Supplementary Methods
advertisement

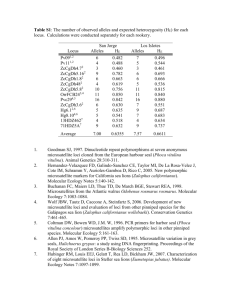
Supplementary Methods Mitochondrial data. A 356-bp fragment of the mtDNA control region (CR) was analyzed for 246 harbour porpoises including: 148 sequences of animals from the northern Bay of Biscay (49 from Tolley & Rosel (2006) and 99 from Walton (1997)), 30 from the Iberian coast (13 from Tolley & Rosel (2006) and 17 newly sequenced in this study), and 72 from the Black Sea (8 sequences from Rosel et al. (1995), 2 from Tolley & Rosel (2006), and 62 new sequences generated in this study) (see table S1). The 79 new sequences were obtained as follows: genomic DNA was extracted from soft tissue using the Qiagen DNeasy Tissue kit. The complete mtDNA CR was amplified by polymerase chain reactions (PCR) using newly designed forward and reverse primers located in the flanking regions (respectively: PPL1: 5’-AAA TAC CTC GGT CTT GTA AAC C-3’ and PPS1: 5’-AAG TTT AAG CTA CAT TAA CGT GTG G3’). PCR was conducted in a 25 µl reaction volume containing 50 ng of genomic DNA, 0.4 U Taq DNA polymerase, thermophilic DNA polymerase buffer (1X), 2 mM MgCl2, 0.5 µM of each primer, and 400 µM of dNTPs. The PCR conditions started with an initial denaturation step at 95 °C for 2 min, followed by 35 cycles of denaturation at 94°C for 1 min, primer annealing at 60°C for 1 min, and an extension step at 72°C for 1 min (30 min for final extension). The sequencing reactions were conducted on a 96-capillary MegaBACE-1000 DNA Analyzer (Amersham Biosciences) using the manufacturer’s protocol and three sequencing primers: PPL1, L15825, and H16265 previously designed (Rosel et al. 1999). Sequences were edited using Sequence navigator (ABI), aligned primarily using Clustal X and then checked visually. 1 IM analyses using microsatellite and mtDNA D-loop. We conducted the IM simulation contrasting the harbour porpoise population from the Black Sea with those in the Atlantic. We first applied the analysis to the mtDNA CR dataset alone, and then pooled all loci together (i.e., the 10 microsatellite loci and the 356 bps mtDNA CR). We used a similar procedure for the mtDNA CR data set as described for microsatellite loci (see the core ms), with a Hasegawa-Kishino-Yano (HKY) mutation model (Hasegawa et al. 1985) for the mtDNA CR, and 5 to 15 independent Metropolis coupled chains. Preliminary runs showed that migration rate posterior density distributions were not different from zero. We therefore simplified the model by assuming equal migration rates between populations after their divergence. We also applied a refined version of the IM model that allows for population size change by adding a supplemental parameter to estimate the proportion (s and (1-s)) of individuals from the ancestral population that founded the first and the second descendent populations, respectively (Hey 2005). With this additional complexity in the parameter space of the model, MCMC convergence was unachievable for our complex multilocus microsatellite dataset. We were therefore only able to apply the seven parameters scenario to the mtDNA dataset. To convert parameter estimates into demographic units, we used a plausible range of mutation rates between 3.3 x 10-8 bp-1 year-1 (µmtL) and 4.3 x 10-8 bp-1 year-1 (µmtH) (i.e. respectively 1.17 x 10-5 and 1.53 x 10-5 year-1 for a sequence of 356 bps), as suggested in Tolley & Rosel (2006). Estimates of were converted into effective population sizes using this range of mutation rate and the same range of generation time as describe above in the microsatellite analysis. 2 Finally, we ran IM simulations pooling all loci together following a similar procedure as that described for microsatellite loci, using a SMM for microsatellite evolution and a HKY substitution model for mtDNA evolution. Supplementary Figures and Tables legends Table S1 MtDNA control region haplotypes used in the IM analyses comparing Atlantic (AT) vs. Black Sea (BS) porpoises, count of each haplotype, and Genebank accession number Table S2 Details of the IM runs’ conditions for the three longest runs and Effective Sample Size at the end of each run Figure S1 Comparison of marginal posterior probability densities for each parameter of the IM model obtained from the replicated runs conducted on the microsatellite data set comparing Iberian (IB) vs. northern Bay of Biscay (NBB) porpoises. Figure S2 Same as figure S2, but for the microsatellite data set comparing Atlantic (AT) vs. Black Sea (BS) porpoises. Figure S3 Comparison of marginal posterior probability densities for each parameter of the IM model comparing Atlantic (AT) vs. Black Sea (BS) porpoises using mtDNA data set, microsatellite data, and all loci combined together (see supplementary methods for 3 IM runs condition with mtDNA). S refers to the model including (or not) population size change during the divergence. Supplementary References Hasegawa, M., Kishino, H. & Yano, T. 1985 Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22, 160-74. Hey, J. 2005 On the number of the New World founders: a population genetic portrait of the poepling of the Amercias. PLoS Biol 3, e193. Hey, J. & Nielsen, R. 2004 Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167, 747-760. Rosel, P., Dizon, A. E. & Haygood, M. G. 1995 Variability of the mitochondrial control region in populations of the harbour porpoise, Phocoena phocoena, on interoceanic and regional scales. Can J Fish Aquat Sci 52, 1210-1219. Rosel, P. E., France, S. C., Wang, J. Y. & Kocher, T. D. 1999 Genetic structure of harbour porpoise Phocoena phocoena populations in the northwest Atlantic based on mitochondrial and nuclear markers. Mol Ecol 8, S41-S54. Tolley, K. A. & Rosel, P. E. 2006 Population structure and historical demography of eastern North Atlantic harbour porpoises inferred through mtDNA sequences. Mar Ecol Progr Ser 327, 297-308. Walton, M. J. 1997 Population structure of harbour porpoise Phocoena phocoena in seas around the UK and adjacent waters. Proc R Soc B-Biol Sci 264, 84-94. 4