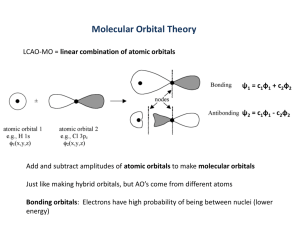
5-1
advertisement

Created by Margret J. Geselbracht, Reed College (mgeselbr@reed.edu) and posted on VIPEr (www.ionicviper.org) on January 22, 2010. Copyright Margret J. Geselbracht 2010. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License. To view a copy of this license visit http://creativecommons.org/about/license/. EXPLORING MOLECULAR ORBITALS WITH SPARTAN 1. In this problem, you will compare the MO diagrams of LiH and HF. (a) First, look up and report the ionization energies of H, Li, and F. Ionization energy of H: 1312 kJ/mol Ionization energy of Li: 520.2 kJ/mol Ionization energy of F: 1681 kJ/mol These ionization energies are a good way to gauge the relative energies of the atomic orbitals comprising the molecular orbital diagrams for LiH and HF. (b) Draw the MO diagrams for LiH and HF side by side to show the relative energies of all of the orbitals involved. That is, make sure that you put the hydrogen 1s orbital at the same energy for both diagrams. Show the electron occupancy. Label the MO's with the appropriate symmetry labels. Think about whether or not there is an inversion center! 2* 2s * 3 1s E 1š nb 2p 1 H LiH Li 2 2s 1nb F HF (c) Spartan models for these two molecules can be found on the computers in the computer lab. Once Spartan launches, under the File menu, choose Open…then browse until you find the folder entitled Local Disk (C:)/Chem212/. Inside this folder are a large number of subfolders and Spartan files. Look in the Diatomics ’03 folder. Examine these models, focusing on the various molecular orbital surfaces listed in the table below. For each of the following MOs, draw what the Spartan surface looks like, being careful to show the location of each nucleus (label which atoms they represent). Also draw a cartoon showing which atomic or hybrid orbitals might be contributing to each MO surface. Draw the cartoon orbital larger if there appears to be a greater contribution from that AO to the MO. Be sure to clearly label which orbitals you are using for each atom. You might find it helpful to display the output and find the relative energies of these MOs. HF LiH HOMO HOMO-1 HOMO-2 LUMO HOMO HOMO-1 LUMO Created by Margret J. Geselbracht, Reed College (mgeselbr@reed.edu) and posted on VIPEr (www.ionicviper.org) on January 22, 2010. Copyright Margret J. Geselbracht 2010. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License. To view a copy of this license visit http://creativecommons.org/about/license/. HF: HOMO This is a nonbonding 2p x (or 2py ) orbital on fluorine. The HOMO –1 looks the same, but is oriented such that it represents the other 2p orbital perpdendicular to the internuclear axis. H F HF: HOMO – 2 This is the bonding combination of 2p z on fluorine with H 1s. HF: LUMO H F HF: LUMO This is the antibonding combination of 2pz on fluorine with H 1s with a greater contribution from H 1s. H F LiH: HOMO Li H At first glance, this would seem to be the bonding combination of the H 1s and the Li 2s. However, note that there is a node running through the Li nucleus (the small blob on the other side is a different color, indicating the wavefunction has changed sign). So, there must be some s-p z hybridization going on on the Li nucleus. This MO is better thought of as the bonding combination of H 1s with a Li sp hybrid. Why would the Li want to use sp hybrids? (By the way, of course Li has 2p orbitals, they are just empty and higher in energy than the 2s orbital.) By mixing in some of the higher energy 2pz orbital, the Li atomic orbital contribution to the bonding MO can achieve greater spatial overlap with the H 1s. The sp hybrid concentrates the electron density in a lobe that is pointed right at the H for maximum overlap. That’s better than just using a pure 2s orbital on Li. LiH: HOMO–1 H Li In this MO, there is obviously no contribution from the hydrogen atom. Most likely this corresponds to the Li 1s orbital, which is too low in energy to effectively overlap with H 1s. Created by Margret J. Geselbracht, Reed College (mgeselbr@reed.edu) and posted on VIPEr (www.ionicviper.org) on January 22, 2010. Copyright Margret J. Geselbracht 2010. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License. To view a copy of this license visit http://creativecommons.org/about/license/. LiH: LUMO H Li This MO could be thought of as a slightly bonding combination of H 1s with a Li sp hybrid in which the majority of the electron density is on the lithium end of the molecule. (d) What is the orientation of the dipole moment in each of these molecules? Rationalize this based on your MO diagrams and pictures of the MOs. Orientation of the dipole moments: F H Li H In HF, the bonding pair of electrons is located in an MO that has a larger contribution from the fluorine 2pz orbital than the H 1s orbital (HOMO-2). Although this is not very obvious from the MO surface in Spartan, it is more obvious when you look at the Slice for HOMO-2 (see below on the left). The Slice shows that there is a greater probability of finding the electrons on the fluorine end of the molecule (the red end), which acquires a partial negative charge relative to the hydrogen end. In LiH, the bonding pair of electrons is located in an MO that has a larger contribution from the hydrogen 1s orbital than from the Li sp hybrid orbital (the HOMO drawn above on the right, the H atom is on the right). The result is that the electrons are more likely to be found on the hydrogen end of the molecule, which acquires a partial negative charge relative to the lithium end. 2. Spartan models for both CO2 and XeF2 can be found in the Triatomics folder. (a) First look at CO2. Examine the output file, and draw an energy diagram showing the energies of the MOs formed from the valence orbitals of carbon and oxygen. Created by Margret J. Geselbracht, Reed College (mgeselbr@reed.edu) and posted on VIPEr (www.ionicviper.org) on January 22, 2010. Copyright Margret J. Geselbracht 2010. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License. To view a copy of this license visit http://creativecommons.org/about/license/. First some comments about the Spartan MO diagram. The 3 lowest energy MOs calculated by Spartan are the core 18.24 eV u orbitals (1s) for both of the oxygen atoms and the carbon atom. Normally, MO diagrams involve only the valence orbital 8.01 eV g interactions, so these core orbitals are left off of the diagram. (b) For each of the MOs in your energy diagram, draw a cartoon showing which atomic or hybrid orbitals might be contributing to each MO surface. E Draw the cartoon orbital larger if there appears to be a greater contribution from that AO to the MO. Be sure to clearly label which orbitals you are using for each atom. 5.74 eV –-14.19 eV –--19.653 eV u gnb u –-19.650 eV The MOs from Spartan indicate u that there is likely some s-p mixing going on in that both s and pz orbitals can overlap in different g –-21.01 eV MOs. So for any of the -type MOs, one might also consider using an sp hybrid on one or more u –-40.91 eV atoms. I have kept my cartoons fairly simple using either 2s or 2pz –-42.15 eV 1g atomic orbitals on the C and O atoms. There are some images of selected -type MOs shown on the next page. A key hint to figuring out that the atom is contributing a 2s orbital to the MO is to look for the inner radial node around the nucleus. For example, see the 1g and 3g MOs below. (c) Assign symmetry labels (such as 1g, 2πgnb, or 6u*) to each MO in your energy diagram. See the diagram above. (d) Fill in the electrons in your MO diagram, write the molecular orbital electron configuration, and derive a bond order for each bond in CO2. (1g)2 (2u)2 (3g)2 (1πu)4 (4u)2 (2πgnb)4 The 2πg MOs are clearly nonbonding, so those 4 electrons do not factor into a bond order calculation. If we count all of the rest as bonding type MOs, which is what appears to be the case from a first glance, then we end up with a bond order of ½ (12) = 6 divided over 2 positions for an average bond order of 3 for each C–O bond. Now that does not seem valid, since the carbon cannot make 2 triple bonds at once! To really decide which of these orbitals are bonding, nonbonding or antibonding, we need to look more carefully at the MO surfaces and slices. The tricky ones are the occupied -type MOs. Here are some images of the MOs where the slices are most helpful: Created by Margret J. Geselbracht, Reed College (mgeselbr@reed.edu) and posted on VIPEr (www.ionicviper.org) on January 22, 2010. Copyright Margret J. Geselbracht 2010. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License. To view a copy of this license visit http://creativecommons.org/about/license/. 1g: Clearly a bonding MO 3g: Although this has bonding character, I would argue it is mostly nonbonding, since the highest probability of finding the e- is in the outer blue lobes. 2u: Clearly a bonding MO 4u: Again, this has bonding character, but highest probability of finding the e- is in the outer two lobes, making it more nonbonding. Perhaps this is a bit of hand-waving, because I want the bond order to be lower, but from my arguments above, I would leave out the 4 electrons in 3g and 4u from my bond order calculation. This leaves me with ½ (8) = 4 divided over 2 positions resulting in an average bond order of 2 for each C–O bond. Sadly, your text does not include a complete MO diagram of CO2. If you look at other texts, you may find a slightly different interpretation of the MOs to arrive at a bond order of 2. Some texts suggest that the lowest two MOs are mainly nonbonding combinations of the O 2s orbitals with no contribution from the carbon atom. This makes some sense from an energy point of view as the O 2s orbitals are going to be much more stable than the C 2s and C 2p orbitals. However, the MOs in Spartan suggest that the carbon does make some contribution to each of the MOs. In some ways, this points out the limitations of the fairly simplistic view of bond order calculations. (e) If we exclude the use of the Xe d orbitals, then the MO diagram for XeF2 should look fairly similar to that of CO2 with a different number of electrons. Using your MO diagram for CO 2 as a guide, write the molecular orbital electron configuration for XeF2, and derive the average bond order for each Xe–F bond. Sometimes these bonds are described as 3-center, 2-electron bonds. Explain what you think this means. Created by Margret J. Geselbracht, Reed College (mgeselbr@reed.edu) and posted on VIPEr (www.ionicviper.org) on January 22, 2010. Copyright Margret J. Geselbracht 2010. This work is licensed under the Creative Commons Attribution Non-commercial Share Alike License. To view a copy of this license visit http://creativecommons.org/about/license/. XeF2 is also linear, and so should have a very similar MO diagram to that of CO2, particularly if we ignore any contribution from the empty Xe 5d orbitals and consider only involvement of the Xe 5s and 5p valence orbitals. The primary difference will be in the number of valence electrons that fill the MO diagram, 22 e- in the case of XeF2. If you did look at the MOs on Spartan, they can look fairly funky and are not necessarily in the same order as in CO2. But let’s assume for the purposes of this question that we can still use the same MO diagram as is shown above. These 22 valence electrons will fill the MOs up to the level of the 5g* MO. This now introduces some antibonding electrons into the picture. So, the total bond order for XeF2 is ½(8 – 6) = 1. However, this must again be divided over two bond locations, resulting in a bond order of ½ for each bond. A 3-center, 2-electron bond means that I have 2 electrons, but these are delocalized over 3 atoms rather than having 4 electrons and 2 bond positions to hold the 3 atoms together. This is a common way to think about covalent bonding in hypervalent (expanding the octet) molecules if you do not consider use of the empty d orbitals. 3. BF3 is a classic example of an electron deficient molecule with only six electrons around boron. However, if the octet rule is strictly obeyed, resonance structures can be drawn with a delocalized B– F π bond and an admittedly unusual distribution of formal charge. Let’s examine the MO diagram for BF3 on Spartan. I think it is in a folder called something like Small Inorganics. To help you understand what is going on, examine the figures in your text that show all of the group orbitals formed from the fluorine atom 2p orbitals and the MO diagram of BF3. (a) Examine the LUMO in BF3 on Spartan. BF3 is best known for its behavior as a classic Lewis acid. Draw a cartoon of the LUMO, showing which AOs contribute to this MO and explaining why it looks like a good electron acceptor. Describe any specific geometry requirements for the interaction of BF3 with a Lewis base. The LUMO of BF3 is the π-antibonding combination of the B 2pz orbitals with the F 2pz orbitals with the greatest contribution coming from the B orbital. If BF3 accepts a pair of electrons, they must go into an empty molecular orbital, most likely this LUMO. The electron pair, when it is donated into this MO, will spend more of it’s time on the B, the electron deficient atom. The Lewis base must approach the LUMO from either the top face or the bottom face with its lone pair pointed directly at the boron atom. (b) You may be somewhat surprised to find that this MO appears to have B–F π* character. Examine some of the lower energy occupied MOs to find the B–F π bonding half of this MO. Which Spartan MO is this? Describe its name and energy in eV. Why is this MO not simply the HOMO? The MO that matches the π bonding half of the MO is MO #11 in the Spartan output file at –20.4 eV. In the surfaces list, it is called HOMO-5. This naming scheme means that it is the 5th orbital below the HOMO in energy (the HOMO energy is –17.6 eV). If you look at the HOMO and the other MO surfaces that are higher in energy, these are different combinations of the 2p orbitals on the F atoms that do not point towards the B atom. The combinations are such that there is no atomic orbital on boron that will provide any net overlap with the fluorine orbitals. So, the HOMO and HOMO-1 (etc) are nonbonding group orbitals of fluorine. Since HOMO-5 has net π-bonding character, it will be lower in energy than these nonbonding combinations.