Chem 340 - Lecture Notes 3 – Fall 2013
advertisement

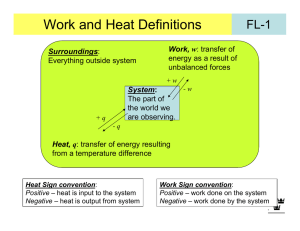
Chem 340 - Lecture Notes 3 – Fall 2013 - Heat and Work and Energy Thermodynamics at its core is about energy, the flow of energy from one substance or system to another i.e. use of energy to do work, to cause electrons to flow in a circuit or to make reactions go; and the result of energy flow evidenced as heat or temperature change. In other words there are many forms of energy that can interconvert. To focus our definitions, we will consider internal energy, U, as that corresponding to: kinetic energy of atoms and molecules in system, potential energy with regard to an applied field, and internal energy of molecules in bonds or in vibrations and rotations First law is experiential: internal energy of an isolated system is constant More interesting when system interact with surroundings, and look at the change, U Utotal = Usystem + Usurroundings = 0 or Usystem = - Usurroundings So if system loses energy, surroundings pick it up, and vice versa (sometimes this is written as: energy of universe is constant, - not so useful) indicates change, but can fix a variable, e.g. T - isothermal, p – isobaric, V - isochoric We need to sum up all those possible energy changes and then we can use this idea. However, simpler, since experience shows all energy changes can be described as heat (q) or work (w) or a combination, which gives the operative First law: U = q + w Now need to define the concepts of heat and work Work, w – something done to system so that energy flows, by nature transitory process rather than state energy remains in system, a state function (see later), which we can analyze by a change in p,V,T Consider system at right, work done on system if piston moves (e.g. gravity causes it to fall when stop removed) w = - ∫ F.dx recall work is force (F) through distance (x) here: F = mg (mass, gravity), distance change - x = h2-h1 (heights) energy change is difference in potential energy, V = mgh Energy change is U = U(p2,V2,T2) - U(p1,V1,T1) just difference in initial and final state energy, not depend on path – state function 1 Sign convention for work: Positive work - done on system, here compression, lower mass in surroundings [V(h) -] Negative work - done by system on the surroundings, here expansion to raise mass In thermodynamics, often more convenient to consider pressure-volume work, pressure is force per unit area against which the mass moves w = - ∫ F.dx = - ∫∫ pext dA.dx = - ∫ pext dV NOTE: pext can differ from psys also have electrical work wel = It, I - current (flow charge), - potential (volts), t – time; or stretching of materials (e.g. membranes), expanding surfaces (bubbles, balloons) in biology - have muscle contraction work, heart motion, artery constrict mechanical cells - mobility (mechanical), charge flow across membranes (electrical, for control and nerve communication), osmotic (concentration change) – all are forms of work Transport Temperature (T), activity/concentration (a) w = ∫ RT.d(ln a) J Examples: Calculate work in: (take care to watch signs) a. Work by person to exhale 0.5 L air (compress lungs) against 1 atm w = -∫PextdV = -Pext(Vf-Vi) = - 1 atm x 0.5 L = 0.5 L-atm (- since person does w) Convert: (1.01x105 Pa/atm)(10-3 m3/L) x 0.5 L-atm = -50 J b. expanding a soap (water) bubble from 1.0 cm to 3.25 cm radius, assume surface tension is 71.99 Nm-1. ( - area, 4r2, tech. 2-D, 1 vari., factor 2, inner-outer surf.) w = -∫d = -24(rf2-ri2) = - 8 x 72 Nm-1(3.252-1.02)cm2 x10-4m2/cm2 = -1.73 J (bubble does work on surroundings, -w) c. transporting 2 g glucose from a 0.01M to a 0.05 M glucose solution through a semi-permeable membrane, at 310 K. Dilute so let a = c MW glucose = 180 g mol-1 n = 2 g/180 gmol-1 = 1.11x10-2 mole w = nRT ∫d(ln c) = nRT ln(c2/c1) (if final c inc., + work done on glucose) -2 = 1.11x10 mole x 310K x 8.314 JK-1mol-1 x ln(0.05/0.01) = 46 J 2 Heat, q, is similar, only a process-transitory, causes a change (i.e. temperature) Transfer of energy due to difference in temperature of system and surroundings Quantity of heat proportional to T compare system to reservoir (source energy) Sign: q positive if deposited in system, Tsur ↓, negative if given off from system, Tsur ↑ Signs — increase Usystem, positive w or q Measure - create isolated composite system where control outer bath temp: Tout = Tin balanced by heating coil-or isolate Composite eliminates “universe” study reaction, determine heat given off by temperature change of inner bath + plus reaction vessel – alternative, closed adiabatic isolated Since T = 0 no heat flow (inout), and since rigid no work, so composite U = q + w = 0 + 0 = 0 isolated Example: A heating coil heats 100 g of liquid water at 100oC in insulated (adiabatic) beaker against 1 bar of external pressure. 10% of liquid is converted to vapor (1 bar) in the process, with 2.00 A from a 12.0 V battery for 103 sec. Densities, liquid: 997 kg m-3, vapor: 0.590 kg m-3 a. diagram at right is model for process –isolated? b. heat equals elect work done on the heating coil q = It = 2.00 A x 12.0 V x 103 s = 24.0 kJ c. work of vaporization, volume inc. vs. const. Pext note: only need to look at 10% which changes w = -Pext (Vf-Vi) = -105 Pa x (0.01 kg/0.590 kg.m-3 - 0.01 kg/997 kg.m-3) = -1.70 kJ = -1.70 x103 J Electrical work for heating much greater than mechanical (P-V) work done - inefficient Heat Capacity Response of system to heat flow is described by heat capacity, depends on material and on phase (solid, liquid, gas) and temperature, and conditions like constant p,V: C = lim(T0) (q/T) = ᵭq/dT - note ᵭq is inexact differential, not integrable ∫ᵭq = q, not q, i.e. there is no q1, q2 Above is an extensive quantity, so often specified as molar quantity, Cm, units JK-1mol-1 Commonly evaluated at constant P or V, for CPm and CVm – note CV or CP (+) 3 Consider diagram for CPm at right for Cl2 The solid phase can be seen as collection of oscillators (lattice that vibrates) and at very low temps the spacing becomes greater than kT, not populated, so cannot take up energy, CPm 0 as T 0 But CPm increases rapidly as kT increase and excite many lattice vibrations, but discontinuity is seen as it becomes liquid. Liquid CPm higher since more low energy vibrations CPm drops on forming a gas, translations cannot take up as much energy. Steady increase after this from putting energy into molecular vibrations (internal states) Use heat capacity to determine heat flow, T1 T2: qp = ∫ Csysp(T)dT = - ∫ Csurp dT see can measure system or surround, water nice bath, Cp ~4.18 Jg-1K-1 from 0 to 100oC Note: system can lose (-) or gain (+) heat (not cold), sign of q relates to process See constant pressure heating must do work, expand V against the pext (pV work) except if pext = 0, free expansion, w = 0 But constant V heating just changes T,p since V = 0, w = 0 For ideal gas this results in relationship of Cp and CV Const V, all heat goes to energy, since w = 0 UV = q Const p, do work, so some heat not used to increase energy, Up < UV Up = q + w - but w(-), system does work Similarly, Tp < TV - since T ~ avg. U leads to Cp > CV – take more heat for same T (“handwave”-ideal: dU = ᵭq + ᵭw = ᵭq - pdV = ᵭq - nRdT (const p) dU/dT = ᵭq/dT – nR (see next page) CV = Cp - nR [Cp = ( ᵭq/dT)p] CP - CV = nR or CPm - CVm = R) Recall: T measure internal energy, U, TU 4 Calorimetry – heat const. V system, dU = dq or U = qV - where w = 0, bomb calorimeter like previous page 3, reaction in bomb changes T of inner bath, but system is adiabatic, no energy flow inner to outer, U = 0, C - system response to heat: dq/dT q = CT where C determined by calibrating, burn known subst. or elect heat: q = It Text example: p.54 – calibrate calorimeter heat with 10A at 12V for 300s q = 10A x 12V x 300s = 3.6x104 J A = Cs-1, C.V = J 4 if calorimeter inc. 5.5 K: C = 3.6x10 J/5.5K = 6.5x103 J/K Increasing temperature increases internal energy, proportionality is heat capacity, however work also done, which will lose some of the heat applied as work on surroundings. So const V system avoids that only heat inc. U w = 0, dU = dq dU/dT = dq/dT Which can be seen to equate to CV = (U/T)V So const. V slope of U vs. T is CV Integrate: U = CVT or qV = CVT (big CV, less T, phase transition, Cv∞) Enthalpy -- Defined as H = U + pV Like U, H is a state function, H independent of path for change Virtue is relation to heat at constant pressure (common for chemical processes) [Consider const. P, dU = ᵭqp – pdV – integrate: Uf - Ui = qp – (pfVf-piVi) or qp=(U+pV)] dH = dqP or for macro change: H = qP justification on p.57 text, consider definition and make infinitesimal change in H (H + dH) = (U + dU) + (p + dp)(V + dV) dH + (H – U – pV) = dU + Vdp + pdV + dVdp (last term, prod. of 2 diff. ~0) dH = (dq + dw) + pdV (use Vdp = 0 const p) so dH = dqp (dw = -pdV for pV work by system on surrounding) measure: isobaric (const. p) calorimeter or differential scanning calorimeter (DSC)Cp (text p. 58 ex: solids, liquids, little change V, H ~ U) Ideal gas: H = U + pV = U + nRT H = U(T) + nRT (n≠0 if rxn produce gas) Clear H depends only on T, ideal, at constant P: response of system to heat, dH = dqp Heat capacity at constant pressure: qp = CpT dqp = CpdT = dH Cp = (H/T)p or Cp is slope of H vs. T (Cp = dH/dT = dU/dT + nR)p – dH depend on dT, like dU So if measure Cp, need to integrate over T to get H - homework or dH = CpdT H = CpT qp = CpT (if Cp constant over T) Cp extrinsic, so often use CPm = CP/n as molar quantity, like CVm 5 ideal: H = U + nRT: Cp = (H/T)p = (U/T)p + nR = CV + nR or Cp -CV = nR (text p.78) Example: Heat water at 100 C and get vapor formed (boil). If 10 g vaporized (lg) and the heat of vaporization at 1 atm is qvap,g = 2.27 kJ/g for H2O, what are H and U for the process (assume ideal gas)? H = qp = qvap,g .m = 2.27 kJ/g x10.0g = 22.7 kJ U = H -nRT = 22.7 kJ – (10g/18gmol-1)x8.314JK-1mol-1x373K = 20.9 kJ DSC analyses are very useful in biochemistry for determining energies of binding of substrates to biomolecules, and of structure changes (folding) p.62 text has a nice discussion of DSC methods and sensitivity it achieves Sample and reference (e.g. solvent) are put in two identical metal holders Each is heated at a constant rate (T=T0+t, t=time) and the difference in heating current is difference in heat capacity sample and reference (qp = CpT, T = t, q = qp + qpex) Cp = qPex/T = qPex/t = Pex/ where Pex - extra power for sample compared to ref. Enthalpy is then: H = ∫ CPexdt Output is directly the Cp need to integrate the curve (area under it) to get H Integration could be done by various methods, setting a series of rectangles under the curve can approximate it, or cutting it out and weighting, or digitizing the curve and integrating (computing area) or fitting to a function with a program. Normally correct for the baseline, i.e. the difference in Cp before and after the transition is Cp for the two states. Assume this data is for a polymer that melts at ~65 C, then Cp between the solid and melted state is ~2mJK-1. To get the H for the transition itself, need H = ∫(CPex – CPbas)dT. To get CPbas normally draw a line between the two ”steady points”, e.g. here at T~38 C and ~85 C. 6 Irreversible work, const pext Reversible processes, pext = pint Above expansions against constant pressure sound “sudden” i.e. release the piston and it goes up or down irreversibly - depend if pext is < or > pint (fig left) wirrev brown area Alternative is to have pext = pint and system is at equilibrium, if infinitesimally reduce pext, system expands, does work, and if then infinitesimally increase p ext can reverse process such a system stays in equilibrium through change and does reversible work, wrev (right) wrev = -∫ pdV - where p is no longer a constant, but changes with V (yellow+brn) Isothermal reversible expansion, would have T = 0, and wrev, use p = nRT/V wrev = -nRT ∫(dV/V) = -nRT ln(Vf/Vi) T = 0, so U = 0 = q+w or q = -w the shaded area in right figure is wrev, yellow is added expansion work: wrev > wirrev or wrev is wmax, cannot obtain more work from system than if pext = pint or wrev Adiabatic expansion, work, w ≠ 0, but no heat flow, q = 0, so U ≠ 0, T ≠ 0, U = wrev all T change is U, but due to w U = CVT = Can show, text p. 84, # 2.1 that Ti/Tf= (Vf/Vi)-1 where = CPm/CVm = 1+ R/CVm Ti/Tf= (Vf/Vi)1/c Tic Vi = TfcVf where c = CVm/R Similarly the pressure change is pi/pf= (Vf/Vi) piVi= pfVf See different behavior isothermal and adiabatic (T) rev. adiabatic compression: T(+), expansion: T(-) Example: Reversibly adiabatic expand 0.02 mol He, Vi =0.50 Vf = 2.00 dm3 at Ti = 25 C; ideal: CV = 3/2 R (c =1.5) Tf = Ti(Vi/Vf)1/c = 298 K (0.5/2.0)2/3 = 118 K, T = -180K q=0 so w = U = CVT = 0.02 molx1.5 x 8.314 Jmol-1K-1x(-180K) w = - 44.9 J = U (note text p.64 diff answer ? different CV?) -2 3 pi = nRT/Vi = 0.02molx8.314x10 dm barK-1mol-1x298K/0.50dm3 = 0.99 bar pf = pi(Vi/Vf)= 0.99 bar (0.5/2.0)5/3 = 0.39 bar = CPm/CVm = 5/3 (Cp = CV + R) 7 State functions U = ∫dU = Uf –Ui - state function only depend on initial and final state, exact differential If integrate steps around a cycle: U = ∫O dU = Uf –Uf = 0 – i.e back where started State determined by relationship of 2 of 3 state variables: U(P,V) or U(P,T) or U(V,T) i.e. P, V, T not independent, coupled by the equation of state Work and heat not state functions Consider example at right Weight held by stops, provides constant Pext Drop piston isolated system get, V1V2, P1P2, T1T2 Then bring into contact with bath, T3, get P2P3, const.V2 Do it again with diff. mass, diff Pext but same final V2, P3, T3 So U same in both processes, but w = -PextV differ Since U = q + w, q must also differ: q=U - w = U + PextV q and w are path functions, depend on process, represent as inexact differentials, ᵭq and ᵭw, ∫ᵭq = q, no qi or qf important to recognize - system does not have heat or work, it has energy that can convert to heat or work Since U is state function, can analyze its change by any process that goes from initial to final state, so can imagine breaking up change into segments, e.g isothermal change to final volume and isochoric (const V) change to final temp. Each is easier to analyze and their sum gives total energy change between states Example: Compress 1.0 mole of ideal gas 10 4 L, from initial T of 800K to final T of 200K. What is U? Assume isothermal step at 800K 104 L U1 = 0 -q=w, w= -∫pdV= -nRT∫dV/V= -nRTln(Vf/Vi) (w= -1.0 molx8.314 Jmol-1K-1x800Kxln(4/10)= 6.1 kJ) then do constant volume step cooling 800200K: U2 = q+w = q = CV∫dT = CVT = (3/2R)(800-200)= 7.48 kJ U =U1 +U2 = 7.48kJ, qtot=(-6.1+7.48)kJ =1.38kJ, wtot= w1= 6.1kJ, U=q+w= 7.48kJ 8 Equilibrium - review Internal equilibrium, system has properties that fit is equations of state, see plot above, if choose any point on the 3-D surface then T,P,V values reflect a system in equilibrium Points off surface do not fit any physically realizable state of system—non-equilibrium Quasi-static process –change in infinitesimal steps - each at an equilibrium, reversible If change is fast or big step, can still do thermodynamics, initial & final, but irreversible Reversible and irreversible work example (review) Engel: fig. 2.9 fig. 2.10 Do cycle: Start P1 = Pext, creates V1 at T1, remove weight, get P’ext=P2, expand to V2 suddenly, irreversible, but keep in diathermal container in a bath, so const T1 Now put weight back on, compress back to V1. Consider cycle: U = 0 = qtot +wtot wtot = -P2(V2-V1) – P1(V1-V2) = -(P2-P1)(V2-V1) wtot > 0 since P2 < P1 (wtot on syst) Fig 2.9 - red rectangle work (-) 1st step, red plus yellow (+) work 2nd step, wtot = yellow Arrows indicate which process (compress, expand) happening (sorts signs) Since U = q + w = 0, then qtot = -wtot and thus qtot < 0 (system loses heat to surr.) Alternative, do reversible isothermal process wrev = -∫pdV = -nRT∫dV/V = -nRT ln V2/V1 Same magnitude both directions (reversible), wtot = -nRT ln V2/V1 - nRT ln V1/V2 = 0 Fig 2.10, both same area, but areas differ in irrev. - one larger one smaller, Fig.2.9 First step, expansion work by system, wrev > wirev - focus on work by system (but total work in cycle, wrev < wirev, which is fct of sign convention) Similarly, qtot = -wtot = 0 reversible conditions, require const. T, if T vary: q≠0, w≠0, case for adiabatic 9 Imagine doing the expansion in two steps, to intermediate Px, Px< P1, Px> P2 Look at work, wirev, expansion work by system, 2 step more w (“more” mean more neg.) w1 + w2 = -Px(Vx-V1) -P2 (V2-Vx) = -(Px-P2)(Vx-V1) -P2 (V2-V1) > -P2 (V2-V1) (neg↑) Engel - Fig 2.11 -- if more steps, get more rectangles, each touches curve of wrev at top, (stops at curve, Pext=Pint, equil.) inc. # steps inc. work possible by the system Expansion work – red+tan reversible, tan only –irreversible in steps Compression – green irreversible in steps, black line is reversible again See increase number of steps approach reversible, so maximum expansion work, maximum work by the system always reversible wmax extract from a system between same initial and final states is always wrev Irrev: wtot >0, qtot < 0, U=0 in cycle system in original state – but surroundings not surroundings did work on system in cycle, gained heat important for entropy Note: engine or battery must not work in infinitesimal steps, work will not be useful 10 Bio example: consider conversion glucose to ethanol C6H12O6 2C2H5OH + 2CO2 In cell this is facilitated by enzyme and uses ADPATP to store some energy C6H12O6 +2HPO42- + 2ADP3- 2C2H5OH + 2CO2 +2ATP4Overall efficiency of bio reaction – break into multiple steps-- is ~30% But if done in single step, only work would be pV expansion, wirrev, of CO2, effic~3% 11