c325 UV Vis COMPLETE notes
advertisement

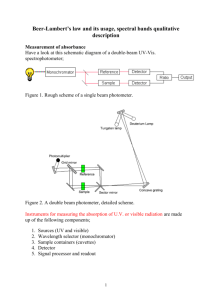
Electromagnetic Spectrum Chem 325 UltraViolet-Visible Spectroscopy λν = c E = hν ν = hc/λ λ Organic Spectroscopy Absorption of different EM radiation produces different molecular energy changes 1. 2. 3. 4. 5. UV-vis Absorption Spectroscopy Light of wavelength 190 to ~800 nm is passed through a sample. Amount of light that makes it through the sample is compared to the amount when the sample is not present. Radiowaves Nuclear Spin Transitions (NMR) Microwaves Electron Spin Transitions (ESR) Infrared Vibrational Transitions Visible-NIR Raman Scattering (Vibrational) Ultraviolet-Visible Electronic Transitions 1 Transmittance and Absorbance Beer-Lambert Law The transmittance is the ratio of the light that is detected when the sample is present to the ratio when the sample is not present. Transmittance is related to concentration in a non-linear way (exponential), so it is usually converted to the much more useful quantity ‘absorbance’, where: T= I I0 Transmittance is measured in the following way: Absorbance = A = log I0 I Absorbance is related to concentration in a linear way according to the Beer-Lambert Law. Absorbance = A = εlc Where l is the pathlength in centimeters And c is the concentration of the absorbing species in Molarity (mol/L) And ε is the molar absorptivity or molar extinction coefficient Molar Absorptivities No absorption gives ε = 0! Often given as logarithmic values e.g. ε = 23,500 equivalent to log ε = 4.37 Units of ε ε= A cl L mol-1 cm-1 or M-1 cm-1 - very rarely stated explicitly! The typical UV-vis spectrometer scans the wavelength range of 190 nm to 800 nm, and the absorption at each wavelength is plotted vs the wavelength in nm. 2 Absorbance Molar absorptivities may be very large for strongly absorbing compounds (ε >10,000) and very small if absorption is weak (ε = 10 to 100). Absorbance Spectra λmax λmax λmax λmax 1.5 1 0.5 0 255 280 305 330 355 380 Wavelength (nm) 2 Electronic Transitions Chromophores UV and Visible light can be absorbed by organic molecules causing an electronic transition. What physically happens is an electron from the Highest Occupied Molecular Orbital (HOMO) is promoted to a higher energy orbital. Solvents • Normal hydrocarbons: no UV-vis absorptions • They do have absorption but in the FAR UV (vacuum UV) • Requires presence of a chromophore – a group with easily promoted electrons • Typically: π-bond systems Solute-Solvent Interactions Solvents generally required due to large absorptivities Must be ‘transparent’, i.e. not absorb OH Must be very pure Solvent cutoffs (where they start to absorb!): acetonitrile chloroform cyclohexane 1,4-dioxane 95% ethanol 190 nm 240 195 215 205 n-hexane methanol iso-octane water trimethylphosphate 201 nm 205 195 190 210 3 Electronic Energy Levels Electronic Transitions Electronic Transitions Ethylene Orbital and Transitions Organics that absorb in the UV and Visible region (200 – 800 nm) generally contain one or more πbonds. 4 Molecular Orbital Diagrams Conjugated molecules such as 1,3-butadiene absorb at longer wavelengths. Excited States Electron Spin Selection Rule When a molecule absorbs light, this energy promotes an electron from an occupied MO to an unoccupied MO. This produces a ‘Singlet Excited State. A number of singlet excited states are possible, and they are labelled S1, S2, …, Sn. The ground state is always S0. S0 → S1 is spin-allowed both states are ‘singlet’ spin multiplicity = 2S+1, S = ½ for each unpaired e- Spin multiplicity = 3 A TRIPLET state α S0 → T1 is spin-forbidden T1 ground state (S0) S1 S2 S3 5 State Diagrams Electronic and Vibrational Transitions The possible excited states are often drawn as a ‘state diagram’ or ‘Jablonski diagram’. The vibrational energy levels are shown for each electronic level. Intersystem crossing Rigid Compounds Non-Rigid Compounds In rigid molecules, like polyaromatic hydrocarbons, excitation to each vibrational level is resolved. These narrow peaks appear in sets, called ‘bands’. If the molecule is flexible, several conformations are possible at any given time. The vibrational ‘fingers’ for each conformation are averaged to give a rounded band. 0.8 ν=1 ν=2 0.4 1.6 S0 S1 ν=3 0.2 Absorbance Absorbance 2 ν=0 0.6 1.2 0.8 S0 S1 0.4 0 275 325 375 Wavelength (nm) 425 0 Vibronic Structure 200 250 300 350 Wavelength (nm) 6 Types of Excitation What Factors Affect Absorption? Only two types of excitation commonly encountered, depending on the orbitals involved. These are: A) π* n (or n π*) Electron from an n orbital (nonbonding or lone pair) is excited into a π* (antibonding) orbital. Commonly the longest wavelength absorption for ketones and aldehydes. π* n transitions are “forbidden”, so these usually give weak bands (small ε) B) π* π (or π π*) Electron from a π-orbital is excited into a π*-orbital. This is commonly the longest wavelength absorption for unsaturated hydrocarbons. Substituent Effects Empirical rules: help predict the λmax of a specific compound from the base chromophore and what substituents are attached to it. H2C λmax nm CH2 Hyperchromic Hypsochromic ε 175 15,000 217 21,000 258 35,000 465 125,000 β-carotene Bathochromic Hypochromic 200 nm Lengthening the conjugation also increases the molar absorptivity (more intense absorption bands). Adding substituents and functional groups will have the same effect but to a much smaller degree. Chromophores and Substituents Substituents may have any of four effects on a chromophore 1. Bathochromic shift (red shift) – a shift to longer λ: lower E 2. Hypsochromic shift (blue shift) – shift to shorter λ: higher E 3. Hyperchromic effect – an increase in intensity: higher ε 4. Hypochromic effect – a decrease in intensity: lower ε ε The major factor that affects absorption is the degree of conjugation. The longer the conjugation, the lower the energy required to excite (and therefore longer wavelength band). 700 nm O n π* 280 π π* 189 12 900 O n π* 280 π π* 213 27 7,100 7 Woodward-Fieser Rules for Dienes For a compound to absorb above 200 nm, generally it will contain some degree of conjugation. The simplest conjugated molecule is a diene. Woodward-Fieser Rules for Dienes Next we add the contribution from any attached groups (substitutents). Group Extended conjugation Each exo-cyclic C=C Alkyl -OCOCH3 -OR -SR -Cl, -Br -NR2 -Ph Woodward and Fieser developed a set of empirical rules to help predict what the λmax will be for a diene-based compound. There are separate rules for cyclic and acyclic dienes. Butadiene is the simplest acyclic diene and has an absorption maximum of 217 nm. acyclic butadiene, λmax = 217 nm Examples Experimental value Allylidenecyclohexane acyclic butadiene = one exocyclic C=C 2 alkyl subs. Experimental value +5 +5 +0 +6 +30 +5 +60 +60 Cyclic Dienes Isoprene acyclic butadiene = one alkyl subs. Increment +30 217 nm + 5 nm 222 nm 220 nm 217 nm + 5 nm +10 nm 232 nm 237 nm There are two major types of cyclic dienes, with two different base values. Heteroannular (transoid): Homoannular (cisoid): ε = 5,000 – 15,000 base λmax = 214 ε = 12,000-28,000 base λmax = 253 Increment table is the same as for acyclic butadienes with one addition: Additional Homoannular: +39 If two dienes are present in a molecule, the base with longer λmax is used. 8 Example heteroannular diene = 214 nm 3 alkyl subs. (3 x 5) +15 nm 1 exo C=C + 5 nm 234 nm Structure Determination? In the pre-NMR era of organic spectral determination, the power of the method for discerning isomers is readily apparent: Consider abietic vs. levopimaric acid: Experimental value 235 nm C OH O abietic acid Types of Excitation Only two types of excitation commonly encountered, depending on the orbitals involved. These are: A) π* n (or n π*) Electron from an n orbital (nonbonding or lone pair) is excited into a π* (antibonding) orbital. Commonly the longest wavelength absorption for ketones and aldehydes. π* n transitions are “forbidden”, so these usually give weak bands (small ε) B) π* π (or π π*) Electron from a π-orbital is excited into a π*-orbital. This is commonly the longest wavelength absorption for unsaturated hydrocarbons. C OH O levopimaric acid Structure Determination! abietic acid heteroannular diene = 214 nm 4 alkyl subs. (4 x 5) 1 exo C=C C OH O +20 nm + 5 nm 239 nm levopimaric acid homoannular diene = 253 nm 4 alkyl subs. (4 x 5) 1 exo C=C C OH O +20 nm + 5 nm 278 nm 9 WARNING !! Woodward-Fieser Rules Three common errors: R This compound has three exocyclic double bonds; the indicated bond is exocyclic to two rings See Pavia, Chapter 7, Section 10 for the Rules and worked examples!! This is not a heteroannular diene; you would use the base value for an acyclic diene Likewise, this is not a homooannular diene; you would use the base value for an acyclic diene Product Analysis Product Analysis ∗ CH3 Base value CH3 H2N 214 Homoannular 39 Alkyl substituents 3 × 5 15 Exocyclic C=C H2N ∗ H2N H2N O CH3 CH3 CH3 Two possible enamines from this ketone. Can UV-vis tell them apart? NH2 group Predicted λmax 5 60 333 nm Base value Alkyl substituents 3 × 5 Exocyclic C=C NH2 group 214 15 5 60 Predicted λmax 294 nm 10 The Carbonyl Group Absorptions Woodward-Fieser Rules for Enones Enones have a strong π* π band and a longer wavelength π* n that is usually around 100 times less intense. •Two possible absorptions – Longer wavelength (lowest E) is n → π* – Symmetry ‘forbidden’, thus low ε If the enones are conjugated enough, the π* π band completely swamps out the π* n band. Also, the positions of π* π bands are much easier to predict with empirical rules than π* n bands. – Shorter wavelength (highest E) is π → π* – Symmetry ‘allowed’, thus high ε – Can conjugate with alkenes π-systems For these reasons, the WoodwardFieser rules for enones ONLY APPLY TO THE π* π TRANSITION!! Ketones Group Increment 6-membered ring or acyclic enone Base 215 nm 5-membered ring parent enone Base 202 nm Acyclic dienone Base 245 nm δ γ β α δ C C C C C O Double bond extending conjugation 30 Alkyl group or ring residue α, β, γ and higher -OH α, β, γ and higher 35, 30, 18 -OR α, β, γ, δ 35, 30, 17, 31 10, 12, 18 α, β, δ 6 -Cl α, β 15, 12 -Br α, β 25, 30 β 95 -O(C=O)R -NR2 Exocyclic double bond 5 Homocyclic diene component 39 Aldehydes and Acids/Esters Unsaturated system Aldehyde With α or β alkyl groups With α,β or β,β alkyl groups With α,β,β alkyl groups Acid or ester With α or β alkyl groups With α,β or β,β alkyl groups Group value – exocyclic α,β double bond Group value – endocyclic α,β bond in 5 or 7 membered ring Base Value 208 220 230 242 208 217 +5 +5 11 Solvent Effects on Enones Examples α For enones, the solvent will also affect the position of λmax. Solvent correction cyclic enone = 2 x β - alkyl subs.(2 x 12) O β Experimental value Increment Water +8 Ethanol, methanol 0 Chloroform -1 Dioxane -5 Ether -7 Hydrocarbon -11 R cyclic enone = extended conj. β -ring residue δ-ring residue exocyclic double bond O Experimental value Product Analysis Br CH3 CH3 Br2 CH3 O H O 215 Base value 215 12 β -alkyl ×2 24 H H H CH3 O O O 215 nm +30 nm +12 nm +18 nm + 5 nm 280 nm 280 nm Product Analysis Bromination a steroid can produce two possible products. Dehydrobromination gives two enones. Can we tell them apart? CH3 215 nm +24 nm 239 nm 238 nm Br -HBr Base value -HBr β -alkyl 227 nm CH3 CH3 Exocyclic C=C 5 244 nm O O H 12 UV-Vis of Aromatics Aromatics Benzene has 6 π-MOs which leads to a number of transitions. π6 ∗ π4 ∗ π5 ∗ π2 π3 π1 Benzene has three main bands, the E, K, and B bands. The E band is also called the primary band. It is strongly allowed (εε = 47,000) but shows up below 200 nm. The K band is called the second primary band, can be observed above 200 nm if substituents cause a red shift. Its molar absorptivity is 7400. The longest wavelength B band (260 nm) is called the secondary band and is forbidden and therefore weak (εε = 230) Aromatics in General Substituent effects Substitution with auxochromes lead to the same general effects as observed for dienes and enones, but in a less predictable way. The formation of rules for predicting the position of the bands is not very useful since there tend to be more exceptions than there are rules. However, we can certianly highlight qualitative trends. G G G G 1. Substituents with lone pairs will red-shift the primary and secondary bands. 2. Protonating or deprotonating functional groups changes how they affect the primary and secondary bands. Primary Secondary Substituent λmax ε λmax ε -H 203.5 7,400 254 204 -OH 211 6,200 270 1,450 -O- 235 9,400 287 2,600 -NH2 230 8,600 280 1,430 -NH3+ 203 7,500 254 169 -C(O)OH 230 11,600 273 970 -C(O)O- 224 8,700 268 560 3. Functional groups that extend the conjugation red shift the primary and secondary bands. 13 Electronic Effects Di-Substituted Aromatics Electron withdrawing groups (EWG) red-shift the primary band and electron donating groups (EDG) red-shift both bands. Primary Substituent Electron withdrawing Electron donating -H 1) If both are EWG or both are EDG, the effect is equal to the stronger of the two. Secondary λmax ε λmax ε 203.5 7,400 254 204 -CH3 207 7,000 261 225 -Cl 210 7,400 264 190 -Br 210 7,900 261 192 -OH 211 6,200 270 1,450 -OCH3 217 6,400 269 1,480 -NH2 230 8,600 280 1,430 -CN 224 13,000 271 1,000 C(O)OH 230 11,600 273 970 -C(O)H 250 11,400 -C(O)CH3 224 9,800 -NO2 269 7,800 2) If one is an EWG and the other is an EDG, the overall effect is additive if they are ortho or meta to one another. 3) If one is an EWG and the other is an EDG, the overall effect is greater than additive if they are para to one another. _ O + O N OH Woodward and Fieser Again… _ O _ N O OH + Woodward-Fieser Rules for Aromatics Woodward and Fieser were able to come up with some rules for predicting the λmax of a subset of aromatic compounds – those containing a carbonyl attached to the ring. A substituent will have a different effect depending on whether it is ortho, meta, or para substituted. Note that the strongest effects are observed for para substituents. Substituent increment The rules are for R = H, R, OH, and OR. The rules are not as accurate as for dienes and enones, but are generally within 5 nm. The following are the possible base structures. O R G o m p Alkyl or ring residue 3 3 10 -O-Alkyl, -OH, -O-Ring 7 7 25 -O- 11 20 78 -Cl 0 0 10 Parent Chromophore λmax -Br 2 2 15 R = alkyl or ring residue 246 -NH2 13 13 58 R=H 250 -NHC(O)CH3 20 20 45 R = OH or O-Alkyl 230 -NHCH3 20 20 G -N(CH3)2 73 85 14 Visible Spectrum Molecules absorbing between 400 and 800 nm can be detected by the human eye. The common colours and their associated wavelengths are given below. UV Vision Many insects have vision that extends into the UV region, which is useful for them since many flowers have UV colourings that are invisible to humans. β -carotene Appearance of Coloured Compounds The appearance of coloured compounds can be determined by looking at the colour wheel below. Consider β-carotene β-carotene, λmax = 455 nm This molecule absorbs at 455 nm (in the blue), therefore the compound will appear orange. Select the λmax of the compound of interest, and the appearance of that compound will be the colour opposite it. What we ‘see’ is the opposite of the colour absorbed. β-carotene is the primary pigment that colours carrots. 15 Lycopene and Indigo Azo Dyes Similarly, lycopene absorbs at 474 nm and appears red. This compound is the main pigment found in tomatoes. One of the most common classes of organic dyes are the azo dyes. They have the following general structure: N N lycopene, λmax = 474 nm O H N EWGs N H EDGs O indigo These dyes have an EDG on one ring and an EWG on the other, similar to disubstituted benzenes. λmax for indigo is at 602 nm – in the orange region of the spectrum – this is absorbed, the compliment is now indigo. Azo Dyes Azo Dyes Many azo dyes are pH sensitive, which makes them useful pH indicators. Azo Dyes are used to colour a variety of materials. NO2 Methyl Orange HO N N H2N OH N N O 3S N N O3S NH2 Yellow, pH > 4.4 SO3 Para Red Fast Brown N N CH3 N CH3 O3S H N N CH3 N CH3 Red, pH < 3.2 Sunset Yellow (Food Yellow 3) 16 Final Notes UV-Vis not especially useful as a primary tool for determining the structures of organic molecules. However, it is useful in certain instances to distinguish between isomers. Main contribution that UV-Vis makes to structure determination is that it can readily identify the presence of conjugated π-systems and unique chromophores. Final Notes • UV-vis most widely used instrumental technique in chemistry and science/medicine! • Detection in chromatography. • Monitoring reaction kinetics (chem, biol, medicine) • Materials science, synthesis, analytical, inorganic, organic, physical chemistry, biochemistry, biology, medical applications, food industry. Polarimetry Chiral Molecules and Optical Activity (R), (S) Nomenclature • Different molecules (enantiomers) must have different names. • Configuration around the chiral carbon is specified with (R) and (S). • Cahn – Ingold - Prelog • • • • • • Use monochromatic light, usually sodium D-line, 589 nm Movable polarizing filter to measure angle Clockwise = dextrorotatory = d or (+) Counterclockwise = levorotatory = l or (-) Optical activity Not related to (R) and (S) => 17 Specific Rotation Observed rotation depends on the length of the cell and concentration, as well as the strength of optical activity, temperature, and wavelength of light. [α α] = α (observed) c•l Racemic Mixtures • • • • Equal quantities of d- and l-enantiomers (or R and S forms). Notation: (d,l), (± ±), (R,S) No optical activity. The mixture may have different b.p. and m.p. from the pure enantiomers! c is concentration in g/mL l is length of path in decimeters. Normal UV-vis spectrum Isotropic radiation (unpolarized) Same spectrum for both enantiomers It is possible to measure the optical rotation (of an enantiomer solution) at different wavelengths by using a monochromator. The variation of optical rotation with wavelength of the (plane) polarized light is called OPTICAL ROTATORY DISPERSION, a.k.a. the ORD spectrum 18 • Optically active compounds rotate the plane of linearly polarized light • Different ε for left and right circularly polarized light, ∆ε = εL - εR ≠ 0 • Related to the difference in refractive index for left and right circularly polarized light Normal UV-vis CD Spectrum • Difference in molar absorptivity ε is called CIRCULAR DICHROISM Chiroptical Spectroscopy • Optical rotatory dispersion (ORD) and circular dichroism (CD) known as CHIROPTICAL methods USES • Deconvolution of overlapping UV-vis bands • Relative chiralities of isomers • Absolute configurations of molecules • Biomolecules: amino acids, proteins, enzymes, DNA • Macromolecules, polymers with ‘handedness’ 19