Jaundice and Cola-Colored Urine in a Young Indian Boy
advertisement

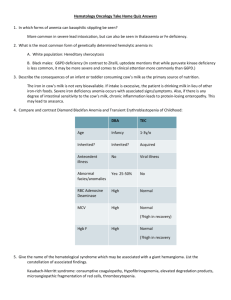
Case Studies Received 2.5.06 | Revisions Received 2.15.06 | Accepted 2.15.06 Jaundice and Cola-Colored Urine in a Young Indian Boy Anureet Kaur, MBBS,1 Menka Doomra, MD,1 Naveen Kakkar, MD,1 Jasbir Dhanoa, MD2 (Departments of 1Pathology and 2Medicine, Christian Medical College and Hospital, Ludhiana, Punjab, India) DOI: 10.1309/2AQ5LAYPQ82RHD3P Clinical History Patient 16-year-old Indian boy. Chief Complaint Fever, jaundice, and passage of cola-colored urine for 1 day. The patient was admitted to the hospital for follow-up. Physical Exam Findings The patient was well built and had severe pallor and icterus. There was no organomegaly. Drug History Intake of an unbranded white powder given by a non-licensed individual. 2. The constellation of this patient’s clinical and laboratory findings (eg, markedly decreased hemoglobin and hematocrit levels with a markedly increased serum LD activity), along with the history of intake of an unknown drug and the presence of bite/blister cells and microspherocytes in the PBS, are characteristic of acute oxidant damage to the red blood cells and hemolysis due to glucose-6-phosphate dehydrogenase (G6PD) deficiency. 3. Most likely diagnosis: acute hemolysis due to G6PD deficiency. Deficiency of G6PD is the most common metabolic disorder of red blood cells. Table 1_Principal Laboratory Findings Test Hematology WBC count Hemoglobin Hematocrit Platelet count Reticulocyte count Corrected reticulocyte count Differential: Myelocytes Metamyelocytes Neutrophils Lymphocytes Eosinophils NRBCs PBS findings Chemistry BUN Creatinine Bilirubin, total Bilirubin, direct ALT AST ALP LD Patient’s Result Reference Range 28.3 4.0-11.0 x 103/µL 3.5 12.0-16.0 g/dL 11.4 38-47% 211 150-400 x 103/µL 50.0 0.2-2% 12.6 – 10 0% 11 0% 71 40-80% 7 20-40% 1 0-6% 5/100 WBCs Anisopoikilocytosis with numerous microspherocytes, bite and blister cells, irregularly contracted cells, few macrocytes and nucleated and polychromatic RBCs; some of the RBCs showed basophilic stippling and Howell-Jolly bodies [Image 1] 139 2.0 12.9 1.0 29 244 97 5165 15-45 mg/dL 0.7-1.5 mg/dL 0.5-1.4 mg/dL 0-0.4 mg/dL 5-50 U/L 5-50 U/L 70-230 U/L 230-460 U/L WBC, white blood cell; NRBCs, nucleated red blood cells; PBS, peripheral blood smear; BUN, blood urea nitrogen; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; LD, lactate dehydrogenase. 4. The incidence of the deficiency state varies from 20% among African Bantu males to 12% in African-American men and 8% in Brazilian blacks. G6PD deficiency is also prevalent among the Greek population with a maximum incidence of 20% to 32% in the low lands. In Cambodia, South China, and India, the incidence is 14.0%, 5.5%, and 2.6%, respectively. It is rare among native Americans. labmedicine.com Downloaded from http://labmed.oxfordjournals.org/ by guest on March 5, 2016 Possible Answers 1. Fever, abdominal pain, passage of cola-colored urine, pallor, icterus, severe anemia, leukocytosis, azotemia, hyperbilirubinemia, markedly increased lactate dehydrogenase (LD), and the presence of microspherocytes, bite/blister cells with polychromasia, and nucleated red blood cells in the peripheral blood smear (PBS) (Table 1 and Image 1). LABMEDICINE 䊏 Volume 37 Number 7 䊏 July 2006 Hospital Course The patient’s hematological findings reverted back to normal within 10 days of hospitalization although he developed renal failure consequent to massive hemolysis. Medical/Family History Unremarkable. Questions 1. What is (are) this patient’s most striking clinical and laboratory findings? 2. How do you explain these findings? 3. What is this patient’s most likely diagnosis? 4. What is the geographical distribution of this condition? 5. What is the inheritance pattern and the subtypes of this condition? 6. What is the pathogenesis of the condition leading to hemolysis in this patient? 7. What are the principal clinical features of this patient’s condition? 8. What are the factors which predispose an individual to the clinical manifestations associated with this patient’s condition? 9. What are the laboratory investigations which can be carried out to diagnose this patient’s condition? 10. What are the other red cell enzymopathies which have hematological manifestations? 422 Principal Laboratory Findings Table 1 and Image 1 Case Studies 5. G6PD deficiency is a sex-linked trait with the gene for G6PD located on the X-chromosome (band Xq28). It is expressed in males carrying the variant of the gene, whereas heterozygous females are usually clinically normal. Because of inactivation of one of the 2 X-chromosomes (Lyon hypothesis), heterozygotes have 2 populations of RBCs—normal and G6PD deficient. The degree of lyonisation and the degree to which the abnormal G6PD variant is expressed decide the enzyme activity in females, which may be normal, moderately reduced, or grossly deficient. More than 400 biochemical variant forms of G6PD are recognized. The normal G6PD enzyme is designated as type G6PD B. The most common variant is G6PD A—encountered primarily in individuals of central African descent. This is followed by G6PD Mediterranean in prevalence. The other commonly occurring variants are G6PD A+ and G6PD Canton. The names for the newer variants are designated according to the location of the individual in whom they were first described. 7. The common clinical features of G6PD deficiency are acute hemolytic anemia, favism, congenital non-spherocytic hemolytic anemia, and neonatal hyperbilirubinemia. Acute hemolytic anemia occurs only after exposure to certain offending agents (drugs, infections, acidosis). After 2 to 4 days of exposure to the offending agent, all signs and symptoms of acute hemolytic anemia are observed which include jaundice, pallor, and dark colored urine, with or without abdominal and back pain. There is an abrupt decrease of 3 to 4 g/dL in hemoglobin concentration. The anemia is self-limited because the older susceptible population of RBCs is replaced by younger RBCs with sufficient G6PD activity. A similar clinical presentation occurs in favism on ingestion of fresh fava beans (Vicia fava; European broad bean) or inhalation of pollen from the fava plant. In congenital nonspherocytic hemolytic anemia, anemia, jaundice, and hyperbilirubinemia occur, along with hemolysis in the absence of a triggering factor. Beyond infancy, signs and symptoms of G6PD deficiency are subtle, and mild to moderate anemia is the rule. Neonatal hyperbilirubinemia occurs due to ingestion of drugs, chemicals, and/or fava beans by mothers in late gestation leading to accelerated RBC destruction or impaired liver clearance of bilirubin. 8. Hemolytic crisis occurs only after exposure to certain offending agents, including drugs, infections, exposure to fava beans, and diabetic acidosis. Drugs associated with hemolysis in labmedicine.com 9. The diagnosis of G6PD deficiency can be made by indirect or direct methods. Indirect methods include hematological and biochemical changes. Hematological changes include a fall in hemoglobin concentration by 4 to 8 g/dL and marked anisopoikilocytosis following an acute hemolytic episode. The peripheral blood smear shows microspherocytes, irregularly contracted cells, and eccentrocytes (blister or bite cells). Blister cells are red blood cells in which the hemoglobin appears to be drawn to one side of the cell, leaving an unstained non-hemoglobin containing cell membrane. (Image 1). Also seen are red cell fragments, polychromatic RBCs, and basophilic stippling. There is associated moderate leukocytosis and thrombocytosis. Reticulocytosis occurs within 5 days. Heinz bodies can be demonstrated by supravital staining early in the hemolytic episode. Biochemical changes that provide indirect evidence of hemolysis in G6PD deficiency include hyperbilirubinemia, raised plasma hemoglobin, markedly elevated serum lactate dehydrogenase (LD) activity, low serum haptoglobin level, and positive tests for urine hemoglobin and hemosiderin. Direct methods involve demonstration of deficient or reduced activity of G6PD. It is important that this testing is not carried out during an acute hemolytic episode as reticulocytosis can give rise to a false negative result, the younger RBCs being rich in G6PD. Accurate estimation of G6PD deficiency is possible if testing is carried out 2 to 3 months following an acute hemolytic episode when reticulocytosis has subsided and steady state hematopoiesis has been restored. The majority of these screening tests demonstrate the presence or absence of G6PD by the ability of the red cells to generate NADPH from NADP with simultaneous oxidation of Image 1_Patient’s peripheral blood smear showing microspherocytes, bite/blister cells (arrows) with hemoglobin drawn to one side of the RBC, irregularly contracted cells, and a polychromatic RBC (arrowhead). A nucleated red blood cell is also present along with a neutrophil. (Leishman stain; 1,000x magnification). July 2006 䊏 Volume 37 Number 7 䊏 LABMEDICINE 423 Downloaded from http://labmed.oxfordjournals.org/ by guest on March 5, 2016 6. G6PD is an enzyme of the hexose monophosphate (HMP) shunt pathway, which metabolizes 5% to 10% of the glucose used by RBCs. It catalyzes oxidation of glucose-6-phosphate (G6P) to 6-phosphogluconolactone with simultaneous reduction of nicotinamide adenine dinucleotide phosphate (NADP) to reduced NADP (NADPH). The latter is an important reducing compound for conversion of oxidized glutathione (GSSG) to glutathione (GSH) which protects red cells against free radical (eg, superoxide anion) oxidative injury. Individuals with G6PD deficiency, especially when exposed to certain drugs, are unable to maintain adequate levels of reduced glutathione in their RBCs leading to oxidation of hemoglobin sulfhydryl groups. Hemoglobin precipitates within RBCs, forming Heinz bodies. These membrane-bound inclusions lead to mechanical trapping of affected red cells in splenic capillaries resulting in hemolysis. Both intravascular and extravascular hemolysis occurs in these patients. G6PD deficiency include antimalarials (Primaquine, pamaquine), sulphonamides (Sulphamethoxazole), nitrofurantoin, analgesics (acetaminophen, aspirin, phenacetin), isoniazid (INH), methylene blue, and nalidixic acid. G6PD deficiency due to accidental exposure of children to naphthalene (present in moth balls) has also been reported. The common infectious agents which trigger hemolysis in G6PD deficient individuals are Salmonella, E. coli, β-hemolytic streptococci, and Rickettsiae.1,2 Case Studies an appropriate substrate. The laboratory techniques used to demonstrate G6PD deficiency include the brilliant cresyl blue dye test, methemoglobin reduction test, and the fluorescent spot test.3 An enzyme assay which spectrophotometrically measures the rate of reduction of NADP to NADPH is also available. Common deficient G6PD variants can also be diagnosed by restriction enzyme digestion of the appropriate PCR product.4 More recently, denaturing high performance liquid chromatography (DHPLC) has been used for rapid detection of G6PD mutations.5 10. Other red cell enzymopathies which have hematological manifestations include those associated with enzymes of the Embden-Meyerhof pathway (pyruvate kinase, hexokinase, phosphofructokinase, phosphoglycerate kinase and aldolase), enzymes involved in nucleotide metabolism (adenylate kinase deficiency, pyrimidine 5-nucleotidase), and other enzymes of the HMP shunt pathway (glutathione reductase, glutathione synthetase). LM Keywords: G6PD, lyonisation, hemolysis, anisopoikilocytosis, blister cell, glutathione 1. Frank JE. Diagnosis and management of G6PD deficiency. Am Fam Physician. 2005;72:1277-1282. 2. Glader B. Hereditary hemolytic anemias due to enzyme deficiencies. In: Greer JP, Foerester J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology, 11th ed. Philadelphia: Lippincott Williams and Wilkins; 2004: 1115-1140. 3. Jiang J, Ma X, Song C, et al. Using the fluorescence spot test for neonatal screening of G6PD deficiency. Southeast Asian J Trop Med Public Health. 2003;34S:140-142. 4. Al-Ali AK, Al-Mustafa ZH, Al-Madan M, et al. Molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Eastern Province of Saudi Arabia. Clin Chem Lab Med. 2002;40:814-816. 5. Tseng CP, Huang CL, Chong KY, et al. Rapid detection of glucose-6phosphate dehydrogenase gene mutations by denaturing high-performance liquid chromatography. Clin Biochem. 2005;38:973-980. Downloaded from http://labmed.oxfordjournals.org/ by guest on March 5, 2016 424 LABMEDICINE 䊏 Volume 37 Number 7 䊏 July 2006 labmedicine.com