Structures of Metals and Ceramics: Chapter 3.
advertisement

Chapter 3
Highlights:
1.
3 types of materials- amorphous, crystalline, and polycrystalline.
2.
Understand the meaning of crystallinity, which refers to a regular lattice based on a
repeating unit cell.
3.
For fcc, bcc, and sc lattice, be able to determine the number of atoms in each cell (n)
and the atomic packing factor (APF), also be able to determine the relationship
between the atomic radius (R), the lattice constant (a) and the density
ρ = ρ (n, a, R).
4.
Know the common ceramic crystal structures and the properties (ionic charge and
ionic size) that determine crystal structure. Be able to use this information to predict
crystal structure.
5.
Same as #3 for the ceramic and diamond crystal structures.
6.
In a cubic unit cell, be able to identify crystallographic directions and draw them.
7.
In a cubic unit cell, be able to identify crystallographic planes and draw them.
8.
In a cubic unit cell, be able to determine linear and planar atomic densities.
9.
Be able to match an x-ray crystallography pattern to the crystal structure and to use
the diffraction pattern to obtain the lattice constant or atomic radius.
Notes:
Metallic Crystal Structures
Metals, ceramics, polymers, etc. may be either:
1.
2.
3.
Crystalline- Si for semiconductor and photovoltaic devices.
Amorphous- corrosion-resistant Ni plating, no grain boundaries.
Polycrystalline- structural steels, other industrial metals (see figure).
c03f35
Figure 3.35 (above) from text, good example of polycrystalline structure.
You might want a different morphology depending on the application.
Example 1: Corrosion resistant Ni coatings are amorphous, since grain boundaries provide
an easy path for corrosion to weaken a material.
Example 2: Si semiconductor devices are constructed from single crystal (crystalline over
long length scales) Si wafers. Show IBM wafer. The required electrical performance of cpu
chips, DRAM chips, etc. depends on using single crystal Si.
Lattice:
Unit cell:
Infinite array of points in a solid.
Smallest repeating configuration of atoms from which lattice can be constructed.
Table below illustrates the different crystal systems, with different combinations of a,b,c,α,β,γ. In
ES 260, we will focus on only one crystal system, the cubic system, which is the most important.
However, many common materials fall in the hexagonal system.
c03tf06
FCC unit cell (Figure 3.1)
c03f01
A)
# of atoms, n = 8(1/8) + 6(1/2) = 4
B)
Coordination number = 12, count nearest neighbors.
C)
Show drawing of one face, relate lattice constant (a) to atomic radius (R), show that
a = 2 2 R . This relies on recognizing that the atoms are touching along the diagonal
of each face, as can be seen in the upper left graphic.
D)
Calculate density ρ from equation (3.5):
ρ=
ρ=
nA
Vc NA
4A
3
(2 2 R ) N A
=
A
4 2 R3 N A
Given A and a or R, be able to calculate ρ and vice versa. Also, be able to determine the
crystal structure given A, a and ρ.
E)
Atomic packing fraction (APF)
APF =
Volume of atoms within unit cell
Total unit cell volume
4
n π R3
4π n R 3
3
APF = 3 =
3 a3
a
APF =
APF =
Example:
4π (4) R 3
3
3(2 2 R )
π
3 2
= 0.74
Calculate the radius of a iridium atom, given that it has a FCC structure, with a
density of 22.4 g/cm3 and an atomic weight of 192.2 g/mol. Start with the equation
that we derived above for the density of a FCC material:
ρ=
4A
3
(2 2 R ) N A
22.4 g / cm 3 =
(2
=
A
4 2 R3 N A
4 (192.2 g / mol )
2R
) (6.023 x10
3
23
/ mol
)
R 3 = 2.518 x10 −24 cm −3
R = 1.36 x10 −8 cm = 0.136 nm
Next two figures illustrate body-centered cubic (BCC) and hexagonal close-packed (HCP) crystal
structures. You should be able to do the above calculations (A-E) for the BCC unit cell, and for all
other cubic unit cells covered in this course.
c03f02
Note that atoms within the BCC unit cell are touching along the body diagonal, meaning the
diagonal that crosses the unit cell in all three coordinates (x,y,z). This must be recognized in
order to determine the relationship between a and R.
c03f03
c03f29
c03f30
c03f31
c03tf01
Something to think about:
Why are there different metal crystal structures?? Remember that metallic bonding is mostly
nondirectional, with ion cores floating in a sea of electrons. Show figure 2.11 (below). You might
expect metallic bonding to give the closest-packed structure, with the maximum APF and maximum
coordination. Both FCC and HCP have APF=0.74 and coordination # of 12, which are the highest
possible, so about 3/4 of metals are FCC or HCP. But why do other metals have the BCC structure
(APF=0.68, coordination # = 8)? This is complicated but involves some degree of covalent bonding
through the d-orbitals of transition metals. Later we will introduce the diamond crystal structure
(silicon, germanium, diamond), where the APF is only 0.34. This structure is favored due to
covalent bonding in these materials, which requires a coordination number of only 4.
c02f11
Ceramic Crystal Structures
Ceramic materials are wholly or substantially ionic. For this reason and because the unit cell must
contain two different species, they form different crystal structures from metals. Ceramic crystal
structure is determined by the relative charge and relative size of the anion and cation. Monitor this
by rC/rA, which is always less than one, since the size is determined by the number of electrons in
the electron cloud. Remember that in a ceramic, the cation gives up electrons to the anion, so the
anion is normally much larger.
Ceramic crystal structure: total cation, anion charges must be equal.
1)
2)
Take the many possible geometrical arrangements of the large anions as in
VSEPR (linear, triangular, tetrahedral, octahedral, etc.)
Choose the structure where the cation just fits into the interstice. This maximizes the
Coulomb attraction and binding energy.
Table 3.3 below is constructed by the above two principles. The ratio rC/rA given in table 3.3 can be
derived from those principles. The most common coordination numbers are 4, 6, 8. Ionic radii are
given in table 3.4 below.
c03tf03
c03tf04
Example:
Show that the minimum cation-to-anion radius ratio for a coordination number of 6
is 0.414.
c03f04
Draw four anions in a square (above) so that they just touch. Now put a cation into the interstice
space. Now you can draw a right triangle with two side of length 2rA + 2rC and the hypotenuse of
length 2rA. Therefore,
cos( 45° ) =
2
2r A
=
2 2rA + 2rC
This can be solved to yield,
1+
rC
2
=
= 1.414
rA
2
rC
= 0.414
rA
This is the borderline between coordination numbers of 4 and 6 in table 3.3.
A-X Crystal Structures
First, consider ceramic crystal structures with an equal number of anions and cations. Figures
below show zinc blende (CN 4), rock salt (CN 6), CsCl (CN 8) structures. The rock salt structure is
like two superimposed FCC structures. The CsCl structure looks like BCC, but is a uniquely
different crystal structure because two different ions are involved. Examples of these structures are
ZnS (rC/rA=0.402, zinc blende structure), NaCl (rC/rA=0.564, rock salt structure), and CsCl
(rC/rA=0.939, CsCl structure).
c03f05
In the figure above, the ions are actually touching along each edge of the cube, so a = 2rA +
2rC. Counting ions, nA = 8(1/8) + 6(1/2) = 4 and nC = 12(1/4) + 1 = 4.
c03f06
For the CsCl unit cell above, ions are touching along the body diagonal, similar to the BCC
unit cell. Counting ions, nA = 8(1/8) = 1 and nC = 1.
c03f07
For the zinc blende structure above, ions connected by dotted lines are touching. If you can
obtain the Cartesian coordinates (xi, yi, zi) of a cation and anion that are touching, their
separation distance (rA + rC) is equal to their separation according to (Δxi2 + Δyi2 + Δzi2)1/2
AmXp Structures
If m and/or p are not equal to 1, the number of cations and anions are unequal. The ceramic crystal
structures are the same as AX, but some positions are now left empty. For CaF2, show figure 3.8
below. Same as CsCl, but half of the cation (Ca) sites are empty.
c03f08
Ceramic density calculations
Similar to metals, the density of ceramic materials can be calculated if one knows the numbers of
the different species in the unit cell, the ionic radii, and the atomic weights. From equation (3.6):
ρ =
n'
(∑ A + ∑ A )
C
A
VC N A
This includes summations over atomic weights, and introduces n’, the number of formula units of
the ceramic material within the unit cell. This is best explained with an example.
Example:
MgO has the rock salt crystal structure and a density of 3.58 g/cm3. What is the
lattice constant, and how does this compare to the sum of the ionic radii in table 3.4?
In the above equation for the density of a ceramic, we need to know the number of formula (MgO)
units in the unit cell. Inspection of figure 3.5 (above) shows that the unit cell contains 4 anions and
4 cations. Therefore, the unit cell contains 4 units of MgO and n’ = 4. The rest is just substitution:
ρ =
n'
3.58 g / cm 3 =
(∑ A + ∑ A )
C
A
VC N A
4 (24.31 + 16.00 g / mol )
a 3 (6.023 x10 23 / mol )
a 3 = 7.478 x10 −23 cm 3
a = 4.21 x10 −8 cm = 0.421 nm
One expects a = 2rA + 2rC = 0.424 nm . This is a discrepancy of 0.7% from the calculated value.
Diamond crystal structure
The diamond crystal structure (below) is the crystal structure of Si, Ge and C (diamond). This is
introduced mainly because of the widespread use of Si to make semiconductor devices (transistors),
from which integrated circuits are built. The semiconductor chips in a personal computer (cpu,
DRAM) and a cell phone are built starting from crystalline Si. This is by far the most widespread
application of a crystalline material, since metals are polycrystalline in most applications. For this
reason, crystalline Si is several orders of magnitude cheaper than most other crystalline materials.
Carbon can also be found as graphite, C60, and nanotubes, as shown in figures below.
c03f16
c03f17
c03f19
c03f18
Point Coordinates
In order to name directions and planes, we need to learn to identify the location of points within the
unit cell. Go through example problems 3.7 and 3.8 on your own. The point coordinates are always
given as a fraction of the lattice constants.
Crystallographic Directions
Any vector drawn in a crystal system can be related to a standard crystallographic direction, this
provides a common language that allows communication regarding materials properties. The book
gives a standard recipe for determining the crystallographic direction, which is denoted by square
brackets []. I am going to give you a slightly different standard recipe that I think is easier.
1.
Redraw the coordinate system so its origin coincides with the origin of the vector of interest.
2.
The 3-dimensional coordinates of the vector endpoint are determined in terms of the unit
cell dimensions.
v = uaxˆ + vbyˆ + wczˆ
3.
Multiply or divide by a common factor to reduce [uvw] to the smallest set of integers.
4.
Refer to this direction as the [uvw] direction. [uvw] = [-u-v-w]
All parallel vectors are considered to represent the same crystallographic direction. For cubic unit
cells, a=b=c.
c03prob
D)
A)
B)
Taking the origin of the coordinate system as the vector origin, the vector endpoint is at
1
1 1
− a , a , − a . In terms of the lattice constant (a), the vector endpoint is at
2
2 2
1
1 1
. Multiply through by 2, and this direction is [1 1 1 ].
−
−
,
,
2 2
2
1
The vector endpoint is at a , a , − a . In terms of the lattice constant (a), the vector
3
1
−
endpoint is at 1, 1, −
. Multiply through by 3, and this direction is 3 3 1 .
3
1
2
The vector endpoint is at − a , 0, − a . In terms of the lattice constant (a), the vector
3
2
1
2
endpoint is at − , 0, −
. Multiply through by 6, and this direction is [4 0 3 ].
3
2
C)
1
1
− a , a , a . In terms of the lattice constant (a), the vector
2
6
1
1
−
. Multiply through by 6, and this direction is 3 6 1 .
−
,
1
,
2
6
The vector endpoint is at
endpoint is at
Crystallographic Planes
The book again gives a standard recipe for determining the crystallographic plane, but mine is
slightly different.
1.
Redraw the coordinate system so that its origin is in a convenient corner of the unit cell that
is shown. This plane of interest cannot intersect with this corner. This corner should be
chosen so that it is easy determine the three axis intercepts of the plane of interest.
2.
Note the three axes intercepts and express them in terms of the lattice constants a,b and c.
(haxˆ , kbyˆ , lczˆ)
3.
Replace h, k and l with their reciprocals.
4.
Multiply or divide (hkl) by a common factor to achieve the smallest possible set of integers.
5.
Express this plane as the (hkl) plane, known as the plane's Miller indices.
All parallel planes represent the same crystallographic plane. For cubic unit cells, a = b = c. If the
plane does not intercept an axis, its intercept is taken as infinity, which has a reciprocal of zero.
c03prob
A)
Take the coordinate system origin at the corner usually labeled (a, a, 0). From that origin,
the axes intercepts are − a , − a , a . In units of the lattice constant, this is − 1, − 1, 1 .
Taking reciprocals has no effect. No need to multiple or divide by a common factor. The
final notation for this plane is 1 1 1 .
(
(
B)
)
)
Take the coordinate system origin at the corner usually labeled (0, 0, 0). From that origin,
the axes intercepts are
1 1
a, a, ∞ .
2 3
In units of lattice constant (a), this is
1 1
, , ∞.
2 3
Taking reciprocals, with no need to multiply or divide by a common factor, the final
notation for this plane is 2 3 0 .
(
)
Families of planes and families of directions
Some planes contain identical configurations of atoms, so for practical purposes they are equivalent.
For example, the BCC 1 1 0 plane has the same atomic configuration as the 1 0 1 , 0 1 1 ,
(
)
(
) (
)
(1 0 1), (1 1 0), and (0 1 1 ) planes. Since materials properties depend on the atoms from which
they are composed, these 6 planes are equivalent. Therefore, these 6 planes are referred to as the
{110} family of planes. The idea of a family of planes is important in x-ray diffraction and slip.
One can also define the <100> family of directions, which includes the directions <100>, <010>,
<001>.
Linear and planar atomic densities
The linear and planar atomic densities are defined as:
Linear Atomic ρ =
# of atoms centered on direction vector
Length of direction vector
Planar Atomic ρ =
# of atoms centered on a plane
Area of plane
Note that even though these two quantities are described as densities, they are really more like
atomic packing fractions in one and two dimensions. We may not have time to cover this in class.
If not, you will be responsible for learning this material on your own. The following two examples
should help.
Example:
What is the planar atomic density in the BCC (110) plane?
First, count the number of atoms in the rectangle ACDF, n = 1 + 4(1 / 4) = 2 . The atoms in the
corner count as1/4 instead of 1/8 since in two dimensions, they are only cut in half by two planes.
We also need to know the area of the rectangle ACDF, which is A = a 2a = 2a 2 . Now,
determine the planar atomic density (PAD):
( )
PAD =
2
2a 2
Remember, in a BCC unit cell a =
4R
3
, so substitution yields:
2
PAD =
Problem:
4R
2
3
2
=
3 2
16 R 2
−
What is the linear atomic density along the BCC [001] and 1 11 directions?
You should be able to draw in these directions in the figure above.
−
Along the 1 11 direction, this is the body diagonal, and:
LAD =
2
3a
=
2
1
=
4R 2R
3
3
Along the [001] direction (this is one side of the unit cell), the linear atomic density (LAD) is:
LAD =
1
1
3
=
=
a 4R / 3 4R
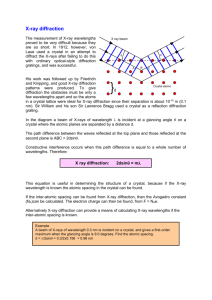
X-ray diffraction
From basic physics, diffraction occurs when light encounters a series of regularly spaced obstacles
with the spacing comparable in magnitude to the light’s wavelength. Since atomic dimensions are
extremely small, you need very short wavelength, high energy x-rays.
The utility of x-ray diffraction is that the line pattern reflects the crystal structure, whereas the line
spacing allows calculation of the atomic radius. In other words, you can determine the crystal
structure by simply looking at the pattern of lines, and then calculate the atomic radius by noting the
specific diffraction angle at which each line occurs. Here are the x-ray diffraction patterns for Pb
(FCC) and W (BCC):
Notice that for FCC metals, the first two diffraction lines are closely spaced, and then there is a
significant gap. On the other hand, the diffraction lines are more equally spaced for BCC metals.
An expert can simply conclude by inspection that the first pattern corresponds to FCC, and the
second to BCC crystal structure.
The figures below illustrate the physics of the diffraction process.
c03f36
c03f37
In the last figure, the extra segment in the light path is SQT. If SQT is an integral # of wavelengths,
constructive interference occurs.
nλ = SQ + QT
nλ = d hkl sin θ + d hkl sin θ = 2 d hkl sin θ
This is Bragg's law for diffraction, where the quantity dhkl is the distance between parallel (hkl)
planes. For cubic systems,
a
d hkl =
2
2
2
h +k +l
Bragg's law is a necessary but not sufficient condition for diffraction to occur. It is only rigorous for
simple cubic (sc) systems, which do not exist. For FCC and BCC systems there are extra scattering
centers that are not at the corners of the cube. These provide additional constraints:
BCC: h+k+l must be even.
FCC: h,k,l either all even or all odd.
All FCC metals have the same x-ray diffraction pattern; it is just expanded or compressed according
to the size of the lattice constant. Similarly, all BCC metals have the same pattern (different from
FCC).
As shown below, if one keeps λ constant and varies θ, you only see a diffracted beam at certain θ.
Note that as the sample rotates by θ, the detector must rotate by 2θ.
c03f38
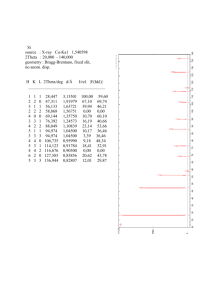
Example:
Label the first 6 lines of the x-ray diffraction pattern of Pb, shown above. Use the
first three lines to calculate dhkl and a. Assume λ = 0.1542 nm.
We will do this problem knowing that Pb has a FCC crystal structure, but nobody has assigned the
diffraction peaks to particular planes. This problem now requires us to name the first 6 peaks in the
x-ray diffraction pattern, and then calculate the interplanar spacing and the lattice constant from the
first 3 peaks.
Remember that for a FCC unit cell hkl must be all odd or all even for diffraction to occur. First, we
must use trial and error to find the 6 peaks which occur at smallest θ in the x-ray diffraction pattern.
To minimize θ in equation (3.15) we must maximize dhkl. From equation (3.16) this occurs for the
minimum values of h,k and l. I will use equation (3.11) to calculate dhkl for several combinations of
h,k and l.
a
a
a
, d 220 =
, d 200 =
d 111 =
4
3
8
a
d 222 =
12
a
d 333 =
27
, d 311 =
, d 400 =
a
11
a
16
, d 331 =
, d 420 =
a
19
a
18
a)
Choosing the 6 maximum values of dhkl from above, the first 6 peaks in order are
(111), (200), (220), (311), (222), and (400).
b)
Using equation (3.15):
d 111 =
d 200 =
d 210 =
c)
λ
2 sin( θ )
=
λ
2 sin( θ )
λ
2 sin( θ )
=
=
0.1542 nm
= 0.22797 nm
32°
2 sin
2
0.1542 nm
= 0.2430 nm
37°
2 sin
2
0.1542 nm
= 0.1728 nm
53°
2 sin
2
After rearrangement, equation (3.16) yields the lattice constant:
a = d hkl
2
2
2
h +k +l
For the first 3 diffraction lines one obtains a = 0.4844, 0.4860, and 0.4888 nm. This
is pretty good agreement given the very crude readings of the diffraction angles. If
we take the average of these values (0.486 nm) and determine the atomic radius (R)
from a = 2 2 R , one obtains R = 0.172 nm, close to that given in the front cover of
the textbook (0.175 nm).
This is similar to how x-ray diffraction is used in real life. From the overall look of a diffraction
pattern, an expert can identify the crystal structure involved (bcc, fcc, diamond, etc.). Then the
lowest few lines are assigned, and the atomic radius calculated for those lines. If all the atomic radii
agree, then the original assumption for the crystal structure was correct. The atomic radius is then
taken as the average from the lowest diffraction lines.
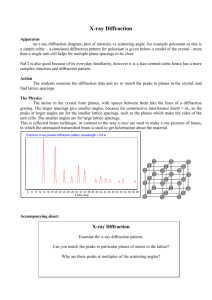
Practical Utility of X-ray Diffraction
X-ray crystallography is a standard method for characterizing samples of metal and semiconductor
materials. This can be used to identify the chemical composition of an unknown mixture, the
degree of crystallinity of a sample, or to determine the crystal structure of a newly fabricated
material. X-ray diffractometers are commonly found in academic and industrial research
laboratories.
One exotic example of the utility of x-ray crystallography is the determination of protein crystal
structures. Known information about protein structures is summarized in the protein data bank:
http://www.pdb.org/pdb/home/home.do
Searching through this database, by far the most common method for studying protein structure is xray diffraction. Determination of protein structure in solution is greatly complicated by the
heterogeneous structure of proteins in solution, and by their continuous rotational and vibrational
motion. Therefore, protein size and shape in solution is often inferred from x-ray diffraction
measurements on protein crystals, despite the obvious possibility of different protein conformations
in the solution and solid phases.
Another example of the utility of x-ray diffraction is to monitor recrystallization of photovoltaic thin
films, which absorb photons and convert them to electrons in solar cells. Here are x-ray diffraction
patterns of particulate CIGS films before (A-C) and after (E-F) annealing:
The x-ray diffraction peaks appear in proportion to the extent of recrystallization of the CIGS thin
films. This is taken from:
G.S. Chojecki, D.H. Rasmusxsen, K.B. Albaugh and I.I. Suni, “Recrystallization of Porous
Particulate CIGS Precursors into Dense Thin Films,” submitted to Sol. Energy Mater. Sol. Cells.
It is sometimes possible to estimate the average grain size (introduced in Chapter 5) from the width
of the x-ray diffraction peaks. The example below illustrates the use of x-ray diffraction to provide
real time monitoring of Cu receyrstallization, where the Cu thin films will be employed as ULSI
interconnect materials.
Immediately following electrodeposition, the Cu thin film above exhibits grain boundaries, which
will increase the Cu film resistance (introduced in Chapter 12). The Cu grain size will increase
slowly at room temperature, and more rapidly with low temperature annealing. This can be
monitored by time resolved x-ray diffraction, as shown below: