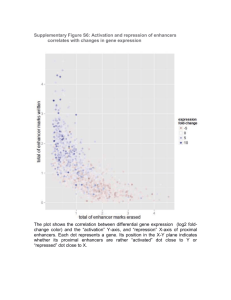
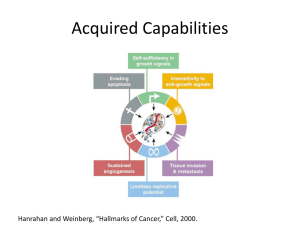
CORE FACILITY III: Multiple inducible gene expression cell model
advertisement

1. 中文摘要 細胞受到生長因子、賀爾蒙及其他刺激物的刺激,會經由細胞內訊息傳遞 來影響某些特殊基因的表現,以達成細胞功能的發揮。基於過去二年的研究成 果,我們在第三年計畫的每一個分項計畫,把研究焦點更為集中來執行。第一 分項乃基於過去我們對 Sp1 在基因轉錄調控得知其可扮演一個 anchor 蛋白的角 色,可把其他轉錄因子如 c-Jun 及 VDR 帶至基因 promoter,因此在本年度我們 把重點放在探討 c-Jun、VDR 及 Sp1 轉譯之後的蛋白修飾,發現 PP2B 可與 c-Jun 做交互作用,來達成 c-Jun C 端脫磷酸的反應。VDR 之脫磷酸反應也能促進其 Sp1 的交互作用。另 Sp1 可被 acetylation,其 acetylated Sp1 可促進 12(S)lipoxygenase 及 p21(CIP1/WAF1) 的基因表現。在第二分項我們把重點放在癌細胞 Met/Ron 及 Eps8 的訊息傳遞,得知 Ron 會轉移至細胞核,另也得知 Eps8 可調控 FAK 的表現。另得知 ER stress 可誘導 cyclin A 的表現導致肝癌細胞的生長。第 三分項研究方向為探討細胞凋亡之訊息傳遞,研究重點放在免疫細胞。得知 Fas 活化 p38 之訊息傳遞在 T 細胞凋亡扮演一個重要角色,同時也釐清鋰抑制 ceramide 誘導之細胞凋亡經由 MEK/ERK/Hsp70 訊息路徑的活化,且 GSK-3β的 活化在 ceramide 誘導之細胞凋亡扮演一個重要角色。另也得知 low substratum rigidity 也可藉由 ERK 路徑的活化,導致細胞游走的抑制。 配合各分項計畫的執行需求,我們在成大已建立一個研究細胞訊息傳遞的 核心設施,包括蛋白質體學、基因微陣列分析、光學影像儀器等設施,協助計 畫成員執行追求學術卓越研究。 1. Executive Summary Gene expression is regulated through intracellular signal transduction upon the stimulation of growth factors, hormones and other stimulants. There are three subprojects in this proposal. Based on the past two years research results, we made more focus on our research directions in each subproject in the third year of this proposal. In Sub-project I, we focused on the novel function of Sp1 that could serve as an anchor protein to recruit other transcription factors (e.g. c-Jun and VDR) to gene promoter in the regulation of gene expression. We found that c-Jun could interact with PP2B, which resulted in dephosphorylation of c-Jun C-terminus. Dephosphorylation of VDR also enhanced its interaction with Sp1. We also found that Sp1 could be acetylated, and the acetylated Sp1 could enhance the gene expression of 12(S)-lipoxygenase and p21(CIP1/WAF1). In Sub-project II, we focused our studies on signal transduction of Met/Ron and Eps8 signaling pathways, and found the nuclear translocation of Ron, and the gene regulation of FAK by Eps8 signaling. We found that ER stress could enhance cyclin A expression, resulting in enhancement of hepatoma cell growth. In Sub-project III, we studied the apoptosis signaling and emphasized on the immuno-active cells. We found that p38 signaling played a 2 functional role in Fas-induced T cell apoptosis, and also elucidated the functional role of MEK/ERK/Hsp70 signaling in ceramide-induced T cell apoptosis, and the functional role of GSK-3β activation in ceramide-induced cell apoptosis. And finally, we found that low substratum rigidity attenuated cell migration through ERK signaling activation. We have established the core laboratory facilities at NCKU, in the past three years, which are essential for the success of this project. Deployment of larger instruments such as mass spectrometers, confocal microscope, fluorescence and chemiluminescence analyzers together with biochips allow the researchers involved in this project to compete internationally and pursuit the research excellency. 2. General Description The normal cellular function is under a sophisticated regulation network, so called “signal transduction”, to support the integrity of the system. When cellular growth control is abnormal, for example, the cell continuously grows until a tumor is formed which may damage the neighboring tissue and cause the organism to die. In addition, when a cell should go to apoptosis but does not, its presence may block the function of the neighboring cells and the whole tissue. Thus, to continuously perform normal cellular function, a cell needs to be cooperatively regulated by thousands of signal transduction processes within itself. Furthermore, the signal transmission is dynamic and cross-talk may occur within the cell. Therefore, it is also necessary for scientists to work with cross-talk in the research field of signal transduction. We proposed this project to integrate into a single research team all the intelligent scientists working in this field in southern Taiwan. So far, our team has been involved extensively in signal transduction and gene regulation research and has provided major contributions to the field. Among them only two of the more significant discoveries will be mentioned here. First, in the study of how c-Jun and Sp1 work cooperatively in the activation of 12(S)-lipoxygenase expression, we discovered a novel function of Sp1 as a carrier to bring the transcription factor c-Jun to the GC-rich box-containing gene promoter. This is amongst the first few discoveries of such a novel transcriptional factor function. Second, in the studies of HBV-related hepato-carcinogenesis, we found that the mutated pre-S proteins of the hepatitis B viral surface antigen are commonly present in liver tissues of chronic hepatitis B viral infection, and the pre-S mutants may result in the down-regulation of small HbsAg in endoplasmic reticulum (ER) resulting in ER stress. Through intimate contact and intergration in this project, we will contribute to address, at the molecular level, the tumorigenesis of the most important cancers in Taiwan. Also, we will be able to provide knowledge about the regulation of transcriptional factors in mediating gene 3 expression and signal transduction in growth and apoptosis control. We divided this proposal, into three sub-proposals; (I) functional interaction of transcription factors in gene expression; (II) novel mechanisms of signal transduction of four cancers prevalent in Taiwan; and (III) studies of signal transduction mechanisms that contribute to cell survival. 3. Objectives Specifically, our aims, which were actualized by three subprojects, were to: 1) Elucidate functional interactions of transcription factors in gene expression regulation; 2) Study receptor tyrosine kinases signaling through Stat3/Eps8 in human cancer; and 3) Elucidate novel signal transduction mechanisms that mediate apoptosis or anti-apoptotic effects in patho-biology. 4. Interface and Integration between Overall and Sub-Projects The study of cellular signaling pathways and gene regulation is our main shaft in this project. Instead of looking at individual signaling pathways (single dimensional studies), we conducted our studies from a multi-dimentional prospective. Through the study of “new mechanism”, in search of “new genes”, hopefully we will discover “new functions”. In order to improve the research infrastructure in the NCKU medical research center and form a technical support base for the whole project, we established six core laboratories in Overall project. They are (1) Mass Spectrometry (2) Microscopic Facility (3) Inducible Gene Expression (4) Functional Genomics and (5) Structural Biology. 5. Project Dr. W.C.Chang is responsible for the project management. In order to achieve our goals, the following strategies were reinforced. 1) Integration:There were frequent, informal intra-subproject interactions among the 4 PIs. A formal progress report meeting for each subprojects was held once every 2~3 months. And the progress report for the whole project was held once every 5~6 months. Through the informal and formal meetings, we discussed about the technical help, insight sharing and discussion on possible relationship with their own projects. 2) Quality control:In order to guarantee success and minimize unnecessary waste of efforts, we have invited four distinguished scientists, three from abroad and one local scientist to form an External Advisor Committee to oversee our research progress annually. They will be responsible for critical appraisal of our research directions and results, and give important recommendations. The External Advisor Committee meetings of the third year project is scheduled to be held on Mar. 17, 2006. 6. Describe in detail the approaches and methodologies to implement the research works CORE FACILITY Ⅰ: Proteomics Research Core Laboratory (PRCL) (Responsible Investigator: Pao-Chi Liao) Objective: To provide the following services: (1) Offer general courses such as, “Introduction to Proteomics” and “Laboratory for Proteomics Methods”. (2) Short (3-day) training courses for 2-D gel electrophoresis (2D-GE) (3) 2-D gel electrophoresis (4) Protein MW measurement/confirmation (5) Protein identification by mass specgtrometry (MS) Major instrumentations: (1) Six sets 2-D gel electrophoresis (two sets with multiple-gel capability) (2) Applied Biosystems DE-PRO MALDI-TOF mass spectrometer (3) Finnigan LCQ liquid chromatography-mass spectrometer (LC-MS) (4) Applied Biosystems QSTAR LC-MS with o-MALDI (funded by NSC) Work completed in year 2005: (1) Proteomics Research Core Laboratory (PRCL) has provided the following service to core lab users: 5 服務項目 單 2002年 2003年 位 2004年 2005年 2002-2005.11 (1-11月) 蛋白質二維凝膠電泳自行操作 人 蛋白質二維凝膠電泳委託操作 次 21(21) 68 (34) 88 (78) 蛋白質分子量測定 次 0 25 (16) 2 (0) 蛋白質身份鑑定 次 0 蛋白質二維凝膠電泳課程 次 22 (6) 成大暑期生物技術課程 人 0 20 16 26 52 南部科學園區產學協會生物技術課程 人 0 0 17 21 38 蛋白質二維凝膠電泳軟體分析課程 人 0 34 (13) 20 (5) 0 54 (18) 43 (0) 501 (139) 383 (73) 137 (30) 1064(242) 103 (89) 280 (222) 96 (92) 231 64 (45) 198 (147) (210) 56 (18) 22 (3) 12 (12) 123(108) 493 (402) 112 (39) (2) PRCL has set up a web page (http://proteomics.med.ncku.edu.tw) to facilitate the promotion and operation of the services provided by the core lab. (3) A new service category for the analyses of protein post-translational modifications has been added. (4) A new fee structure has been implemented in 2005, as posted on the core lab web page. The PRCL cut the costs of services and provide reduced rates to NCKU users. Plans for year 2006: (1) Continue to provide services listed above. (2) Enhance the service capibilities in the analyses of protein post-translational modifications through personnel training. (3) Prepare the transition period for the PRCL may not be supported by MOE projects after the projects end in April, 2006. CORE FACILITY II: Time-lapse video microscopy/Biological imaging systems (Responsible investigator: Tzeng-Horng Leu/Meng-Ru Shen) Objective: The main purpose of this core is to provide instrumentation support of (1) time-lapse video microscopy and (2) biological imaging systems for researchers in the MOE Program for Promoting Academic Excellence of Universities. (1) Time-lapse video microscopy: The Leica AS MDW system was set up and started to provide service since the July of 2003. This instrument provides recording of intracellular proteins/organelle translocation as well as long-time observation of cellular movement. In this year, there are 34 times (calculated from Dec. 26, 2004 to Dec. 30, 2005) people have utilized this machine in their research in this year. This has added up total 70 times 6 that have been used since it was established in the July 0f 2003. (2) Biological imaging systems: We have set up a core laboratory of optical imaging with the financial support from MOE Program for Promoting Academic Excellence of Universities and Center for Bioscience and Biotechnology, National Cheng Kung University. This core laboratory is well equipped with (1) a new generation of confocal microscope for live cell imaging system; (2) an atomic force microscope (AFM) coupled with a confocal laser scanning biological microscope; (3) an inverted research microscope coupled with high speed cooling CCD and fluorescence illuminators. The function of the core laboratory is to analyze the dynamic processes in living cells, such as cytoskeleton dynamics, secretory membrane trafficking, cellular interactions, chromatin dynamics, intracellular pH and calcium measurement with simultaneously electrophysilogical recording. Up to date (i.e. December 22, 2005), this core laboratory has already provided 5 times of training course and has serviced 100 hr/month. CORE FACILITY III: Multiple inducible gene expression cell model laboratory (Responsible Investigator: Hsiao-Sheng Liu) Objective: The objectives of this core facility are to assist PIs in each subproject to utilize our inducible systems to regulate the genes of interest. Facility: GenePulser XcellTM (BioRad) is an electroporator, which is extremely powerful for DNA, RNA and protein delivery. Accomplishments and service: For training more people to use this electroporator, we hold a lecture in September, 2005. A total of 63 services have been conducted in 2005. Thirty of the 63 services belong to this project. In this core facility, the following inducible systems are available for use: 1. the lactose repressor system (Lac system); 2. the insect hormone ecdysone-dependent expression system (Ecd system); 3. the tetracycline-dependent expression system (Tet system). Furthermore, a Cre/lox P system has been used to construct Tet inducible system. This system further simplifies the procedure of cloning. At least three papers have been published using this instrument. CORE FACILITY IV: DNA Microarray (Responsible Investigator: Hsiao-Sheng Liu) The basic equipments for running this core facility have been purchased and 7 implemented. Supported by the MOE program, the equipments include a deep freezer for preserving the cDNA clones, two PCR machines, one fluorescence scanner with two laser beams, a server computer and a PC for the implantation of yearly-subscribed microarray analysis software. So far we have a collection of 2,600 human cDNA clones and 228 mosquito cDNA clones in this core facility. Most of the clones selected for further analysis have been re-sequenced and re-annotated during the past year. Currently, the arrayer and the scanner are located at our Medical school (building room 82-1039) (the microarray center). The total numbers of services exceed 69 in 2005. So far, we have produced four versions of chips, and the chip 4 “oncogene and kinase” chip is under active service now. Ras signal pathway chip is our version V chip and starts being used now. Cell cycle and apoptosis chip has been designed, and will be released in the future. To encourage more people to use our chips and facility, we had held three lectures to introduce our chip service in 2005. The detailed information regarding the microarray core facility is on the website http://140.116.247.183/smallfat/website1/index2.htm. We will continue our missions to support every PI whether he is in the MOE program or not, to better use this core facility to increase the academic excellencies in their research domains. CORE FACILITY Ⅴ: Structural Biology Core A: Lab for NMR and Protein Expression (Responsible Investigator: Woei-Jer Chuang) Objective: The main purpose of this core is to provide instrumentation support and service to MOE investigators. The aims of this structural core lab are to: (1) determine the 3D structures of proteins involving this project; (2) produce large quantities of proteins for NMR studies; and (3) model protein structure and analyze chemical and physical properties of proteins involving this project. Facilities and Equipments: Server: Sun Fire 6800 Workstation: SGI Octane Fuel Softwares: SRS7, EMBOSS, Artemis, InsightII, Xplor, CNS, Seqfold, Homology, Modeller, Consensus, and Autodock Fermentors: NBS Celligen (protein production in CHO cell) NBS Bioflo 101 (mass protein production in Pichia pastoris) NBS Bioflo 101 (mass protein production in E. coli) Rhoche ProteoMaster: mass protein production in cell free expression system. 8 The Core insures that these instruments are maintained in good working condition. 2005 Workplan Results: A number of important results have been achieved in the past year. (1) The facility and equipments have been installed and the services were provided starting from November, 2002. (2) Seven papers were published in 2005 by using this core facility. (3) One paper was published and sponsored by this grant. Wang, C.-C., Chen, J.-H., Yin, S.-H., and Chuang, W.-J. (2006) “Predicting the Redox State and Secondary Structure of Cysteine Residues in Proteins Using NMR Chemical Shifts", Proteins, in press. 2006 Workplan and Future Outlook: This work will continue from the activities that have taken place in 2005 in the respective working groups. This core provides instruction on the proper set up and use of the equipment and will advise on the best procedures to accomplish the goal of MOE. B: Laboratory of Combinatorial Chemistry and Peptides Synthesis (Responsible Investigator: Wai-Ming Kan) Objective: The main purpose of this core is to provide instrumentation support and service to MOE investigators. The aims of this structural core lab are to: (1) Peptide and Phosphopeptide Synthesis (2) Combinatorial Peptide/Small Molecule Libraries Preparation -- for the identification of substrate selectivity, kinase recognition sequences, and enzyme inhibitors or receptor ligands. Facilities and Equipments: Organic Synthesizers: (1) EYELA solid phase organic synthesizer CCS-1200V (2) EYELA ChemiStation Model PPW-2000 Workstation: SGI O2+ Softwares: Cerius2 C2•Visualizer; C2•DMol3 Interface; C2•DMol3-Molecular; C2•Dynamics; C2•Minimizer; C2•MOPAC Interface and MOPAC program; C2•Open Force Field The Core insures that these instruments are maintained in good working condition. 9 2005 Results: Preparation of commercially unavailable prostacyclin antagonist, 3-[4-(4,5-dihydro-1H-imidazol-2-ylamino) phenyl]-1-(4- morpholino-phenyl)propan1-one (US6417186B1), and their respective conformational restricted derivatives. These derivatives show increased IP antagonistic activity in comparison with the parent compound. 10 Sub-project (I) Functional interaction of transcription factors in gene expression (Principal Investigator: Wen-Chang Chang) Our previous studies indicate a novel function of Sp1 that could be as an anchor protein to recruit other transcription factors c-Jun, and deacetylation of Sp1 plays a functional role in 12(S)-lipoxygenase gene expression (Fig. 1). In the past year of this Sub-project, we focus our studies on the functional role of post-translational modification of c-Jun, VDR and Sp1 in the transcriptional regulation of cellular genes. In Sub-project I, we found that calcineurin-mediated dephosphorylation of c-Jun C-terminus was required for c-Jun/Sp1 interaction in phorphol ester-induced 12(S)-lipoxygenase gene expression. We also found that Sp1 could be acetylated, and the acetylated Sp1 could enhance the 12(S)-lipoxygenase gene expression. These results indicated that dephosphorylation of c-Jun C-terminus and acetylation of Sp1 play a pivotal role in 12(S)-lipoxygenase gene transcription. In Sub-project Ⅱ, we identified a number of potential genes that could be regulated by VDR/Sp1 complex. For example, vitamin D3 repressed the expression of Skp2, an F box protein that is involved in the degradation of p27Kip1. We also found that dephosphorylation of VDR enhanced its interaction with Sp1. Based on these findings, we focused our studies on the effects of post-translational modifications(acetylation and sumoylation)of Sp1 on its binding with c-Jun and VDR. PMA cytoplasm c-Jun nucleus c-Jun HDAC1 HDAC1 c-Jun Acetyl Sp1 H3 Step 1 p300 c-Jun Sp1 H3 Acetyl Step 2 p300 c-Jun Sp1 H3Acetyl Step 3 Figure 1. A scheme illustrating the mechanistic model of PMA-induced activation of the 12(S)-lipoxygenase transcription (Hung, J. J. et al., Mol. Cell. Biol. 2006) 11 I-1-1: Calcineurin-mediated dephosphorylation of c-Jun C-terminus is required for c-Jun/Sp1 interaction in phorbol ester-induced 12(S)-lipoxygenase gene expression (Ben-Kuen Chen and Wen-Chang Chang) BACKGROUND We previously demonstrated that EGF- and PMA-induced gene expression of 12(S)-lipoxygenase is regulated by the functional interaction between c-Jun and Sp1 (1, 2). These studies also showed that c-Jun, induced by extracellular stimulators such as PMA and EGF through an activation of mitogen-activated protein kinase, can cooperatively interact with Sp1, and that Sp1 can serve, at least in part, as an anchor protein to recruit c-Jun to the promoter, thus transactivating the transcriptional activity of the 12(S)-lipoxygenase gene. Furthermore, we recently found that EGF-induced gene expression of keratin 16 is also regulated by c-Jun/Sp1 interaction (3). Because PMA induces serine/threonine dephosphorylation of c-Jun at the C-terminal domain (4), the functional role of this dephosphorylation in regulating the c-Jun/Sp1 interaction was investigated in this study. METHODS In order to study whether the dephosphorylation of c-Jun C-terminus is required for the interaction between c-Jun and Sp1 in PMA-treated cells, we constructed TAM-67-T231A, TAM-67-S243A, TAM-67-S249A and TAM-67-M3A and studied the effect of TAM mutants on the PMA-induced promoter activity. We then addressed the binding affinity between TAM-67, TAM-67 mutants and Sp1 by immunoprecipitation and GST pull-down assays. In addition, we also identified one kind of phosphatases, PP2B was involved in the regulation of c-Jun/Sp1 interaction by an in vitro phosphorylation and GST protein interaction assay. Furthermore, the functional role of PP2B in PMA-induced interaction between c-Jun and Sp1 was confirmed in PP2B knockdown cells by PP2BsiRNA. To directly examine the contribution of endogenous PP2B to activate the transcription of c-Jun/Sp1-regulated genes, DNA affinity precipitation assay and chromatin immunoprecipitation assay was used. Direct binding of c-Jun to PP2B in culture cells was also studied by using fluorescence resonance energy transfer (FRET) assay. RESULTS Treatment of cells with PMA induced dephosphorylation of c-Jun C-terminus. PMA-induced promoter activity of 12(S)-lipoxygenase was dose-dependently inhibited by a specific calcineurin (PP2B) inhibitor, cyclosporin A. Overexpression of PP2B also enhanced the promoter activity of the 12(S)-lipoxygenase in a 12 dose-dependent manner. Moreover, in PMA-treated cells, cyclosporin A inhibited the interaction between c-Jun and Sp1. Overexpression of TAM-67, an N-terminally truncated c-Jun, inhibited the PMA-induced promoter activity of 12(S)-lipoxygenase. Overexpression of TAM-67-M3A, which contains three substitutive alanines at Thr-231, Ser-243, and Ser-249, was about twice as efficacious as TAM-67 in inhibiting the PMA-induced promoter activity of the 12(S)-lipoxygenase gene. Coimmunoprecipitation experiments indicated that, compared to TAM-67, TAM-67-M3A bound more efficaciously with Sp1. Furthermore, cyclosporin A inhibited the PP2B-induced enhancement of the interaction between TAM-67 and Sp1. Overexpression of PP2B siRNA expression vector pSUPER-PP2B-A in cells caused knockdown of endogenous PP2B and resulted in attenuating the PMA-induced promoter activity and c-Jun/Sp1 interaction, indicating that the PMA response was mediated through the endogenous PP2B. PMA also enhanced the association of c-Jun and PP2B with the promoter of 12(S)-lipoxygenase gene. PMA-induced binding of c-Jun/PP2B to the promoter was inhibited in cells treated with cyclosporine A. Moreover, coimmunoprecipitation experiments indicated that PMA could induce interaction between c-Jun and PP2B. The phospho-TAM-67 was dephosphorylated by PP2B in an in vitro kinase assay. In addition, FRET experiments also indicated that PP2B directly bound to c-Jun in culture cells. Taken together, these results indicated that PP2B plays an important role in regulating c-Jun/Sp1 interaction in PMA-induced gene expression. Specifically, the dephosphorylation of Thr-231, Ser-243, and Ser-249 at the c-Jun C-terminus was essential for the protein-protein interaction between c-Jun and Sp1. The PP2B/c-Jun/Sp1 complex also shows the significant role in the regulation of gene expression. DISCUSSION In this series of study, we found that dephosphorylation of c-Jun C-terminus regulated by PP2B was required for c-Jun/Sp1 interaction in PMA-induced gene expression. In addition to 12(S)-lipoxygenase, recent studies documented that c-Jun/Sp1 interaction is essential for growth factor and/or phorbol ester-induced expression of genes, including mouse neuronal nicotinic acetylcholine receptor 4 (nAChR4), human cytosolic phospholipase A2, p21WAF1/CIP1 and human keratin 16. Functions of these c-Jun/Sp1-regulated genes have been well documented. For example, 12(S)-lipoxygenase plays an important physiological role in regulating apoptosis. nAChRs are important for synaptic transmission in the nervous system. cPLA2 hydrolyzes membrane phospholipids to release arachidonic acid and mediates cell injury. Keratin 16 is the marker of keratinocyte hyperproliferation in psoriasis, and p21WAF1/CIP1 modulates cyclin-dependent kinase activity resulting in cell growth 13 arrest or progression. It is apparently that PP2B also participated in other gene regulation through c-Jun/Sp1 interaction in addition to 12(S)-lipoxygenase. Indeed, EGF-induced promoter activity of K16 gene was inhibited by cyclosporine A in HaCaT cells and overexpression of PP2B in cells also induced promoter activity of K16. Since genes regulated by c-Jun/Sp1 interaction play important cell functions, elucidation of the underlying mechanism of c-Jun/Sp1 interaction could provide pivotal clue to explain how theses genes are regulated in cells. REFERENCES 1. Chen, B. K. and Chang, W. C. (2000) Functional interaction between c-Jun and promoter factor Sp1 in epidermal growth factor-induced gene expression of human 12(S)-lipoxygenase. Proc. Natl. Acad. Sci. USA 97, 10406-10411. 2. Chen, B. K., Tsai, T. Y., Huang, H. S., Chen, L. C., Chang, W. C., and Tsai, S. B. (2002) Functional role of extracellular signal-regulated kinase activation and c-Jun induction in phorbol ester-induced promoter activation of human 12(S)-lipoxygenase gene. J. Biomed. Sci. 9, 156-165. 3. Wang, Y. N. and Chang, W. C. (2003) Induction of disease-associated keratin 16 gene expression by epidermal growth factor is regulated through cooperation of transcription factors Sp1 and c-Jun. J. Biol. Chem. 278, 45848-45857. 4. Boyle, W. J., Smeal, T., Defize, L. H., Angel, P., Woodgett, J. R., Karin, M., and Hunter, T. (1991) Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 64, 573-584. I-1-2: Histone deacetylase 1 recruited by Sp1 positively regulates p21(CIP1/WAF1) expression (Jan-Jong Hung and Wen-Chang Chang) BACKGROUND Two types of enzymes, the histone deacetylases (HDACs) and histone acetyltransferases (HATs), are known to control the acetylation of histones and other proteins, leading to the chromatin remodeling that modulates the transcription activity of numerous genes (1). Many transcription factors have been shown to associate with HDAC1 to regulate the gene transcription. HDAC1 can be recruited to or dissociated from the affected promoter to repress or induce respectively the transcription activity of regulated genes (2). For instance, HDAC1 recruited by Sp1 to the promoter of Sp1-regulated genes can cause histone deacetylation, leading to the Ras-induced down-regulation of the metastasis suppressor RECK (3). In analogy, another study proposed a novel mechanism by which STAT5 mediates the deacetylation of C/EBPbeta, thus allowing the transcriptional activation (4). However, several reports 14 have revealed a positive role for histone deacetylase in cytokine-dependent gene regulation (5). These studies imply that HDACs can have a positive or a negative regulation of different genes. Recently, we found that HDAC1 recruited by Sp1 is required for the induction of 12(S)-lipoxygenase upon PMA treatment (6). These results support a notion that HDAC1 may have a positive role in some gene regulations. METHODS To investigate the role of HDAC1 in gene regulation, HDAC1 was co-transfected with the reporter gene driven by promoter of 12(S)-lipoxygenase or p21(CIP1/WAF1) into the cells, and the luciferase activity was then detected. In addition, the mRNA and protein of 12(S)-lipoxygenase and p21(CIP1/WAF1) were also determined under HDAC1 overexpression. Next, HDAC1 was knockdown by siRNA of HDAC1, and the effect on p21 and 12(S)-lipoxygenase expression was studied. In order to study whether this effect is Sp1-dependent, reporter activity driven by promoter, in which the Sp1-binding sites was mutated, on 12(S)-lipoxygenase and p21(CIP1/WAF1) was determined under HDAC1 knockdown or overexpression. The interaction between Sp1 and p300, and the recruitment of related transcription factors to the promoter of p21 by immunoprecipitation assay and ChIP assay were studied. And the effects of p21 under HDAC1 overexpression on G1arrest, Rb hyperphosphorylation and cell growth were performed. RESULTS In this study, we demonstrated that histone deacetylase 1 recruited by Sp1 to regulate p21(CIP1/WAF1) positively. The level of p21(CIP1/WAF1) was increased under HDAC1 overexpression in various cell lines including p53-dependent and –independent cells. In contrast, the level of p21(CIP1/WAF1) was decreased by knockdown of HDAC1, but not by knockdown of other HDACs. We also found that this effect of HDAC1 on p21(CIP1/WAF1) expression was Sp1-dependent. The increase fold of luciferase activity decreased when the Sp1-binding sites localized in the promoter of p21(CIP1/WAF1) was mutated. In addition, the acetylation of Sp1 was reduced obviously under HDAC1 overexpression, which led to the recruitment of p300 to the promoter to induce the gene expression. Because we have known that deacetylation of Sp1 can induce the transcriptional activity of 12(S)-lipoxygenase (6), herein, we also found that deacetylation of Sp1 could induce the transcriptional activity of p21(CIP1/WAF1) by Sp1 site-directed mutant Sp1(K703/A). We then studied the effect of HDAC1 overexpression on G1arrast and cell growth. Our results indicated that percentage of G1 is increased about 10% under HDAC1 overexpression 15 in HeLa cells, and the cell growth rate is significantly decreased. DISCUSSION Chromatin package level by histones demonstrates the transcription level (7). It is well-known that histones could be acetylated and dissociated from chromatin by charge interaction. Therefore, the chromatin remodel results in increasing the transcription activity of regulated gene(s). In contrast, deacetylated histones enhance the association with chromatin to turn off the gene transcription. Both of the HAT and HDACs control the acetylated level of histones. Recently study has also shown that many protein factors are also regulated by proteins containing HAT activity and HDACs (6). In this study, we found that not all of the genes were regulated by HDACs negatively, but several genes such as p21(CIP1/WAF1) and 12(S)-lipoxygenase were affected by HDAC1 positively through the deacetylation of Sp1 by HDAC1 recruitment. REFERENCES 1. Zhao, Q., Cumming, H., Cerruti, L., Cunningham, J. M. and Jane, S. M. (2004) Site-specific acetylation of the fetal globin activator NF-E4 prevents its ubiquitnation and regulates its interaction with the histone deacetylase, HDAC1. J. Biol. Chem. 279, 41477-41486. 2. Varshochi, R., Halim, F., Sunters, A., Alao, J. P., Madureira, P. A., Hart, S. M., Ali, S., Vigushin, D. M., Coombes, R. C. and Lam, E. W. (2005) ICI182, 780 induces p21Waf1 gene transcription through releasing histone deacetylase 1 and estrogen receptor alpha from Sp1 sites to induce cell cycle arrest in MCF-7 breast cancer cell line. J. Biol. Chem. 280, 3185-3196. 3. Chang, H.C., Liu, L. T. and Hung, W. C. (2004) Involvement of histone deacetylation in ras-induced down-regulation of the metastasis suppressor RECK. Cell. Signal. 16, 675-679. 4. Xu, M., Nie, L., Kim, S. H. and Sun, X. H. (2003) STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J. 22, 893-904. 5. Shankaranarayanan, P., Chaitidis, P., Kuhn, H. and Nigam, S. (2001) Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene.J. Biol. Chem. 276, 42753 6. Hung, J. J., Wang, T. T. and Chang, W. C. (2006) Sp1 deacetylation induced by phorbol ester recruits p300 to activate the 12(S)-lipoxygenase transcription Mol. Cell. Biol. In press. 7. Legube, G. and Trouche, D. (2003) Regulating histone acetyltransferases and 16 deacetylases. EMBO Rep. 4, 944-947. I-2: Molecular mechanism of interaction between vitamin D receptor and Sp1 in gene regulation (Wen-Chun Hung) BACKGROUND Recent studies demonstrate that steroid hormones may activate the expression target genes in which the promoter regions lack steroid receptor response elements. It is possible that steroid hormone/receptor complexes, instead of directly binding to DNA, may interact with transcription factors to modulate gene expression. We have previously demonstrated that vitamin D receptor (VDR) may physically interact with Sp1 transcription factor (1). GST-pull down assay indicated that Sp1 interacted with VDR via its N-terminal (a.a. 1-283) and C-terminal (a.a. 611-778) domain. In addition, our results demonstrate that the VDR/Sp1 complex binds to the Sp1 binding sites corresponding to the sequence –544 to -512 bp in p27Kip1 promoter to trigger gene expression. In this study, we addressed two issues: (1) Whether the VDR/Sp1 complex activates gene expression via the transactivation domain of VDR and (2) Identification of the target genes which expressions are regulated the VDR/Sp1 complex. METHODS We first addressed whether VDR functions as the transcriptional component of the VDR/Sp1 complex to induce expression of target genes. Full-length and AF-2 deletion mutants of VDR were created and were co-transfected with Sp1 expression vector and p27Kip1 promoter-luciferase construct into SL2 cells to test the transactivation ability of the various VDR/Sp1 complexes by using luciferase assay. In addition, we also transfected low VDR-expressing SW620 colon cancer cells with full-length and AF-2 deletion mutants of VDR and studied vitamin D3-stimulated p27Kip1 promoter activity. We next performed DNA affinity precipitation assay (DAPA) to investigate the interaction between VDR or its deletion mutants with some co-activators (including SRC-1, TRAP250 etc) after they bind to the DNA probe corresponding to the region of the p27Kip1 promoter. We used microarray to identify the target genes that may be regulated by the VDR/Sp1 complex. Expression of Sp1 in cells was inhibited by Sp1-specific siRNA. Cells transfected with control or Sp1-specific siRNA were then stimulated with vitamin D3 and mRNA were isolated for analysis by microarray analysis. RESULTS 17 Results of promoter activity assays in SL2 and SW620 cells demonstrated that deletion of AF2 domain of VDR significantly attenuated the transactivation of p27Kip1 by the VDR/Sp1 complex. The interaction of SRC-1 and TRAP205 with VDR was also abolished in after truncation of AF2 domain. These results are accepted for publication (2). Our microarray study identified a number of potential genes that could be regulated by the VDR/Sp1 complex. We found that vitamin D3 repressed the expression of Skp2, an F box protein that was involved in the degradation of p27Kip1. RT-PCR analysis was also performed to verify the results of microarray and our data indeed confirmed that inhibition of Skp2 by vitamin D3. We next cloned the human Skp2 promoter and tested the effect of vitamin D3 on promoter activity. Our data indicated that vitamin D3 inhibited Skp2 via Sp1 binding sites in the promoter. DNA affinity precipitation assay (DAPA) showed that VDR interacted with Sp1 to bind to Skp2 promoter and to repress gene expression. In addition, CHIP assays also confirmed the binding of VDR and Sp1 to the Sp1 sites in human Skp2 promoter. Results of this study are submitted for publication. Taken together, these data support the notion that VDR and Sp1 may interact with each other and the VDR/Sp1 complex may bind to DNA via the Sp1 consensus sequence to regulate gene expression. DISCUSSION We have previously demonstrated that vitamin D3 may stimulate the interaction between VDR and transcription factor Sp1 to activate the expression of a cyclin-dependent inhibitor p27Kip1 and to suppress proliferation of cancer cells (1). In this study, we find that Sp1 functions as a carrier protein to bring VDR to the Sp1 binding site in the p27Kip1 promoter and VDR then recruits the co-activators via its activation domain to stimulate p27Kip1 gene expression. Microarray analysis identifies several potential target genes which expression may be controlled by the VDR/Sp1 complex. In addition, we find that post-translational modification of VDR may affect its interaction with Sp1. Our study provides a new mechanism by which VDR regulates gene expression. REFERENCES 1. Huang YC, Chen JY, and Hung WC. (2004) Vitamin D3/Sp1 complex is required for the induction of p27Kip1 by Vitamin D3. Oncogene 23, 4856-4861. 2. Cheng HT, Chen JY, Huang YC, and Hung WC. (2005) Functional role of VDR in the activation of p27Kip1 by the VDR/Sp1 complex. J. Cell. Biochem. (in press) 3. Huang YC, and Hung WC. (2006) Vitamin D3 transcriptionally represses p45Skp2 expression via Sp1 sites in human prostate cancer cells. (submitted) 18 Sub-project (II) Studies of receptor tyrosine kinases signaling through Stat3/Eps8 in human cancers (Principal Investigators: Ih-Jen Su and Tzeng-Horng Leu) Within this year, as previously, each subproject has performed experiments to see whether Stat3 and/or Eps8 are involved in the tumorigenesis of bladder cancer, colorectal cancer, lung malignant plural effusion, and HBV-associated liver cancer. In the subcomponent I, Drs. Chow and Liu have continued to pursue the mechanism involved in the nucleus localization of Ron in bladder cancer cell line TSGH8301. They have observed importin 1, 1 and de-phosphorylated EGFR are all involved in RON nucleus localization. Furthermore, the potential NLS sequences in RON were demonstrated and indicated to be important for cell proliferation and antiapoptosis. Furthermore, nuclear RON/EGFR complex was demonstrated to bind to AP1-binding site of the promoter by DAPA assay indicating that nuclear RON could function as a transcription factor. In the second subcomponent, Dr. Leu has demonstrated the importance of overexpression of p97Eps8 in regulating cell growth and motility of human colorectal cancer cells. Furthermore, he has demonstrated this is probably through upregulation of the expression and activity of FAK, but not Rac. However, activation of Stat3 was not associated with Eps8 expression in colon cancer cells indicating Eps8-independent pathway is required for Stat3 activation in colon cancer cells. In the third subcomponent, Drs. Su and Lai have observed activation of Stat3 by paclitaxel could be through ROS generation followed by Jak2 activation in PC14PE6/AS2 cells. Furthermore, PKC-kinase activity is also required for both baseline and paclitaxel-induced Stat3 tyrosine phosphorylation. In addition, by microarray analysis of gene expression in lung adenocarcinoma, they have identified ER- expression is associated with active Stat3. These results were further confirmed by utilizing PC14PE6/AS2 cells transfected with either dominant-active Stat3 (S3C) or dominant-negative Stat3 (S3D and S3F). In the fourth subcomponent, Dr. Su Ih-Jen observed cyclin A induction in HBV preS2 overexpressing cells and in ground glass hepatocytes (GGHs). They observed retinoblastoma (RB) hyperphosphorylation occurs in cells with S2-LHBs. This is probably through S2-LHBs interacting with Jun activation domain-binding protein 1 (JAB1), dissociating JAB1 from the JAB1/IRE1 complex, and JAB1 nuclear translocation and targeting p27Kip1 for degradation. Furthermore, they identify ER stress-induced activation of calpain is responsible for the formation of cyclin A truncated form, which contributes to cytosolic cyclin A accumulation and centrosome overduplication in hepatoma cells. 19 II-1: The novel mechanisms of tumor stroma and cellular oncogenes in modulation of bladder carcinogenesis (Nan-Haw Chow and Hsiao-Sheng Liu) BACKGROUND This study was designed to explore the molecular mechanisms underlying nuclear translocalization of RON receptor, a distinct receptor tyrosine kinase of c-met receptor family, in human bladder cancer. Our cohort study revealed that over-expression of RON is positively related to cancer progression, and co-expression of RON/Met is an important poor prognostic indicator, supporting that RON-associated signaling plays an important role in the progression of bladder cancer (1). A similar conclusion was also observed in human breast cancer (2). As for biological significance, over-expression and increased phosphorylation of Stat-3 is involved in RON-associated signaling. Moreover, MSP induces the phosphorylation of RON and enhances the migration, proliferation and anti-apoptosis of cancer cells in vitro. Intriguingly, de-phosphorylated RON was also detected in the nuclear fraction of cancer cells, and the aberrant nuclear localization of RON was ligand-independent. METHODS To verify the ligand-independent nuclear localization of RON, both immunoblotting and immunofluorescence assay were performed on TSGH8301 cells together with function-blocking assay. Then, immunoprecipitation was chosen to analyze the potential factor(s) involved in translocalization of RON to the nucleus, and RNAi technology was utilized for confirmation. Site-directed mutagenesis assay was carried out on N- and/or C-terminal nuclear localization signals (NLSs) of RON to clarify the involvement of NLS in the nuclear translocalization. The cell proliferation status was measured by cell counting and XTT assay. The anti-apoptosis effect was assessed by MTT assay after treatment with methotrexate (MTX). Finally, DNA affinity precipitation assay (DAPA) was used for examination the role of RON as transcriptional factor. RESULTS We demonstrated that nuclear RON was co-localized with importin 1, 1 and de-phosphorylated EGFR. However, MSP treatment results in EGFR phosphorylation followed by dissociation from RON in the nucleus, implying a cross-talk between RON and EGFR (3). The siRNA experiments confirmed the importance of EGFR and importin 1 on nuclear translocalization of RON. Then site-directed mutagenesis assay revealed that both basic clusters of NLS in RON are important for nuclear 20 translocalization, suggesting the involvement of NLS in the phenomenon. Moreover, both proliferation and anti-apoptotic effects were suppressed in NLS mutants compared with wild-type transfectants, suggesting the importance of nuclear RON in cell proliferation and anti-apoptosis. Given that heat shock proteins (HSPs) were reported to be the chaperones in regulating stress-induced signaling and nuclear transport of cell cycle kinases, expression of HSPs was analyzed by immunoblotting on subcellular fractions. Levels of HSP70, HSP90 and heat shock factor 1 (HSF 1) were all increased in the cell nuclei. The DAPA revealed that nuclear RON/EGFR complex binds to AP1-binding site of the promoter, suggestive of nuclear RON as a transcription factor. DISCUSSION On the basis of findings described above, we hypothesize that RON receptor transduces two distinct signaling pathways. One is MSP-dependent activation of ERK/Elk and PI3K/Akt signaling pathways. The other is ligand-independent pathway, in which full-length of RON receptor co-localizes with EGFR, importin 1 and 1 into the nucleus. MSP seems to impose a higher affinity to membrane RON receptor with associated dissociation of RON from EGFR in the nucleus. The nuclear RON/EGFR complex appears to bind to AP1 or SP1 binding site in modulation of gene expression. Whether these pathways are redundant or divergent in controlling of biological activities remains to be elucidated. Given that co-expression of RON/Met also plays a role in the metastatic progression of bladder and breast cancers (1, 2), interaction between EGFR and RON may play an important role in the progression of epithelial carcinogenesis. As a result, key issues under intensive investigation include the mechanism underlying nuclear translocation of RON/EGFR complex, the roles of RON/EGFR in transcriptional regulation, and its biological implication in human bladder carcinogenesis. FUTURE WORK 1. Clarify the potential role of nuclear RON as a transcription factor 2. Define the biological significance and the related signaling events of nuclear RON 21 Current hypothetical model of nuclear localization of RON in human bladder cancer cells REFERENCES 1. Cheng, H. L., Liu, H. S., Chen, Helen H. W., Lin, Y. J., Chang, T. Y., Tzai, T. S., Ho, C. L., Hsu, P. Y., Chow, N. H. Co-expression of RON and MET as a prognostic indicator for patients with transitional cell carcinoma of the bladder. Br. J. Cancer 92: 1906-1914 ( 2005). 2. Lee, W. Y., Chen, Helen H. W., Chow, N. H., Su, W. C., Lin, P. W., Guo, H. R. Co-expression of RON and MET receptors in patients with lymph node-negative breast cancer: Synergistic effect in metastasis. Clin Cancer Res 11: 2222-2228 (2005). 3. Hsu, P. Y., Liu, H. S., Chow, N. H. Cross-talk between RON receptor and epidermal growth factor receptor in human epithelial carcinogenesis. Proceedings of the American Association for Cancer Research, 46: 397 (2005). II-2: Aberrant expression of Eps8 in human colorectal tumors (Tzeng-Horng Leu) BACKGROUND Our previous studies have indicated that Src, and FAK are parallel overexpressed in human colorectal tumors (1). Furthermore, p97Eps8 overexpression could transform murine fibroblast (2). Interestingly, human colorectal cancer cell SW620 exhibits higher expression of both Eps8 and FAK and they grow faster than SW480 cell. To address the importance of p97Eps8 in the formation of colorectal cancer, we have 22 generated eps8 siRNA-overexpressing cells from SW620 and WiDr cells. RESULTS AND DISCUSSION First, we observed a correlation between p97Eps8 reduction and their decreased ability of cell proliferation. Similar results were observed in another high Eps8 expression colorectal cancer cell line WiDr. Accompanying with Eps8 knock down in these cell lines is the reduction of FAK expression. We hypothesized that Eps8 might modulate FAK expression in colorectal cancer cells. Indeed, overexpression of p97Eps8 in low Eps8-expressing SW480 cells results in the elevation of FAK expression. Next, we want to know the role of FAK expression in Eps8-mediated cell proliferation. To address this question, dominant negative FAK (i.e. FRNK)-expressing construct was transfected into SW620 or WiDr cells and the potential of BrdU incorporation of these cells was measured and compared. We observed overxpression of FRNK could abrogate BrdU incorporation in both SW620 and WiDr cells. To substantiate the importance of FAK expression in cell growth of colorectal cancer cells, ectopical overexpression of either mouse Myc-p97Eps8 or Myc-FAK could rescue the growth suppression exerted by the overexpression of eps8 siRNA in SW620 cells. P97Eps8 overexpression not only elevates FAK expression but also increases Src-mediated FAK phosphorylation on Tyr-397 and -861. Furthermore, phosphorylation of Src-Tyr-416 and Shc-Tyr-317 occurs in Eps8-dependent manner and may explain why overexpression of Eps8 could potentiate serum-induced ERK activation in SW620 cells. Albeit, Src activation depends on Eps8 overexpression, activation of Stat3 was not observed in a similar manner. Eps8 overexpression in SW620 and WiDr is also required for its motility. Overexpression of eps8 siRNA decreased cell migration of these cells in wound healing assay. Albeit Eps8 has been demonstrated to participate in Ras-mediated Rac activation, no correlation of Eps8 expression and Rac activation was observed in these cells indicated that Rac pathway was not involve in Eps8-mediated cell movement of SW620 and WiDr cells. Instead, our observation of Eps8-mediated Src and FAK activation in these cells may provide an explanation for Eps8-regulated cell motility in these cells. Further experiments are required to answer this issue. Since Eps8 can promote colon epithelial cells to be tumorigenic, its expression and implication in human colorectal tumors are attempted. A total number of 76 tumor specimens derived from primary colon carcinomas along with their normal counterparts were analyzed by Western immunoblotting. Statistical analysis of fold induction of p97Eps8 in advantage stage (i.e. Duke Stages C and D; n= 37) is significantly greater than that in early stage (i.e. Duke Stages A and B; n=39) by Mann-Whitney test (3.1+2.0 vs. 2.3+1.7; P=0.024). Interestingly, the levels of Eps8 23 were positively correlated to the levels of FAK (r = 0.762, P < 0.001) in tumor specimen. Thus, both in SW480, SW620, and WiDr cell lines and in human colon tumor specimen all indicated that Eps8 might regulate FAK expression. Examining Eps8 and FAK expression of several other colorectal cancer cell lines including LS174T, HT29, and HCT 116 further supported the fact of FAK expression regulated by Eps8. Thus, it is possible that there is a common pathway regulating Eps8 and FAK expression in colorectal cancer cells. In summary, we have identified Eps8 protein as a protein regulating FAK expression and activity in human colon cancer cells, which are important for cell growth and motility. Identification of the pathway that regulates Eps8 expression may highlight a potential alternative method to cure colon cancer in the future. We will continue to search molecules that regulate Eps8 expression in colon cancer cells. REFERENCES 1. Lee, J.-C., M.-C. Maa, H.-S. Yu, J.-H. Wang, C.-K. Yen, S-T. Wang, Y.-J. Chen, Y. Liu, Y.-T. Jin, and T.-H. Leu. Butyrate regulates the expression of c-Src and focal adhesion kinase and inhibits cell invasion of human colon cancer cells. Mol. Carcinog. 43, 207-214. (2005) 2. Maa, M.-C., C.-Y. Hsieh, and T.-H. Leu. Overexpression of p97Eps8 leads to cellular transformation: implication of pleckstrin homology domain in p97Eps8-mediated ERK activation. Oncogene 20, 106-112. (2001) II-3: Study of the pathogenesis of malignant pleural effusion associated lung adenocarcinoma in Taiwan and Stat3 as a model gene (Wu-Chou Su and Ming-Derg Lai) BACKGROUND 1. Stat3, one of the 7 known STAT (Signal Transducers and Activators of Transcription) family members, is frequently constitutively activated in malignant 24 cells1. Activation of Stat3 is involved in regulating many genes such as cell cycle progression, cellular proliferation and survival. In our system, constitutively activated Stat3 in human lung adenocarcinoma cell line --PC14PE6/AS2, mediated by IL6/gp130/Jak2 pathway, was demonstrated. Upon treatment with anti-cancer agents, the activation of Stat3 in PC14PE6/AS2 cells was enhanced at earlier period (around 3 hours), and then declined gradually. We therefore are interested the underlying mechsnisms. 2. Lung cancer with its high prevalence, recurrent rate and metastatic potential becomes the leading cause of cancer death in Taiwan. High prevalence of female lung adenocarcinoma is a hall marker of lung cancer in Taiwan. Estrogen status is a recognized factor in lung-cancer risk in women. Estrogens could act as lung-tumor promoters, through a receptor-mediated mechanism. Using Affimatrix oligonucleotide array experiments to study genes that differentially expressed in PC14PE6/AS2 and PC14P6/AS2/dnStat3 cells, we found that ER- expression was higher (4.7 folds) in Stat3 active cells. We further identified a Stat3 binding site in the promoter of ER- [AGT GAG TTC GAG GAA GTA CCT GGG ATC TTT (-550)]. Whether ER-is regulated by Stat3 was studied. METHODS RT-PCR was used to evaluate changes of mRNA. Activation of Stat3 and expression of other proteins were analyzed by Western blot. Cell survivals after treatment with drugs were measured by MTT colorimetric assay. Affymatrix oligonucleotide microarray was used to study the gene expression profile of neoplastic cells in MPE. The intracellular ROS generation was measured by DCFH-DA fluorescent assay. RESULTS 1. The further activation of Stat3 in PC14PE6/AS2 cells by paclitaxel could be abolished by adding Jak2 inhibitor – AG490 – indicates that Jak2 mediates the reaction. For the mechanisms underlying the activation of Jak2 by anticancer agents, we suspect the induction of ROS and subsequent deactivation of tyrosine phosphatase may play a role. Using the redox-sensitive fluorescence probe DCFH-DA, we found that anti-cancer agent, paclitaxel, stimulates ROS production in PC14PE6/AS2 cells. Generation of ROS was suppressed by diphenylene iodonium (DPI), an inhibitor of flavoprotein-dependent oxidases, Rotenone, an inhibitor of mitochondrial respiratory chain complex I, and Antimycin A, an inhibitor of mitochondrial respiratory chain complex III. The activation of Stat3 in PC14PE6/AS2 cells by paclitaxel is also inhibited by the pretreatment of the above three compounds. Other inhibitors of NAC 25 or catalase did not abolish Stat3 activation by paclitaxel. Taken together, paclitaxel induces ROS generation is likely through a mitochondrial-mediated pathway. In the other part, we suspect that generation of ROS by paclitaxel may transiently inactivate PTP-1B and then switch the equilibrium towards autophosphorylation of Jak kinases. Although this phenomenon was not observed, we found another pathway may partially involved STAT3 activation induced by paclitaxel. H2O2-induced activation of PKC- is reported to be independent from tyrosine phosphatase inhibition. Inhibition of PKC- - kinase activity is required for both baseline and paclitaxel-induced STAT3 tyrosine phosphorylation. The paclitaxel-induced STAT3 activation resulted in upregulation of Bcl-2 protein, which may contribute to the relatively resistant to paclitaxel in PC14PE6/AS2 than in A549 cells. 2. Our preliminary data showed that the ER- protein is expressed in the majority of lung cancer cell lines. The mRNA and protein levels of ER- increased in PC14PE6/AS2 cells transfected with dominant-active Stat3 (S3C) but declined when transfected with dominant-negative Stat3 (S3D and S3F). The ER- protein was also induced in PC14PE6/AS2 cells at 24 hours after treatment with IL-6 but decreased when Stat3 activation was inhibited by AG490. In other human lung cancer cell lineH661, ER- protein expression was also induced by IL-6 treatment. The preliminary data suggested that ER- might be a Stat3 targeting gene. DISCUSSION 1. Paclitaxel enhance STAT3 activation in PC14PE6/AS2 cells. The reaction is mediated mainly by mitochondria-respiratory-chain-generated ROS. Paclitaxel-induced STAT3 activation may be partially mediated by PKC-kinase but not by deactivation of PTP-1B. Paclitaxel-induced Stat3 activation induces cellular expression of Bcl-2 and contributes to cellular survival and cellular context may determine whether Stat3 be activated by anti-cancer drugs or not. Since STAT3 is an important target for cancer therapy, a comprehensive understanding of the interaction between STAT3 activation and anti-cancer drugs will provide better treatment of malignant diseases. 2. In lung tissue, ER-, but not ER-, plays the major role in regulating physiological and pathological functions. Therefore lung epithelial cells may response differentially from mammary epithelial cells to estrogen stimulation. For example, E-cadherin is reported to be a direct transcriptional target of ER. In MCF-7 breast cancer cells, ER binds directly to the E-cadherin promoter, recruits corepressors, and suppresses E-cadherin expression [50]. In contrast, in vivo administration of E2 increases E-cadherin transcreption in the mouse ovary [51] as it does in lung cancer 26 cells in vitro [20]. It is possible that the effect of estradiol exposure on E-cadherin expression depends on which ER subtype predominates in a given tissue, and which of the coactivators and corepressors are expressed in that tissue. Whether activated Stat3 plays a role in modulating ER performance is intriguing and warrant further studies. FUTURE WORK We plan to study how Stat3 regulate TF (tissue factor) and ER and their interactions in the next year. We will also study why lung metastases decrease in PC14PE6/AS2 cells transfected with plasmid containing dominant-negative Stat3. Actually, NSCLC cell lines had diverse expression of STAT3 tyrosine phosphorylation both in baseline and in response to paclitaxel. We would like to explore underlying mechanisms further. We plan to study how Stat3 regulate TF (tissue factor) and ER and their interactions in the next year. We will also study why lung metastases decrease in PC14PE6/AS2 cells transfected with plasmid containing dominant-negative Stat3. Actually, NSCLC cell lines had diverse expression of STAT3 tyrosine phosphorylation both in baseline and in response to paclitaxel. We would like to explore underlying mechanisms further. II-4: The biological role of ER stress in HBV pre-S2 mutant-mediated hepatocarcinogenesis (Ih-Jen Su) BACKGROUND During chronic hepatitis B virus (HBV) infection, the pre-S mutants have been linked to hepatocellular carcinoma, especially the pre-S2 mutants1. In past few years, we have demonstrated that pre-S2 mutant protein (S2-LHBs) could serve as an unfolded protein which accumulates in endoplasmic reticulum (ER) and thus induces ER stress in hepatocytes1. ER stress signal induced by S2-LHBs may induce COX-2 expression2 and enhance oxidative stress which leads to genomic instability3. Furthermore, the upregulation of cyclin A was demonstrated both in vitro and in vivo suggesting the potential role of S2-LHBs in cell cycle regulation4. This year, we focus on the regulation of cyclin A in both gene regulation and protein modification by S2-LHBs-induced ER stress. RESULTS AND DISCUSSION (I) For the study of gene regulation on cyclin A, we firstly test the status of retinoblastoma (RB) phosphorylation in cells with S2-LHBs. By immunoblotting, the induction of RB hyperphosphorylation is demonstrated in HuH-7 cells expressing 27 S2-LHBs and transgenic mice livers. Furthermore, our studies reveals that S2-LHBs may interacted with Jun activation domain-binding protein 1 (JAB1), dissociating JAB1 from the JAB1/IRE1 complex in endoplasmic reticulum, and thus resulted in JAB1 nuclear translocation and target p27Kip1 for degradation. The degradation of p27Kip1 may be responsible for activations of cyclin A/Cdk2 and cyclin E/Cdk2 complexes, which subsequently lead to RB hyperphosphorylation. In addition, the induction of RB hyperphosphorylation can be blocked by ER stress inhibitors, indicating the essential role of ER stress in this process5. (II) Previously, we have frequently observed aberrant cytoplasmic cyclin A in hepatocytes expressing S2-LHBs4. Here, we show that the induction of cytoplasmic cyclin A can be induced by ER stress and S2-LHBs protein. The induction of cytoplasmic cyclin A is due to N-terminal cleavage by calpain, which is calcium dependent, and provided by ER stress. Furthermore, we find that the resulted N-terminal truncated cyclin A interacts with Cdk2 in the cytoplasm and thus contribute to centrosome overduplication and nuclear abnormalities6. In conclusion, the interaction with JAB1 and the induction of cytoplasmic cyclin A/centrosome overduplication provide us a new concept of ER stress in HBV-related hepatocarcinogenesis. In this respect, HBV pre-S2 mutant proteins may be used as a model to study the contributing role of ER stress in tumorigenesis, especially in solid tumors. The understanding of ER stress-mediated tumorigenesis may provide new therapeutic targets and shed some light on their biological implications. REFERENCES 28 1. 2. 3. 4. 5. 6. Wang, H. C., Wu, H. C., Chen, C. F., Fausto, N., Lei, H. Y., and Su, I. J. (2003) Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am J Pathol 163, 2441-2449. Hung, J. H., Su, I. J., Lei, H. Y., Wang, H. C., Lin, W. C., Chang, W. T., Huang, W., Chang, W. C., Chang, Y. S., Chen, C. C., and Lai, M. D. (2004) Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem 279, 46384-46392. Hsieh, Y. H., Su, I. J., Wang, H. C., Chang, W. W., Lei, H. Y., Lai, M. D., Chang, W. T., and Huang, W. (2004) Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 25, 2023-2032. Wang, H. C., Chang, W. T., Chang, W. W., Wu, H. C., Huang, W., Lei, H. Y., Lai, M. D., Fausto, N., and Su, I.J. (2005) Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 41, 761-770. Hsieh, Y. H. et al. Pre-S2 hepatitis B virus surface antigen mutant induces retinoblastoma hyperphosphorylation through interaction with JAB1 protein in endoplasmic reticulum: a novel mechanism for HBV-induced hepatocellular carcinoma. In submission. Wang, H. C., Lai, M. D., Huang, W., and Su, I. J. ER stress induces cytoplasmic cyclin A and centrosome overduplication. In submission. 29 Sub-project (III) Novel signal transduction mechanisms that mediate anti-apoptotic effects in patho-biology (Principal Investigator: Ming-Jer Tang) Apoptosis and anti-apoptosis have become important issues for modern biomedical science. Mechanisms that trigger apoptosis or anti-apoptosis in cell play very important roles in morphogenesis during development or in patho-physiological conditions, such as carcinogenesis or regeneration of specific organ. Apoptosis signals may come from outside of the cell, work on cell membrane receptor, trigger intracellular death machinery and finally degrade the integrity of cell structure. In this subproject, we have collaboratively made some progress in studying signaling mechanisms regarding to interactions of Fas/Fas-L, substratum rigidity-controlled cell behaviors, such as regulation of integrin activation, FAK phosphorylation, MAPK and cell migration, and counteractions of lithium to ceramide-induced cell death. Dr. B. C. Yang demonstrates that Fas cross-linking could directly activate p38 in T cells in a caspase 8/3-dependent way and p38 MAPK pathway is an auto feed-back switch in Fas signaling that dampens the death event in T cells. Collagen gel via the physical property induced down-regulation of focal adhesion complex proteins in all cells examined, which is mediated by 21 integrin. Dr. M. J. Tang and C. Y. Chou demonstrate that low rigidity of collagen fibrils suppresses activation of 1 integrin as well as FAK397 phosphorylation. On the other hand, low substratum rigidity triggers activation of ERK1/2, which results in augmented cell migration. Finally, Dr. Y. S. Lin works on the novel mechanism by which lithium serves to counteract ceramide-induced apoptosis in immune cells and shows that lithium confers protection from ceramide-induced apoptosis via activation of MEK/ERK/Hsp70 and inhibition of mitochondrial activation. Lithium promotes cell survival by inhibiting PP2A activity and caspase-2 activation. Furthermore, GSK-3 is required in ceramide-induced mitochondrial apoptosis. Roles that lithium may play in PP2A-mediated Bcl-2 dysfunction and initiator caspases activation will be further investigated. Results of our studies should have impact in cancer biology, immunology, and developmental biology. III-1: Suppression of the Fas-mediated death signal in T cells upon contact with tumors (Bei-Chang Yang) BACKGROUND Fas/Fas-L system is one of the major apoptosis-mediating signals and plays an important role in various immune functions. Previously, we have shown a tissue-dependant effect of Fas-L on tumorigenecity (1, 2). Accumulating data reveal that extracellular matrix (ECM), constituting mainly the tissue/tumor 30 microenvironments, can directly initiate a variety of signals. For instance, integrin binding activates the MAPK/ERK pathway in T lymphocytes (3, 4). We hypothesize that ECM also contribute to the immune responses against tumor. In the first part of this project, we evaluate the Fas signaling in the presence of tumor/ECM. Specifically, we try to answer how contact with tumor blocks the Fas-mediated death in T cells. Second part of this project is on novel Fas signaling pathway. Some Fas-expressing cells do not undergo cell death but proliferation and differentiation upon Fas stimulation. In agreement with those findings, we have found that Fas signaling stimulated IL-10 expression in T cells through a PKA-independent pathway (5). Comparing to the Fas-mediated apoptotic pathway through caspase activation, how Fas signal activates kinase activities and what is its biological significance are still poorly understood. We evaluate how Fas signal activate p38 pathway in T cells and its association with autoimmunity. In addition, if components of Fas signaling pathway involve in tumorigenesis, specifically the caspase 3 are evaluated. Caspase-3 has been suggested in positive correlation with tumor malignance in several breast carcinomas and melanoma (6). We try to explore the mechanism for the caspases -associated tumor metastasis. STUDY DESIGN AND METHODS Fas cross-linking is done by CH-11 antibody or recombinant FasL. Fas signal induced gene expression in Jurkat T cells is done by microarray screening and two-dimensional gel/silver staining. Possible non-apoptotic bioactivity of caspase-3 is explored either by over-expression and siRNA gene knock-down method. To study the relevance of ECM signaling pathway in shaping T cell responses, two complementary strategies will be undertaken. First, integrin/ECM-associated signal in T cells will be determined. Mechanism for apoptosis-suppression in T cells upon contact with tumor cells/or in the presence of distinct ECM will be analyzed in conventional culture system. Second, we try to establish 3-dimentional cell culture system, e.g. cell spheroids to mimic tumor microenvironment in vivo. RESULTS AND DISCUSSION Part I: In study on signaling in the presence of tumor/ECM, we demonstrated that the cell contact-induced reduction in the Fas-cross linking-induced apoptosis in T cells is tumor cell line-dependant. Decreases in the characteristics of apoptosis including caspase-9, and -3 activation, loss of plasma asymmetry, cell shrinkage, and loss of mitochondria membrane potential were observed in Jurkat cells upon coculture with glioma cells, which have the ability to suppress apoptosis (manuscript in preparation). Recent studies show that extracellular matrix (ECM) can regulated 31 kinase/mitogen-activated protein kinase (ERK/MAPK), which will modulate the trafficking of immune cells by activating integrin signaling in them. In following up the dynamic change of ERK activity, we found that direct cell-cell contact with MCF-7, a breast tumor cell line, or with U118MG, a glioblastoma cell line, induced a quick and transient phosphorylation of ERK in Jurkat T cells. However, prolonged cell contact reversed the elevated ERK phosphorylation in Jurkat cells in a protein kinase A (PKA)-dependant way. Furthermore, prolonged contact also enhanced the activity of ERK-specific phosphatases. We concluded that T cell chemokines, such as RENTES, will activate RAF/MEK/ERK in T cells. However, prolonged integrin activation by ECM engagement increased cAMP level and activated PKA, which subsequently phosphorylated the Raf at 259 a.a. site leading to inhibition of Raf activity and shut down the MEK/ERK. These findings suggest that anchorage on ECM of tumor can dynamically regulate T cell recruitment. Part II: In search of death-regulating kinases, we found that Fas signaling intrinsically activated p38 MAPK pathway by itself to suppress caspase-cascade, which in turn reduced cell death. Inactivation of caspase 8 by Z-IETD eliminated the phosphorylation of p38MAPK in response to Fas treatment. The expression of pp38, reflecting the activity of p38 kinase, was higher in synovial T cells from Rheumatoid arthritis (RA) as compared with that in T cells isolated from normal peripheral blood. Moreover, synovial fluid stimulated the p38 in T cells implicate a role of this kinase in RA (manuscript in preparation). In this study, we further examined whether caspase-3 may stimulate tumor metastasis. When caspase-3 was introduced into the MCF-7 breast cancer cell line, which was originally caspase-3 deficient, cell motility and lung metastasis were significantly enhanced. In parallel, the caspase-3 expression of A549 lung carcinoma cells was reduced by RNA interference (RNAi) strategy. MCF-7 cells overexpressing caspase-3 did not show significant traits of apoptosis. Overexpression of caspase-3 in MCF-7 cells reduced the expression of paxillin while the cells remained viable. In addition, the caspase-3 overexpressing MCF-7 cells possessed higher motility and secreted more MMP-2. On the contrary, transfection with caspase-3 siRNA reduced motility and invasiveness of A549 cells. In addition, by using the in vivo experimental lung metastasis model, we observed that caspase-3 increased the severity of metastasis. In both MCF-7 and A549 cells, we observed a caspase-3 mediated extracellular signal-regulated kinase (ERK) activation, which is independent of caspase-3 activity. In addition, Erk phosphorylation is dominant in caspase-3 mediated cell migration enhancement. Inhibition of Erk activation strongly decreased cell motility. Currently, we try to constructe caspase 3 mutants with deletion spanning different domains. Characterization of those mutants will help us to answer how caspase 3 involves in tumor metastasis. 32 REFERENCES 1. Chen YL, Wang JY, Chen SH, and Yang BC (2002) Granulocytes mediates the Fas-L-associated apoptosis during lung metastasis of melanoma that determines the metastatic behavior. Br. J. Cancer 87, 359-365. 2. Chen YL, Chen SH, Wang JY, and Yang BC (2003) FasL on tumor cells mediates inactivation of neutrophils. J Immunol 171, 1183-1192. 3. Gendron S, Couture J, and Aoudjit F (2003) Integrin alpha2/beta1 inhibits Fas-mediated apoptosis in T lymphocytes by protein phosphatease 2A-dependent activation of the MAPK/ERK pathway. J Biol Chem 278, 48633-48643. 4. del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RGW, and Schwartz MA (2004) Integrins Regulate Rac Targeting by Internalization of Membrane Domains. Science 303, 839-842. 5. Yang BC, Hor WS, Lin HK, Hwang JY, Lin YP, Liu MY, and Wang YJ (2003) Stimulation of IL-10 expression in T cell lines upon contact with human glioma cells, that is mediated by Fas signaling through a PKA-independent pathway. J Immunol 171, 3947-3954. 6. Woenckhaus C, Giebel J, Failing K, Fenic I, Dittberner T, and Poetsch M. (2003) Expression of AP-2α, c-kit, and cleaved caspase-6 and -3 in nevi and malignant melanomas of the skin. A possible role for caspases in melanoma progression? J. Pathol. 201, 278-287. III-2: Signaling mechanisms of low rigidity-regulated protein synthesis, focal adhesion and cell migration (Ming-Jer Tang and Cheng-yang Chou) BACKGROUND Physical environment has been considered as an important factor in regulating cellular behavior. How mechanical impacts affect cell behaviors is a less studied issue. We have shown that collagen gel, as a soft substratum, down-regulates the level of focal adhesion complex proteins through 21 integrin (1). In this study, we wish to explore cell responses to low rigidity, particularly to examine whether the substratum rigidity of collagen gel affects protein synthesis rates and cell migration, and to delineate the signal transduction mechanisms involved in low rigidity-induced cell responses. METHODS We first investigated the mechano-sensing mechanisms whereby cells utilize to detect the rigidity of collagen fibrils. MDCK cells were cultured under different dimensional conditions (collagen gel, collagen gel-coated dish and dish) to investigate how rigidity of collagen fiber affected the cellular physiology. We checked the level 33 of 1 integrin activation, FAK phosphorylation and activation of MAPK signaling pathways. To elucidate whether the internal force provided from actin filaments and microtubules affected 1 integrin activation and FAK397 phosphorylation, we employed cytochalasin D and colcemide. In addition, wild type and dominant negative FAK or DDR1 stably transfected MDCK cells were employed (5, 6). Furthermore, we examined whether the membrane microdomain provided signal leading to 1 integrin activation by fractionation and MCD, a lipid raft inhibitor. RESULTS AND DISCUSSION When MDCK cells were cultured on collagen gel, phosphorylation sites (407, 577, 861, and 925) of FAK were activated, only FAK397 phosphorylation levels remained low or unaltered. Low rigidity-induced decrease in FAK397 phosphorylation could be observed in various cell lines including fibroblasts and transformed cells. In addition, 1 integrin activation was down-regulated under low substratum rigidity. Low rigidity-induced down-regulation of 1 integrin activation or FAK397 phosphorlyation was not mediated through FAK or DDR1. Disruption of actin cytoskeleton by cytochalasin D blocked FAK397 phosphorylation but not 1 integrin activation, while disruption of microtubules by colcemide had no effect. MCD, a lipid raft inhibitor, inhibited 1 integrin activation in cells cultured on rigid substratum. However, the level of FAK397 phosphorylation remained unaffected. Taken together, our data provides a new concept that FAK397 phosphorylation needs internal force provided from actin filaments, but 1 integrin activation requires preferentially external force from rigid substratum and lipid raft may regulate this process (2). We further showed that low rigidity of collagen gel triggered cell migration in all cell lines examined. Low rigidity-induced cell migration was mediated by a delayed onset of Erk-1/2 phosphorylation which was localized on focal adhesions (3). Collagen gel induced phosphorylation of ERK1/2 within 1 h and the induction could last for more than 8 h. Inhibition of collagen-induced ERK1/2 phosphorylation by MEK inhibitor, UO126, resulted in round up morphology and completely abolished cell migration. Interestingly, the collagen gel-induced ERK1/2 phosphorylation was present in focal adhesions. Moreover, filipin III, a specific inhibitor for caveolae, completely alleviated the collagen gel-induced ERK1/2 phosphorylation. Taken together, low rigidity of collagen fiber induces activation of ERK1/2, which serves to facilitate cell spreading and migration on collagen gel. In addition, low rigidity of collagen gel suppressed protein synthesis rates in all cells examined. We found that the low rigidity-induced decrease in protein synthesis rate could be mediated by down-regulation of transcription factor IID (TFIID) (4). 34 REFERENCES 1. Y. K. Wang, Y. H. Wang, C. Z. Wang, J. M. Sung, W. T. Chiu, S. H. Lin, Y. H. Chang and M. J. Tang. (2003) Rigidity of collagen fibrils control collagen gel-induced down-regulation of focal adhesion complex proteins mediated by 21 integrin. J. Biol. Chem. 278, 21886-21892. 2. W. C. Wei, Y. K. Wang and M. J. Tang. Substratum rigidity controls 1 integrin activation and FAK phosphorylation. (manuscript in preparation) 3. Y. C. Hsu, W. C. Wei, W. T. Chiu, Y. K. Wang and M. J. Tang. Low substratum rigidity inhibits FAK 397Y phosphorylation, but induces activation of ERK1/2 to trigger cell spreading and migration. (submitted) 4. H. H. Lin, Y. K. Wang, Y. H. Wang and M. J. Tang. Low rigidity of collagen fibrils decreases protein synthesis rate via down-regulation of transcription factor IID. (manuscript in preparation) 5. C. Z. Wang, Y. M. Hsu and M. J. Tang. (2005) The function of Discoidin Domain Receptor 1 in HGF-induced branching tubulogenesis of MDCK cells in collagen gel. J. Cell Physiol. 203: 295-304. 6. C. Z. Wang, H. W. Su, Y. C. Hsu, M. R. Shen and M. J. Tang. (2006) SHP-2 mediates DDR1-induced suppression of STAT3 tyrosine phosphorylation, cell migration and branching tubulogenesis. Mol. Biol. Cell (accepted with revision) III-3: Molecular mechanism of ceramide-induced apoptosis: the anti-apoptotic role of lithium (Yee-Shin Lin) BACKGROUND The role of ceramide in caspase-2 and -8 activation and mitochondrial apoptosis was recently demonstrated (1). Ceramide caused Bcl-2 dephosphorylation by protein phosphatase-2A (PP2A), which led to Bcl-2 inactivation and caspase-2 activation (2). Lithium has been shown to confer protection against neuronal apoptosis. In this study, the molecular mechanisms of apoptosis induced by ceramide were further explored, and the effect of lithium on ceramide-induced apoptotic signaling pathways was investigated. Our results showed the requirement of glycogen synthase kinase-3 (GSK-3) in ceramide-induced caspase-2 activation and mitochondrial apoptosis (3). In addition, lithium confers cell survival against ceramide by inhibiting PP2A methylation and activation (4). METHODS 10I T hybridoma cells were treated with ceramide in the presence or absence of lithium, SB260763, and GSK-3 siRNA. Cell apoptosis was detected using 35 propidium iodide staining and caspase activity assay. Mitochondrial membrane potential was assessed by rhodamine 123 staining. Protein expression, phosphorylation, and cleavage were analyzed by Western blotting. The activity of PP2A was analyzed after immunoprecipitation. Overexpression of PP2A was performed by protein transfection technique. RESULTS AND DISCUSSION Caspase-2 was shown to act upstream of mitochondria in stress-induced apoptosis. Our study showed sequential activation of caspase-2 and -8 before mitochondrial damage during ceramide- and etoposide-induced apoptosis (1). Interestingly, the pro-apoptotic function of caspase-2 in TRAIL-mediated apoptosis by processing procaspase-8 has been reported (5). In an attempt to investigate the mechanism of caspase-2 activation, our study showed a regulatory role of Bcl-2 on caspase-2 (2). Furthermore, a role of PP2A that causes Bcl-2 dephosphorylation was demonstrated involving in the apoptotic signaling of ceramide, although its underlying mechanism remains unclear. A recent study has demonstrated the regulation of calcium/calmodulin-dependent protein kinase II (CaMKII) on caspase-2 activation (6). The involvement of endoplasmic reticulum stress and CaMKII activation in ceramide-induced caspase-2 and mitochondrial apoptosis is currently investigated. Previous studies in our laboratory showed that lithium conferred protection from ceramide-induced apoptosis via activation of MEK/ERK (7). During ceramide stimulation, PP2A activation may inactivate survival kinases such as MEK/ERK and Akt. Moreover, our study revealed the involvement of PP2A and Bcl-2 in caspase-2 activation before mitochondrial damage during ceramide-induced apoptosis. Interestingly, lithium promotes cell survival by inhibiting PP2A activity and caspase-2 activation (4). Furthermore, the requirement of GSK-3 in ceramide-induced mitochondrial apoptosis was demonstrated (3). The involvement of PP2A and PI3K/Akt in GSK-3-mediated apoptotic signaling was shown, which was also inhibited by lithium treatment. Our preliminary results indicated that the regulatory role of lithium on PP2A methylation was, at least in part, via PP2A methylesterase. Whether lithium also acts on PP2A methyltransferase needs further investigation. REFERENCES 1. Lin, C. F., C. L. Chen, W. T. Chang, M. S. Jan, L. J. Hsu, R. H. Wu, M. J. Tang, W. C. Chang, and Y. S. Lin. (2004). Sequential caspase-2 and caspase-8 activation upstream of mitochondria during ceramide- and etoposide-induced apoptosis. J. Biol. Chem. 279, 40755-40761. 36 2. Lin, C. F., C. L. Chen, W. T. Chang, M. S. Jan, L. J. Hsu, R. H. Wu, Y. T. Fang, M. J. Tang, W. C. Chang, and Y. S. Lin. 2005. Bcl-2 rescues ceramide- and etoposide-induced mitochondrial apoptosis through blockage on caspase-2 activation. J. Biol. Chem. 280, 23758-23765. 3. Lin, C. F., C. L. Chen, C. W. Chiang, M. S. Jan, and Y. S. Lin. Glycogen synthase kinase-3 acts downstream of PP2A and PI3K/PKB and upstream of caspase-2 in ceramide-induced T cell mitochondrial apoptosis. (manuscript in preparation) 4. Chen, C. L., C. F. Lin, C. W. Chiang, M. S. Jan, M. J. Tang, W. C. Chang, and Y. S. Lin. Lithium inhibits ceramide- and etoposide-induced protein phosphatase 2A methylation, Bcl-2 dephosphorylation, caspase-2 activation and apoptosis. (manuscript in preparation) 5. Shin, S., Y. Lee, W. Kim, H. Ko, H. Choi, and K. Kim. 2005. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 24, 3532-3542. 6. Nutt, L. K., S. S. Margolis, M. Jensen, C. E. Herman, W. G. Dunphy, J. C. Rathmell, and S. Kornbluth. 2005. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 123, 89-103. 7. Jan, M. S., L. J. Hsu, C. F. Lin, C. L. Chen, and Y. S. Lin. Lithium confers protection from ceramide-induced apoptosis via activation of MEK/ERK. (manuscript in preparation) 7. List of Research Results & Achievements (2002 ~ 2006) Sub-project Ⅰ: 37 1) Yi-Wen Liu, Hui-Ping Tseng, Lei-Chin Chen, Ben-Kuen Chen, and Wen-Chang Chang. (2003) Functional cooperation of simian virus 40 promoter factor 1 and CCAAT/enhancer-binding protein β and δ in lipopolysaccharide-induced gene activation of IL-10 in mouse macrophages. J. Immunol. 171, 821-828. 2) Ying-Nai Wang and Wen-Chang Chang. (2003) Induction of the diseaseassociated keratin 16 gene expression by epidermal growth factor is regulated through cooperation of transcription factors Sp1 and c-Jun. J. Biol. Chem. 278, 45848-45857. 3) Wen-Chang Chang. (2003) Cell signaling and gene regulation of human 12(S)lipoxygenase expression. Prostaglandins Other Lipid Mediat. (Invited Review) 71, 277-285. 4) Te-Hsiu Lee, Hui-Chiu Chang, Lea-Yea Chuang, and Wen-Chun Hung. (2003) Involvement of PKA and Sp1 in the induction of p27Kip1 by tamoxifen. Biochem. Pharmacol. 66, 371-377. 5) Yu-Chun Huang, Jing-Yi Chen, and Wen-Chun Hung. (2004) Vitamin D3 receptor/ Sp1 complex is required for the induction of p27Kip1 expression by vitamin D3. Oncogene 23, 4856-4861. 6) Lei-Chin Chen, Ben-Kuen Chen, Jia-Ming Chang, and Wen-Chang Chang. (2004) Essential role of c-Jun induction and coactivator p300 in epidermal growth factor-induced gene expression of cyclooxygenase-2 in human epidermoid A431 cells. Biochim. Biophys. Acta (Mol. Cell Biol. L.) 1683, 38-48. 7) Lei-Chin Chen, Ben-Kuen Chen, and Wen-Chang Chang. (2005) AP1-mediated cyclooxygenase-2 expression is independent of N-terminal phosphorylation of c-Jun. Mol. Pharmacol. 67, 2057-2069. 8) Ju-Ming Wang, Joseph T. Tseng, and Wen-Chang Chang. (2005) Induction of human NF-IL6β by epidermal growth factor is mediated through p38 signaling pathway and cAMP response element-binding protein activation in A431 cells. Mol. Biol. Cell 16, 3365-3376. 9) Shwu-Jen Tzeng, Wen-Chang Chang, and Jin-Ding Huang. (2005) Transcriptional regulation of the rat Mrp3 gene promoter by the specificity protein (Sp) family members and CCAAT/enhancer binding proteins. J. Biomed. Sci. 12, 741-761. 10) Wen-Chang Chang and Ben-Kuen Chen. (2005) Transcription factor Sp1 functions as an anchor protein in gene transcription of human 12(S)-lipoxygenase. Biochem. Biophys. Res. Commun. (Invited Review) 338, 117-121. 11) Jan-Jong Hung, Chih-Ying Wu, Pao-Chi Liao, and Wen-Chang Chang. (2005) Hsp90 recruited by Sp1 is important for transcription of 12(S)-lipoxygenase in A431 cells. J. Biol. Chem. 280, 36283-36292. 12) Ying-Nai Wang, Yung-Ju Chen, and Wen-Chang Chang. (2006) Activation of 38 ERK signaling by epidermal growth factor mediates c-Jun activation and p300 recruitment in keratin 16 gene expression. Mol. Pharmacol. 69, 85-98. 13) Yi-Wen Liu, Chun-Chia Chen, Hui-Ping Tseng, and Wen-Chang Chang. (2006) Lipopolysaccharide-induced transcriptional activation of interleukin-10 is mediated by MAPK- and NF-κB-induced CCAAT/enhancer-binding protein δ in mouse macrophages. Cell. Signal. (in press) 14) Jan-Jong Hung, Yi-Ting Wang, and Wen-Chang Chang. (2006) Sp1 deacetylation induced by phorbol ester recruits p300 to activate the 12(S)-lipoxygenase transcription. Mol. Cell. Biol. (in press) 15) Ju-Ming Wang, Chiung-Yuan Ko, Lei-Chin Chen, Wen-Lin Wang and Wen-Chang Chang. (2006) Functional role of NF-IL6β and its sumoylation and acetylation modifications in promoter activation of cyclooxygenase 2 in A431 cells. Nucleic Acids Res. (in press) 16) Hsuen-Tsen Cheng, Jing-Yi Chen, Yu-Chun Huang, Hui-Chiu Chang, and Wen-Chun Hung. (2006) Functional role of VDR in the activation of p27Kip1 by the VDR/Sp1 complex. J. Cell. Biochem. (in press) 17) Ming-Chuan Hsu, Hui-Chiu Chang, and Wen-Chun Hung. (2006) HER-2/neu represses the metastasis suppressor RECK via ERK and Sp transcription factors to promote cell invasion. J. Biol. Chem. (in press) 18) LunYu Kuo, Hui-Chiu Chang, Tzeng-Horng Leu, Ming-Chie Maa, and Wen-Chun Hung. (2006) Src oncogene activates MMP-2 expression via the ERK/Sp1 pathway. J. Cell. Physiol. (in press) 19) Ben-Kuen Chen, Chi-Chen Huang, Wei-Chiao Chang, Yung-Ju Chen, Ushio Kikkawa, and Wen-Chang Chang. (2006) Calcineurin-mediated dephosphorylation of c-Jun C-terminus regulates phorbol ester-induced c-Jun/Sp1 interaction. Mol. Biol. Cell (in revision) Sub-project Ⅱ: 1) Tzeng-Horng Leu and Ming-Chei Maa. (2002) Tyr-863-phosphorylation enhances focal adhesion kinase autophosphorylation at Tyr-397. Oncogene 21, 6992-7000. 2) Jih I Chuang, Tsuey Y Chang, and Hsiao S Liu. (2003) Glutathione depletioninduced apoptosis of Ha-ras-transformed NIH3T3 cells can be prevented by melatonin. Oncogene 22, 1349-1357. 3) Tsui-Lien Hung, Fen-Fen Chen, Jacqueline M. Liu, Wu-Wei Lai, Ai-Li Hsiao, Wen-Tsung Huang, Helen H.W. Chen, and Wu-Chou Su. (2003) Clinical evaluation of HER-2/neu protein in malignant pleural effusion-associated lung adenocarcinoma and as a tumor marker in pleural effusion diagnosis. Clin. Cancer Res. 9, 2605-2612. 39 4) Tzeng-Horng Leu and Ming-Chei Maa. (2003) Functional implication of the interaction between EGF receptor and c-Src. Front. Biosci. 8, s28-38. 5) Tzeng-Horng Leu, Shu Li Su, Ya-Chun Chuang, and Ming-Chei Maa. (2003) Direct inhibitory effect of curcumin on Src and focal adhesion kinase activity. Biochem. Pharmacol. 66, 2323-2331. 6) Tsuey-Yu Chang, Wen-Jiuan Tsai, Chao-Kai Chou, Nan-Haw Chow, Tzeng-Horng Leu, and Hsiao-Sheng Liu. (2003) Identifying the factors and signal pathways necessary for anchorage-independent growth of Ha-ras oncogene-transformed NIH/3T3 cells. Life Sci. 73, 1265-1274. 7) Tzeng-Horng Leu, Hsu-Hua Yeh, Ching-Chung Huang, Ya-Chun Chuang, Shu-Li Su, and Ming-Chei Maa. (2004) Participation of p97Eps8 in Src-mediated transformation. J. Biol. Chem. 279, 9875-9881. 8) Jui-Hsiang Hung, Ih-Jen Su, Huan-Yao Lei, Hui-Ching Wang, Wan-Chi Lin, Wen-Tsan Chang, Wenya Huang, Wen-Chang Chang, Yung-Sheng Chang, Ching-Chow Chen, and Ming-Derg Lai. (2004) Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-κB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 279, 46384-46392. 9) Shu-Jem Su, Trai-Ming Yeh, Woei-Jer Chuang, Chung-Liang Ho, Kee-Lung Chang, Hsiao-Ling Cheng, Hsiao-Sheng Liu, Hong-Lin Cheng, Pei-Yin Hsu, and Nan-Haw Chow. (2005) The novel targets for anti-angiogenesis of genistein on human cancer cells. Biochem. Pharmacol. 69, 307-318. 10) Yuan-Chang Dai, Chung-Liang Ho, Yi-Chang Tsai, Yung-Hsiang Hsu, Yu-Chuang Chang, Hsiao-Sheng Liu, Helen Hai-Wen Chen, and Nan-Haw Chow. (2005) Allelic loss of 14q32 in the pathogenesis of gastrointestinal and ampullary malignancies: mapping of the target region to a 17 cM interval. J. Cancer Res. Clin. 131, 94-100. 11) Hui-Ching Wang, Wen-Tsan Chang, Wen-Wei Chang, Han-Chieh Wu, Wenya Huang, Huan-Yao Lei, Ming-Derg Lai, Nelson Fausto, and Ih-Jen Su. (2005) Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 41, 761-770. 12) Hong-Lin Cheng, Hsiao-Sheng Liu, Yan-Ju Lin, Helen Hai-Wen Chen, Pei-Yin Hsu, Tsuey-Yu Chang, Chung-Liang Ho, Tzong-Shin Tzai, and Nan-Haw Chow. (2005) Co-expression of RON and MET as a prognostic indicator for patients with transitional cell carcinoma of the bladder. Br. J. Cancer 92, 1906-1914. 13) Wen-Ying Lee, Helen Hai-Wen Chen, Nan-Haw Chow, Wu-Chou Su, Pin-Wen Lin, and How-Ran Guo. (2005) Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin. Cancer Res. 11, 2222-2228. 40 14) Jeng-Chang Lee, Ming-Chei Maa, Hsiu-Shan Yu, Jung-Hui Wang, Chia-Kuang Yen, Shan-Tair Wang, Yen-Jen Chen, Yuan Liu, Ying-Tai Jin, and Tzeng-Horng Leu. (2005) Butyrate regulates the expression of c-Src and focal adhesion kinase and inhibits cell invasion of human colon cancer cells. Mol. Carcinog. 43, 207-214. 15) Te-Jung Lu, Chi-Ying F. Huang, Chiun-Jye Yuan, Yuan-Chii Lee, Tzeng-Horng Leu, Wen-Chang Chang, Te-Ling Lu, Wen-Yih Jeng, and Ming-Derg Lai. (2005) Zinc ion acts as a cofactor for serine/threonine kinase MST3 and has a distinct role in autophosphorylation of MST3. J. Inorg. Biochem. 99, 1306-13. 16) Tzeng-Horng Leu, Suparat Charoenfuprasert, Chia-Kuang Yen, Chiung-Wen Fan, and Ming-Chei Maa. (2006) Lipopolysaccharide-induced c-Src expression plays a role in nitric oxide and TNFα secretion in macrophages. Mol. Immunol. 43, 308316. 17) Hsuan-Heng Yeh, Wu-Wei Lai, Helen H.W. Chen, Hsiao-Sheng Liu, and Wu-Chou Su (2006) Autocrine IL-6 induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene (in press) 18) Pei-Yin Hsu, Hsiao-Sheng Liu, Chung-Liang Ho, Hong-Lin Cheng, Tzong-Shin Tzai, Tsuey-Yu Chang, How-Ran Guo, and Nan-Haw Chow. (2006) Collaboration of RON and EGFR in human epithelial carcinogenesis. (manuscript in submission, J Urol.) 19) Chen-Yun Yeh, Nan-Haw Chow, and Hsiao-Sheng Liu. (2006) Axl and PDGFR- associated with c-Met overexpression in bladder tumoregenesis. (manuscript in submission, Oncogene) 20) Hui-Ching Wang, Ming-Derg Lai, Wenya Huang, and Ih-Jen Su. (2006) ER stress induces cytoplasmic cyclin A and centrosome overduplication. (manuscript in submission, Nature Cell Biology) 21) Yi-Hsuan Hsie, Hui-Ching Wang, Jui-He Tsai, Ih-Jen Su, Wen-Wei Chang, C-C Yun, Ming-Derg Lai, Huan-Yao Lei, Wen-Tsan Chang, and Wenya Huang. (2006) Pre-S2 hepatitis B virus surface antigen mutant induces retinoblastoma hyperphosphorylation through interaction with JAB1 protein in endoplasmic reticulum: a novel mechanism for HBV-induced hepatocellular carcinoma. (manuscript in submission, Nature Medicine) Sub-project Ⅲ: 1) Wei-Shio Hor, Wei-Lune Huang, Yee-Shin Lin, and Bei-Chang Yang. (2003) Cross-talk between tumor cells and neutrophils through the Fas(APO-1, CD95)/FasL system: human glioma cells enhance cell viability and stimulate 41 cytokine production in neutrophils. J. Leukocyte Biol. 73, 363-368. 2) Yi-Ling Chen, Shun-Hua Chen, Jiu-Yao Wang, and Bei-Chang Yang. (2003) Fas ligand on tumor cells mediates inactivation of neutrophils. J. Immunol. 171, 1183-1191. 3) Bei-Chang Yang, Heng-Kai Lin, Wei-Shio Hor, Jun-Yen Hwang, Yu-Ping Lin, Ming-Yie Liu, and Ying-Jan Wang. (2003) Mediation of enhanced transcription of the IL-10 gene in T cells, upon contact with human glioma cells, by Fas signaling through a protein kinase A-independent pathway. J. Immunol. 171, 3947-3954. 4) Meng-Ru Shen, Cheng-Yang Chou, Keng-Fu Hsu, Yueh-Mei Hsu, Wen-Tai Chiu, Ming-Jer Tang, Seth L. Alper, and J. Clive Ellory. (2003) KCl cotransport is an important modulator of human cervical cancer growth and invasion. J. Biol. Chem. 278, 39941-39950. 5) Soon-Cen Huang, Ming-Jer Tang, Ya-Min Cheng, Keng-Fu Hsu, Chung-Liang Ho, and Cheng-Yang Chou. (2003) Enhanced polyadenosine diphosphate-ribosylation in GnRH agonist-treated uterine leiomyoma. J. Clin. Endocrinol. Metab. 88, 5009-5016. 6) Keng-Fu Hsu, Soon-Cen Huang, Jenn-Ren Hsiao, Ya-Min Cheng, Saprina PH Wang, and Cheng-Yang Chou. (2003) Clinical significance of serum human papillomavirus DNA in cervical carcinoma. Obstet. Gynecol. 102, 1344-1351. 7) Yang-Kao Wang, Yao-Hsien Wang, Chau-Zen Wang, Junne-Ming Sung, Wen-Tai Chiu, Shu-Han Lin, Yung-Hen Chang, and Ming-Jer Tang. (2003) Rigidity of collagen fibrils controls collagen gel-induced down-regulation of focal adhesion complex proteins mediated by α2β1 integrin. J. Biol. Chem. 278, 21886-21892. 8) Chun-I Sze, Meng Su, Subbiah Pugazhenthi, Purevsuren Jambal, Li-Jin Hsu, John Heath, Lori Schultz, and Nan-Shan Chang. (2004) Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer's disease. J. Biol. Chem. 279, 30498-30506. 9) Meng-Ru Shen, Ai-Chien Lin, Yueh-Mei Hsu, Tsui-Jung Chang, Ming-Jer Tang, Seth L. Alper, J. Clive Ellory, and Cheng-Yang Chou. (2004) Insulin-like growth factor 1 stimulates KCl cotransport, which is necessary for invasion and proliferation of cervical cancer and ovarian cancer cells. J. Biol. Chem. 279, 40017-40025. 10) Chiou-Feng Lin, Chia-Ling Chen, Wen-Tsan Chang, Ming-Shiou Jan, Li-Jin Hsu, Ren-Huang Wu, Ming-Jer Tang, Wen-Chang Chang, and Yee-Shin Lin. (2004) Sequential caspase-2 and caspase-8 activation upstream of mitochondria during ceramide- and etoposide-induced apoptosis. J. Biol. Chem. 279, 40755-40761. 11) Li-Jin Hsu, Lori Schultz, Jeffrey Mattison, Yee-Shin Lin, and Nan-Shan Chang. (2005) Cloning and characterization of a small-size peptide Zfra that regulates the 42 cytotoxic function of tumor necrosis factor by interacting with JNK1. Biochem. Biophys. Res. Commun. 327, 415-423. 12) Chau-Zen Wang, Yuei-Mei Hsu and Ming-Jer Tang. (2005) Function of Discoidin Domain Receptor 1 in HGF-induced branching tubulogenesis of MDCK cells in collagen gel. J. Cell. Physiol. 203, 295-304. 13) Nan-Shan Chang, Lori Schultz, Li-Jin Hsu, Jennifer Lewis, Meng Su, and Chun-I Sze. (2005) 17β-estradiol upregulates and activates WOX1/WWOXv1 and WOX2/WWOXv2 in vitro: potential role in cancerous progression of breast and prostate to a premetastatic state in vivo. Oncogene 24, 714-723. 14) Chiou-Feng Lin, Chia-Ling Chen, Wen-Tsan Chang, Ming-Shiou Jan, Li-Jin Hsu, Ren-Huang Wu, Yi-Ting Fang, Ming-Jer Tang, Wen-Chang Chang, and Yee-Shin Lin. (2005) Bcl-2 rescues ceramide- and etoposide-induced mitochondrial apoptosis through blockage on caspase-2 activation. J. Biol. Chem. 280, 23758-23765. 15) Nan-Shan Chang, Joan Doherty, Amy Ensign, Lori Schultz, Li-Jin Hsu, and Qunying Hong. (2005) WOX1 is essential for TNF-, UV light-, staurosporine-, and p53-mediated cell death and its tyrosine 33 phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J. Biol. Chem. 280, 43100-43108. 16) Meng-Ru Shen, Yueh-Mei Hsu, Keng-Fu Hsu, Yih-Fung Chen, Ming-Jer Tang, and Cheng-Yang Chou. (2006) Insulin-like growth factor 1 is a potent stimulator of cervical cancer cell invasiveness and proliferation which is modulated by v3 integrin signaling. Carcinogenesis (in press) 43