child’s:
- Family background.
- Social background.
- Development.
- Associated behaviours.
- Past medical history.
What goes with it?
Conduct disorder.
- Anxiety.
- Global learning disability.
- Scholastic skills disorder.
o (1/3 have specific problems with reading, spelling or maths).
- Specific language impairment.
- Developmental motor function disorder.
- Tourette’s Syndrome.
Treatment:
Psychological
- Information for child & parent.
- Family support & interventions.
Focusing on behavioural management:
- Structured tasks.
- Simple instructions.
- Praise for concentrating and completing tasks.
- Time-out regimes for unacceptable behaviour.
- School intervention programmes.
- Individual cognitive approaches to reduce impulsiveness.
- Diet?
o Popular with support groups.
o Works for a few children but hard to follow.
o Risk of dietary insufficiency.
Medication
- Well validated, but response not specific to ADHD.
- Does not ‘cure’ disorder but improves some of the behaviours/symptoms.
- May need to continue for years.
- Most common drug is methylphenidate, (ritalin).
Methylphenidate:
Side-effects
- Reduced appetite or nausea, (50-70%).
- Occasionally slowed growth.
- Difficulty sleeping in 30-50%.
- Stomach pains & headaches, often transient.
- Potential for abuse of stimulant medication either as recreational drug or
overmedication by parents seeking to control child’s behaviour.
- Effects last only a few hours only so need several doses throughout the day - aim to
target times when in school.
Recent guidelines from NICE:
www.nice.org.uk
Autism
Main diagnostic frameworks:
o ICD10 & DSM-IV.
- Abnormalities must be present before 36 months for diagnosis.
3 areas of deficit required for diagnosis:
- Communication.
- Reciprocal social interaction.
- Restricted and repetitive behaviours, interests and activities.
Communication
- Delay or lack of speech development.
-
- 143 -
-
o (*1/2 fail to develop functional speech).
Failure in reciprocal conversation.
Stereotypies or idiosyncrasies of speech,
o e.g. repeating phrases from TV out of context.
abnormalities in pitch, rate, rhythm of speech e.g. sing-song or monotonous.
Lack of imaginative play.
Other special characteristics of speech:
- Pronoun reversal e.g. refer to self as ‘you’.
- Delayed echolalia, (also may see immediate but is also found in other language
problems).
- Neologisms, (made up words).
Reciprocal Social Interaction
- Failure or delay in use of gaze or gesture.
- Failure in peer relationships.
- Rarely seeking others or offering affection at times of stress.
- Lack of shared enjoyment of own or others happiness.
- Deviant response or lack of modulation of behaviour to fit social context.
Restricted & repetitive interests and behaviours
- Preoccupation with restricted interests:
o e.g. maps, birthdates, shoe-sizes.
- Attachments to unusual objects
o e.g. vacuum cleaner nozzle.
- Apparently compulsive, non-functional rituals.
- Stereotyped repetitive motor mannerisms.
- Restricted & repetitive interests and behaviours
- Preoccupation with part objects, (wheels of car), or elements, (smell, texture, noise), of
play materials.
- Distress over small environmental changes e.g. furniture moved. Also may become
distressed if daily routines not followed e.g. bath water not up to certain level.
Management
Medical
- Treatment of medical conditions.
- Correction of hearing/visual defects.
- Dental care.
- Genetic counselling.
- Education
- Speech Therapy
- Family Support
- Information.
- Behavioural methods.
- Counselling.
- Practical help.
- Respite.
Medication (limited role)
- Anticonvulsants for epilepsy.
- Antidepressants, lithium for mood disorder.
Disability
Who definition:
Disorder
Impairment
Disability
A medically definable condition or disease
is the lack of a body part or function – a loss or abnotmility of
psycohological, physiological or anatomical structure or function
is a lack/restriction of ability to undertake an activity ro a level or in a
manner that is considered within the range or formal for a child of that
age
- 144 -
Handicap
is a disadvantage for a given individual resulting from impairment or a
disability that limits or prevents the fulfilment of a role that is normal
(depending on age, sex, social or cultural factors) for that indivudal. It is
the impact of the disability or impairment on the individual
Examples
- 145 -
- 146 -
Assess needs: Checklist for treatment/management
- Diagnosis/prognosis
- Medical management
- Surgical management
- Nutritional
- Mobility/contractures
- Positioning
- Bladder/bowels
- Chest
- Spine
- Vision
- Hearing/language/communication
- Family – siblings, support
- Financial
- Schooling
- Psychology
- Growing up –immunisation, sexuality, jobs and training
Paeds Surgery
Please note when interpreting infants AXR, large and small bowel cannot be distinguished.
Haustrations on the large bowel are undetectable as are the small bowel markings.
-
Elective Surgery
Emergency Surgery
Neonatal conditions
Elective
- Inguinal Hernias
- Hydrocele
- Umbilical Hernia
- Undescended Testis
- Phimosis
Inguinal Hernias
-
-
Prevalence of hernia
o Term 3.5-5%
o Preterm 9-11%
o Male: Female 9:1
Indirect: sac passes through internal inguinal ring and along the inguinal canal
o Usually paeds
Direct: rare, usually prem baby or connective tissue disorder
o Usually adult
What’s the problem with paeds hernia
Male: Not so much strangulation of bowel, but can cause ischaemia of testis
Female: Can pull ovary into hernia (you can feel a round lump in hernia)
Hydrocele
- Scrotal swelling of variable size
- O/E can get above swelling, transilluminable
- Due to persistence of Processus vaginilis
- Resolution with time
- 147 -
Hernia Vs. Hydrocele
Hernia, had bowel, but other than that anatomically the same
Hernia: Can get below it, but not above
Hydrocele: Can get above
Umbilical Hernia
-
Incidence 1:6
Black: White 9:1
Spontaneous closure rule: 90% where defect 0.5cm at 3/12
If present at 3-5 not likely to close
Rarely Symptomatic
Hernia repair for cosmetic reason pre-school
Undescended Testis
- Incidence 1:300
- Most differentiate true undescended testes from retractile testis (will come down if
pulled, may go back up with cremasteric reflex)
- Some descend over first 12 months
- Risk of decreased fertility, torsion and malignant change
Phimosis
- Phimosis: “Muzzled”
- Separation of foreskin from glans occurs from birth releases “smegma”
- Differentiate physiological non-retractile foreskin from pathological phimosis
Paediatric Surgical Emergencies
-
Acute
-
Acute Abdo
Testicular Torsion
Appendices
Most common emergency
Lifetime risk ~7-9%
Male: Female 3:2
1/3 perforated at presentation
Pre-school 1-5%
Intussusception
- Invagination of bowel into itself
- Inflammed/enlarged peyers patches
- Specific lead point in 5%
- 5-18 montyhs, peak ~9/12
- Incidence 1-4:1000
- Clinical features:
o Colicky abdo pain
o Usually white, legs drawn up to chest
o Then OK
o Then “redcurrant jelly” stool, bile stained vomit
70% - Air enema, PR to push out intussusception
- 148 -
Testicular torsion
- Acute unilateral scrotal pain
- Systemic upset
- Scrotal swelling and erythema
- Bell-clapper testis, and contraction of cremaster spins the testis
- 24 Hr – 100% ischaemic
- Clinical Features:
o 10/10 Pain
o Never comfortable
Adhesion Obstruction
- History of previous surgery, 2% of all laparotomies
- 80% within 3/12 of op
- Bile stained vomiting ± abdo distension
- Drip and suck: Rarely successful
Pyloric Stenosis
- First born male child
- 6/52 of age
- Strong family relationship
- Non-bilious pojectile vomiting
- Poor/No wt gain
- Hypochloraemic, hypokalaemic, hyponatraemic, alkalosis
Neonatal
- Necrotising Entercolotis (NEC)
- 1:400
- Unknown aetiology
- Risk Factors
o Prematurity
o Asphyxia, hypoxia
o Maternal drug abuse
o Early feeding
- Oesophageal atresia ± Tracheoesophageal fistula
o 1:3000
o Associated with polyhydraminios during pregnancy. Antenatally this may be
picked up if atresia has caused absence of fluid in bowel/stomach, or
accumulation of fluid above atresia.
o May be picked up at birth, with inability to perform Nasogastric tube pass on
resus.
o Postnatally, aspiration, or poor feeding can occur.
o 86% AO and distal TOF
o 5% pure AO
86%
Atresia with fistula
between distal oesop-
8%
Atresia without
fistula
4%
H-Type (N-type) fistula
without atresia
- 149 -
hagus and trachea
o
o
o
o
o
50% of all cases have associated anomalies
VACTERL, VACTER
VACTERL
Vertebral
Anorectal
Cardiac
Tracheo-oEsophageal
Renal
Radial Limb abnormalities
The VATER acronym signifies
V - vertebral anomalies
A - anal malformations
TE - tracheo-oesophageal fistula/atresia
R - radial and renal anomalies
Investigations
CXR/AXR would show inability to pass N.G tube
Management
Nil by mouth; IV fluids
Closure TOF: fistula brought up to atresia, using a thoractomy surgery or
thoracoscopy.
Oesophageal anastamosis
Umbilical Hernia
An ‘outy’ belly button, protrusion of bowel through umbilicus into a skin contained sac.
Reducable. Repaired cosmetically
Exomphalos
Often diagnosed antenatally. The abdominal contents protude through the umbilical ring,
covered with a transparent sac (whartan’s jelly) formed by the amniotic membrane and
peritoneum. If left the sac would develop into skin. This condition is associated with other
major congenital abnormalities.
Gastroschisis
In this condition, the bowel protrudes through a defect in the anterior abdominal wall, adjacent
to the umbilicus, and there is no covering sac. It is not associated with other congenital
abnormalities. It carries a much greater risk of dehydration and protein loss, so the abdomen
of affected infants should be wrapped in several layers of cling film to minimise fluid and heat
loss. A nasogastric tube is passed and aspirated frequently and an IV infusion of dextrose
established. Colloid support is often required to replace protein loss. Many lesions can be
repaired by primary closure of the abdomen. With large lesions, the intestine is enclosed in a
silastic sac sutured to the edges of the abdominal wall and the contents gradually returned into
the peritoneal cavity.
Malrotation with Volvulus
- IMPORTANT TO KNOWN –Bile stain vomit, is Malrotation until proven otherwise
- Bile staining vomit
- Acutely unwell collapsed child
- AXR: Often gasless
- Upper GI contrast: DJ Flexure to R of midline, corkscrew appearance of jejunum
Large Bowel obstruction
- Hirschprung’s disease: Absence of the myenteric nerve plexus in the rectum which
may extend along the colon. The baby often does not pass any meconium within 48H of
birth and subsequently the abdomen distends. About 15% present as an acute
enterocolitis.
- Rectal atresia: Absence of the anus at the normal site. Lesions are high or low,
depending whether the bowel ends above or below the levator ani muscle. In high
- 150 -
lesions there is a fistula to the bladder or urethra in boys, or the vagina or bladder in
girls. Rx is surgical. Please not meconium is sterile, as is the bladder, so any rectalvesico fistula would not present with UTI etc, but meconium passing in urine.
Paeds Emergency
-
Neonate
o Resus
Paeds
o Anaphylaxis:
Anaphylactic shock is a medical emergency; it is a life- threatening type I
hypersensitivity reaction.
The main characteristics are circulatory collapse and airway impairment
in response to an allergen to which the patient has previously been
sensitized.
The commonest signs of anaphylaxis in children are:
pallor (shock)
limpness
apnoea
Other signs include:
upper airway obstruction - hoarseness and stridor
lower airway obstruction - dyspnoea and wheeze
sinus tachycardia, profound hypotension with tachycardia
Skin:
angioedema - swelling of lips, face, neck, tongue
urticaria - itchy weals with erythmatous edges and pale blanched
centres
diffuse erythema
o CPR:
Cardiorespiratory arrest in children is very different from arrests in
adults. In the latter, the main cause is primarily cardiac in origin though
in the paediatric population this is not the case.
Moreover the nature of causes of cardiorepiratory arrest in children is
such that early recogntion and treatment of the predisposing cause is
important and will prevent an arrest occuring.
The commonest cause of arrest in children is due to respiratory failure.
This may result from a variety of causes which vary according to age. At
birth, asphyxia may occur, whilst in infancy, respiratory illness such as
croup, bronchiolitis and pneumonia occur more frequently. Foreign body
inhalation, asthma and trauma or pneumothorax are also important
causes of respiratory insult.
Respiratory depression may also result from epilepsy, raised intracranial
pressure, neuromuscular problems or poisoning. The second commonest
cause for arrest is due to circulatory failure caused by fluid loss, bleeding
or sepsis.
o DKA:
Diabetic ketoacidosis may be defined to occur in patients with positive
serum ketones and an arterial blood pH of less than or equal to 7.30
and/or a serum bicarbonate less than or equal to 15 mmol/l.
Diabetic ketoacidosis occurs almost exclusively in type I diabetes, and is
stimulated by severe insulin deficiency coupled with absolute or relative
increases in glucagon. It should be regarded a medical emergency; its
rapid recognition and accurate treatment are essential to prevent
morbidity and mortality. It should be noted that there are subtle
differences in the management in children and in adults.
o Hypoglycaemia
o Hyponatraemia:
- 151 -
Hyponatraemia is a low plasma sodium, defined as sodium less than 135
mmol/l.
Epiglottis:
Acute epiglottitis is the result of localized infection of the supraglottic
larynx, usually by Haemophilus influenzae. This results in swelling of the
epiglottis that obstructs the laryngeal inlet.
If there is suspicion of acute epiglottitis the child should be admitted
immediately because of the danger of airway obstruction. Acute
epiglotitis is a paediatric emergency. Management and treatment is as
follows.
DO NOT:
panic
alarm the parents or child
examine the child in any way - especially do not try to visualize
the epiglottitis using a tongue depressor
DO:
call a senior anaesthetist, paediatrician and ENT surgeon
alert theatres or ICU; the child needs to be admitted immediately
Meningitis
Raised intracranial pressure:
Raised intracranial pressure is caused by intracranial lesions causing an
increased volume of blood, CSF or parenchymal tissue.
Raised ICP is monitored with a ventricular catheter or surface pressure
recording device; treatment is instituted when the mean ICP exceeds 30
mm Hg. This is a specialist area and local neurosurgical advice should be
sought.
Management approach depends upon the source of the raised pressure:
space occupying lesion - remove lesion surgically
increased CSF - burr hole with an external drain or shunt
cerebral oedema - osmotic diuretics such as mannitol are now no
longer routinely used because of the danger of reduced cerebral
perfusion; steroids are good for cerebral oedema secondary to
tumours and abscesses, poor for trauma. Other measures include
the use of barbiturates and hyperventilation.
Status asthmaticus:
Status asthmaticus is a severe, prolonged, immobilising attack of asthma
that is unresponsive to normal bronchodilator treatment. Treatment of
this condition may be started at home but hospital treatment is indicated.
Patient management is based upon administering large doses of
corticosteroid drugs and bronchodilators and ensuring that the patient is
receiving adequate oxygenation until the attack is completed.
Status epilepticus:
Tonic-clonic status epilepticus is a condition in which prolonged or
recurrent tonic-clonic seizures occur over 30 minutes without the patient
regaining consciousness.
Status epilepticus is a medical emergency because of the 20% mortality
and the high rates of neurological and systemic morbidity.
o
o
o
o
o
Paediatrics
Appearance
Pulse
Grimmace
Activity (muscle tone)
Resp
- 152 -
Acid-Base (ABG’s)
Henderson-Hasselbach
[H+] [HCO3-] = K [CO2] [H2O]
Normal values in an ABG report. (Table 1)
paO2 in kPa
(mm Hg)
Arterial Blood
Venous Blood
paCO2 in kPa
(mm Hg)
pH
11-13 (804.7-5.9 (35-45) 7.36 - 7.44
100)
5 - 5.6 (37-42) 5.6-6.7 (42-50) 7.34 - 7.42
HCO3
BD/BE
mmols/L
21-28
+/-2
Sat O2
(%)
>95%
The primary defect in an acid-base disorder is defined by its initiating process, which can be
metabolic (changes in Bicarb) or resp (changes in PaCO2). A compensatory response describes
the secondary physiological response to the primary disturbance. Over-compensation does not
occur.
- 153 -
Normal Ranges
Respiratory Rate
Heart Rate
Systolic BP
Infants
40-30
160-110
70-90
Young children
30-25
140-95
80-100
Older children
25-20
120-80
90-110
- 154 -
Heamoglobin
Age
Birth
2 weeks
2 months
1 year
2-6 years
6-12 years
12-18 years
Male
Female
Hb (g/dl)
14.5-21.5
13.4-19.8
9.4-13.0
11.3-14.1
11.5-13.5
11.5-15.5
MCV (fl)
100-135
88-120
84-105
71-85
75-87
77-95
WBC (x109/L)
10-26
6-21
6-18
6-17.5
5-17
4.5-14.5
13.0-16.0
12.0-16.0
78-95
78-95
4.5-13
4.5-13
Platelets (x109/L)
150-450 at all ages
Reference Ranges
- 155 -
- 156 -
Newborn Life Support Algorithm
- 157 -
Paediatrics Life Support Algorithm
- 158 -