DBP - HAA Procedure
advertisement

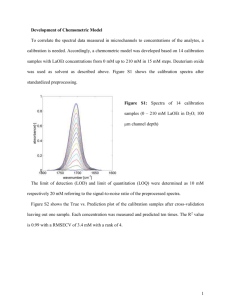
CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Haloacetic Acids – Standard Operating Procedure PARAMETER This SOP for the measurement of haloacetic acids (HAAs) follows a modified version of Standard Method 6251B.1 It is required that Standard Method 6251B and “Quantification of Nine Haloacetic Acids Using Gas Chromatography with Electron Capture Detection” by Brophy, et. al., be studied along with this SOP prior to performance of the analysis. This SOP applies to samples of surface water, ground water, finished water and water at any intermediate drinking water treatment stage. Haloacetic Acid Analyte Bromochloroacetic Acid (BCAA) Bromodichloroacetic Acid (BDCAA) Chlorodibromoacetic Acid (CDBAA) Dibromoacetic Acid (DBAA) Dichloroacetic Acid (DCAA) Monobromoacetic Acid (MBAA) monochloroacetic Acid (MCAA) Tribromoacetic Acid (TBAA) Trichloroacetic Acid (TCAA) CAS No. 5589-96-3 7113-314-7 5278-95-5 631-64-1 79-43-6 79-08-3 79-11-8 75-96-7 76-03-9 PRINCIPLE The determination of HAAs in water samples is accomplished by liquid-liquid extraction, derivatization and gas chromatography with micro electron capture detection. A 30 mL volume of sample is adjusted to pH of less than 0.5 to extract the nondissociated acidic compounds with 3 mL of methyl tert-butyl ether (MtBE). The extracted compounds are methylated with diazomethane solution to produce methyl ester or ether derivatives. The target analytes are identified and measured by capillary column gas chromatography using a micro electron capture detector (GC/ECD). Analytes are quantified using procedural standard quantification. SAMPLE COLLECTION, PRESERVATION AND STORAGE Vial Preparation Use a clean and labeled 40 mL amber EPA vial with a TFE-lined screw cap. A clean vial has been soap washed, rinsed with MQ and then baked. Add 65 mg of ammonium chloride to the clean vial. This amount produces an ammonium chloride concentration of 1625 mg/L in the sample. Page 1 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Sample Collection Collect all samples in duplicate. Completely fill the vial with sample (headspace free) to just overflowing, but take care not to flush out the buffer/dechlorination reagents. No air bubbles should be trapped in the sample when the vial is sealed. This can be best achieved by filling the sample vial just above the top and slip the septa on from the side. Cap vials tightly. Sample Storage Until analysis, samples must be stored at 4°C with minimal exposure to light and atmosphere. The sample storage area must be free of organic solvent vapors. Store extract samples in a freezer until analysis. Extract all samples within 14 days of collection and analyze within 7 days following extraction. APPARATUS AND REAGENTS Apparatus Sample containers/extraction vials: 40 mL amber EPA vial with a TFE-lined screw cap. Autosampler Vials: 2.0 mL vials with screw or crimp cap and a teflon-faced seal. Standard solution storage containers: 10-20 mL glass vials with teflon lined-screw caps. Pasteur pipets: Glass, disposable. Solvent Repipetor: up to 10.0 mL delivery range Volumetric flasks: 2mL, 10 mL. Micro syringes: 10 µL, 25 µL, 50 µL, 100 µL, 250 µL and 500 µL. Transfer pipets: 10 mL pipetor with disposable tips. Orbital Mixer Balance: analytical, capable of weighing to 0.001 g. Gas chromatograph: Analytical system complete with gas chromatograph equipped for electron capture detection, split/splitless capillary or direct injection, temperature programming, differential flow control, and with all required accessories including syringes, analytical columns and ultra high purity nitrogen gas. A data system is recommended for measuring peak areas. An autoinjector is recommended for improved precision of analyses. The gases flowing through the electron capture detector should be vented through the laboratory fume hood system. Primary GC column: DB-1 [fused silica capillary with chemically bonded (Dimethlypolysiloxane)] column, 30m x 0.45m ID, 1.27 µm film thickness. Diazomethane reaction chamber Diazomethane reaction chamber holder: 500 mLerlenmeyer flask Diazomethane reaction chamber ice bath: 800-1000 ml plastic beaker & 3 L crushed ice Plastic syringe: 1 ml Luer lock needle: 25 gauge, 5/8” Erlenmeyer flask: 25 mL Teflon tape Page 2 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Reagents Reagent water -- Reagent water is defined as a water in which an interference is not observed > than the MDL of each analyte of interest. A Millipore Milli-Q water system is used to generate deionized reagent water. Methanol -- High purity, demonstrated to be free of analytes and interferences. Maintain records of the reagent’s manufacturer. HAA standards – The stock standard solution used is purchased as an ampulized solution in methanol from Supelco (Catalog # 4-7787) Surrogate – The stock standard solution (2-bromopropionic acid) used is purchased as an ampulized solution in MTBE from AccuStandard Inc. (# M-552.1-SS). Ammonium chloride, NH4Cl -- ACS reagent grade, used to convert free chlorine to monochloramine. Sodium sulfate, Na2SO4 – ACS reagent grade (granular, anhydrous). If interferences are observed, it may be necessary to heat the sodium sulfate in a shallow tray at 400°C for up to 4 hours to remove phthalates and other interfering organic substances. Store in a capped glass bottle rather than a plastic container. Methyl tert-butyl ether – High purity, demonstrated to be free of analytes and interferences. Maintain records of the reagent’s manufacturer. MNNG - 1-methyl-3-nitro-1-nitrosoguanidine Sodium hydroxide, NaOH – ~5 N NaOH QC PROCEDURES Initial Demonstration of Capability (IDC) The IDC is performed at least once, by each analyst, before analysis of any sample, to demonstrate proficiency to perform the method. The following procedures are based on Section 6 of the DBP/ICR Analytical Methods Manual2. Initial Demonstration of Low System Background: Analyze a Method Blank (MB) to verify that no contamination exists above ½ the minimum reporting levels (MRLs). The MRL for each of the HAAs is listed in the following table. Analyte MBAA DCAA TCAA BCAA DBAA BDCAA MCAA CDBAA TBAA MRL 1.0 g/L 2.0 g/L 4.0 g/L Page 3 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Initial Demonstration of Precision: Analyze a total of five samples of reagent water fortified with each of the haloacetic acids at a concentration of 20 µg/L (Required concentration non given in the ICR Methods Manual for the three additional HAAs: BDCAA, CDBAA and TBAA). These five samples must be analyzed on five separate days and must be of separate extraction and analysis batches. The relative standard deviation (RSD) must be less than 20%. Initial Demonstration of Accuracy: Calculate the average recoveries of the replicates in the Initial Demonstration of Precision. The averages must be within +/- 20% of the theoretical amount. Method Detection Limit (MDL) Determination Prepare a sample with fortifying concentrations at the MRLs for each of the HAAs. The following procedure is based from Standard Methods 1030 C1. Analyze seven portions of this solution over a period of at least 3 d to ensure that MDL determination is more representative than measurements performed sequentially. Calculate the standard deviation to the replicate analysis. From a table of the one-sided t distribution select the value of t for 7 – 1 = 6 degrees of freedom and at the 99% confidence level. This value is 3.14. The product of 3.14 times the standard deviation is the MDL. Method Blank Each time a set of samples is extracted or reagents are changed, a method blank (MB) must be analyzed. If the MB (Milli-Q) produces an interference peak within the retention time window of any analyte that would prevent the determination of that analyte or a peak of concentration greater than ½ the MRL for that analyte, the analyst must determine the source of contamination and eliminate the interference before processing samples. Field samples of an extraction set associated with an MB that has failed the specified criteria are considered suspect. Matrix Spike Chlorinated water supplies will usually contain significant background concentrations of several method analytes. The concentrations may be equal to or greater than the fortified concentrations. Relatively poor accuracy and precision may be anticipated when a large background must be subtracted. For many samples, the concentrations may be so high that fortification may lead to a final extract with instrumental responses exceeding the linear range of the electron capture detector. If this occurs, the extract must be diluted. In spite of these problems, sample sources should be fortified and analyzed as described below. By fortifying sample matrices and calculating analyte recoveries, any matrix induced analyte bias is evaluated. The laboratory must add known concentrations of analytes to 10 % of the samples. The concentrations should be equal to or greater than the background concentrations in the sample selected for fortification. Acceptable fortification concentrations according to the ICR Methods Manual are 20 µg/L and 40 µg/L. Page 4 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Calculate the mean percent recovery, R, of the concentration for each analyte, after correcting the total mean measured concentration, A, from the fortified sample for the back-ground concentration, B, measured in the unfortified sample, i.e.: R = 100 (A - B)/C where C is the fortifying concentration. U.S.EPA Method 551.1 requires the recoveries of all analytes must fall within 80-120%. If a recovery falls outside of this acceptable range, a matrix induced bias can be assumed for the respective analyte and the data for that analyte must be reported to the data user as suspect. Surrogate The surrogate standard is fortified into the aqueous portion of all samples and laboratory reagent blanks. The surrogate is used to measure the efficiency of the sample derivatization and extraction. U.S.EPA Method 552.2 lists acceptance criteria of 70130%. When surrogate recovery from a sample, blank or calibration check is outside these limits check (1) calculations to locate possible errors, (2) standard solutions for degradation, (3) possible sources for contamination, and (4) instrument performance. If those steps do not reveal the cause of the problem, reanalyze the extract. If the extract reanalysis meets the surrogate recovery criterion, report only data for the reanalyzed extract. If the extract reanalysis fails the recovery criterion, the analyst should check the calibration by analyzing the most recently acceptable calibration check standard. If the calibration check fails criteria, recalibration is in order. If the calibration check is acceptable, it may be necessary to extract another aliquot of sample. If the sample reextract also fails the recovery criterion, report all data for that sample as suspect. Duplicate Analysis Ten percent (10%) of samples will be analyzed in duplicate. Duplicate results must not reflect a relative percent difference (RPD) greater that 25% for any one analyte and the RPD for 90% of the analytes being determined must be less than 20%. If this criteria is not met the analysis must be repeated. Solvent Blank Each analysis run must be started with an MTBE solvent blank. This is a check on the extraction solvent as well as on the instrument system. Second Source Calibration An additional calibration standard is extracted and analyzed alongside every calibration curve that is to be used. This additional calibration standard is purchased from a supplier different from the one used to purchase the standards used in preparation of the calibration curve. Percent recoveries are calculated for the second source calibration standard. These recoveries must fall within the range of 70-130 % in order for the initial calibration curve to be considered valid. Page 5 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Calibration Check If an initial calibration is not run on a particular analysis day, the calibration check must be run and matched to the previous calibration curve to ensure that the instrument is still within calibration. In order for the calibration check to be considered valid, recoveries must fall between 50% and 150% for all the target analytes when quantified using the previous calibration curve. Furthermore, a calibration check must be analyzed after every tenth sample analysis, after the final sample analysis and recoveries must fall between 80% and 120% for all the target analytes to be considered acceptable. At least one calibration check must be extracted with each set of samples. Calibration check standards need not necessarily be different extracts but can be injections from the same extract as long as the holding time requirements are met. STANDARDS AND CALIBRATION Preparing a Primary Standard The primary dilution standard is prepared by diluting the ampulized stock standard solution in methanol. The species concentrations in the ampulized stock standard solution are listed in the following table. HAA Species TCAA, DBAA MBAA, BCAA, BDCAA MCAA, DCAA DBCAA TBAA Ampulized Stock Standard Concentration (g/L) 200,000 400,000 600,000 1,000,000 2,000,000 Primary Standard Concentration (g/L) 20,000 40,000 60,000 100,000 200,000 Rinse syringe with small amount of high concentration standard. Dispense 500 µL into a 5 mL volumetric flask partially filled with methanol. Ensure the needle is submerged in methanol at bottom of flask. Fill the flask with methanol to the 5 mL mark. Mix by inversion. The species concentrations in the primary dilution standard are listed in the above table. Methanol is used due to its miscibility with water. But the primary standard is only good for 24 hours due to the spontaneous methylation of haloacetic acids when stored in methanol. Preparing Secondary Standards Secondary standards are prepared by the addition of specific volumes of the primary dilution standard to separate 72 mL volumes of reagent water. These aqueous standards are treated, including extraction, in the same manner as the samples. Fill clean and labeled 72 mL clear bottles with Milli-Q reagent water to top so no headspace is present. This can be best achieved by filling the sample bottle just above the top and slip the septa on from the side. Cap bottles. Bottles do not exactly contain 72 mL. So to determine exact volumes, weigh the capped labeled bottle (w/ septa) before and after adding reagent water and then determine the actual contained volume Page 6 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT by using the density of H2O. Place a 5/8” – 25 guage needle in the septa of the vial. The following table lists the required primary standard volumes assuming the total secondary standard volume in the vials are 72 mL. Level Level 0 Level 1 Level 2 Level 3 Level 4 Level 5 Level 6 Level 7 Standard Volume (mL) 72 72 72 72 72 72 72 72 Primary Standard Volume to add (L) 0 1.8 3.6 18 36 72 144 288 Syringe to Use TCAA DBAA N/A 10 10 25 50 100 250 500 0 0.5 1.0 5.0 10 20 40 80 Secondary Standards (g/L) MBAA MCAA DBCAA BCAA DCAA BDCAA 0 0 0 1.0 1.5 2.5 2.0 3.0 5.0 10 15 25 20 30 50 40 60 100 80 120 200 160 240 400 TBAA 0 5.0 10.0 50 100 200 400 800 Level 1 represents concentrations near the MRL for each analyte. Levels 3 and 4 are used as calibration checks. Inject the appropriate dose with the injection needle plunged to the bottom of the sample bottle. The liquid injected will rise, due to initial density differences between methanol and H2O, so start the injection with the bottle inverted. Then rotate the bottle during injection to ensure better mixing and to ensure that no dosing solution exits the exhaust needle. The dose volume will displace an equal amount of sample out the 5/8” needle. 5/8” exhaust needle Injection syringe Page 7 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Surrogate Standard Solution The surrogate stock standard solution (2-bromopropionic acid) is purchased from AccuStandard (# M-552.1-SS) at a concentration 1,000,000 g/l. in MTBE. From this stock standard solution, a primary dilution standard is prepared in methanol at a concentration of 25,000 µg/l. A volume of 30 µL of this primary dilution standard is spiked into each blank, sample, calibration standard and QC sample, to a final concentration of 25 µg/l. Matrix Spiking Standard Solution A volume of 20 or 40 µL of the primary standard is used in the fortification of the duplicate sample. Consideration should be given to attempting to fortify at concentrations above those expected in the sample. Calibration of Instrument Each analysis run should be started with an MtBE solvent blank. This is a check on the extraction solvent as well as on the instrument system. If this run is acceptable, the extracts for the calibration curve are analyzed (2 µL injection volume). The Chemstation Chromatography Software System is used to generate a calibration curve by plotting the areas (Aan) against the concentrations (Can). The curve can be defined as either first or second order. Correlation coefficients must be greater than 0.990. ANALYTICAL PROCEDURE Diazomethane Preparation Procedure PRECAUTIONS!!! MNNG and the gas produced are very dangerous! ALWAYS handle in hood with proper personal protection equipment and training! 1. Tilt reactor so hole in reactor stem is facing up. It is important to make sure all additions to the reactor stem do not go through the hole. 2. Add the MNNG to the base of the reactor stem up to the fill line (~1 g) Reactor Stem Fill line MNNG Page 8 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT 3. Add ~1 ml MQ water to reactor stem to moisten the MNNG and firmly screw on cap w/ septa. 4. Add 6 ml MtBE to reactor base 5. Place reactor stem in the reactor base with O-ring sandwiched in between glass flange. Wrap Teflon tape around the reactor at the O-ring and clamp tightly. Wrap Teflon tape around the cap. 6. Prepare an ice bath in a 800 ml plastic beaker. Place reactor in ice bath and allow reactor to reach thermal equilibrium 7. Plunge ~25 gauge needle through septa in reactor stem. Add ~1 ml of NaOH very slowly in a drop wise fashion (until all MNNG is gone). [Reaction is Extremely Exothermic!]. Be sure to keep pressure on the syringe since significant pressure builds up inside reactor. Be sure to maintain good heat transfer to the ice bath. 8. Allow ~30 minutes for reaction completion while in the ice bath. The outside MtBE solution should turn a moderate or deep yellow. If this does not occur, the solution is no good. 9. Allow the pressure to slowly release by loosening the cap first. Transfer diazomethane to 25 ml erlenmeyer flask. Keep the diazomethane cold and use as soon as possible after preparation. If bubbles form following addition of the diazomethane to the sample, then diazomethane solution is good! Extraction Procedure 1. Remove the samples from storage and allow them to equilibrate to room temperature. 2. Remove the aqueous sample to the prescored 30 mL mark on each sample vial. 3. Add 30 L of the surrogate standard (25,000 g/L 2-bromoacetic acid in methanol). Mix by slowly and carefully inverting the sample vial two times with minimal sample agitation. 4. Adjust the pH to less than 0.5 by adding 1 ml of concentrated sulfuric acid via a glass pipette. 5. Add 13.0 g of sodium sulfate and 3.0 ml of MTBE (via bottle top pipetor). Sodium sulfate is added to increase the ionic strength of the aqueous phase and thus further drive the haloacetic acids into the organic phase. The addition of the salt should be done quickly so that the heat generated from the addition of the acid will help dissolve the salts. 6. Recap vial and mix for 30 seconds on an orbital mixer (until most of the salts are dissolved). 7. Let stand for ~ 15 minutes for separation. 8. Transfer exactly 2 ml of the top organic layer with a pasteur pipette and add to a 2 ml volumetric flask. Organic Layer Page 9 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT 9. Place the 2 mL volumetric flask containing the sample into the freezer for 25-30 minutes. 10. Add 250 mL of diazomethane via the glass syringe and mix gently by inverting once. 11. Place sample in 4C refrigerator for 15 minutes during esterification. 12. Remove sample and allow it to reach room temperature for 15 minutes. 13. Add approximately 10 mg of silica gel to sample to stop reaction (when bubbles stop forming upon the addition of silica gel, the reaction is complete) 14. Transfer sample to GC vial and cap. 15. Analyze samples as soon as possible. Remove 10 ml 13 g Na2SO4 1 mL H2SO4 3 ml MtBE 30 L Surrogate 1) 2) 3) 1) 2) ~2 ml organic layer Cap vial Mix 30 sec. Stand 15 min. 250 l diazomethane mix by 1 inversion 25-30 min. in freezer 15 min. in 4oC frig. ~2 ml organic layer ~10 mg silica gel 15 min. room temp equilibration transfer to GC vial Gas Chromatography 1. The temperature program utilized is as follows: 50°C initial temperature, 1 minute hold time, then a 4°C/minute ramp to 158°C with a 0 minute hold time, and finally a 4°C/minute ramp to 230°C final temperature with a 5 minute hold time. This represents a 34.8 minute run time. 2. Calibrate the system daily by either the analysis of a calibration curve or a continuing calibration check. 3. Inject 2 µL of the sample extract. Use the Chemstation chromatography system to record the resulting peak sizes in area units. Page 10 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT 4. If the response for the peak exceeds the calibration curve or the working range of the detector, dilute the extract and re-analyze. The analyst must not extrapolate beyond the calibration range established. DATA REDUCTION, VALIDATION AND REPORTING Identify sample components by comparison of retention times to retention data from the calibration standard analysis. If the retention time of an unknown peak corresponds, within limits, to the retention time of a standard compound, then the identification is considered positive. Calculate analyte concentrations from the calibration curve. Retention time windows are given as +/- three times the standard deviation obtained in calculating the average retention time of the initial calibration curve. However, the experience of the analyst should weigh heavily in the interpretation of the chromatogram. PREVENTATIVE MAINTAINANCE Gas purge flow should be left on at all times, with or without GC use to prevent the inflow of oxygen and resulting contamination/oxidation of parts. The flow can be lowered though in times of dormancy. The oven can be shut off if the GC is not used for an extended period of time (i.e. months). Short term Maintenance (Every time) Compressed ultra high purity nitrogen gas tanks are checked daily for content (pounds per square inch-psi) and are replaced when the contents fall below 500 psig. Every time a gas tank is changed out, this should be noted on the oxygen trap, which has a limited capacity. Long term Maintenance (Months) Glass liner: The glass liner, located in the injection inlet, should be replaced ~ once a year or when used up. When it is used the glass wool inside it will have a darker color Rubber injection seal: The rubber seal, located at the top of the injection inlet, should be replaced every 3-4 months Gold seal: The gold seal, located at the bottom of the injection inlet under the glass liner, should be checked ~ every 6 months for carbon build-up. It is accessed from inside the oven. Column Burn: The column should be burned off every 3-12 months, depending on use. This elutes off any constituents that may be more difficult to remove. A chromatogram is produced showing eluted constituents. A 5-7 hour burn is sufficient. The burn temp should be ~50o C less than the max column temperature reported on the box. Very Long term Maintenance (Years) Moisture Trap: will need regeneration when exhausted Oxygen Trap: the high capacity (2 L of oxygen) trap will need replacement after ~18 tanks of gas used ECD Detector: will need servicing every 3-10 or so years. Page 11 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT Gas flow rates: should be checked annually. Specific maintenance on an improperly functioning part is recorded on the GC Maintenance Form. SAFETY 1. The toxicity or carcinogenicity of each reagent used in this method has not been precisely defined, however, each chemical compound must be treated as a potential health hazard. From this viewpoint, exposure to these chemicals must be minimized. The laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets should also be made available to all personnel involved in the chemical analysis. 2. The toxicity of the extraction solvent, MtBE, has not been well defined. Susceptible individuals may experience adverse affects upon skin contact or inhalation of vapors. Therefore protective clothing and gloves should be used and MtBE should be used only in a chemical fume hood or glove box. The same precaution applies to pure standard materials. INTERFERENCES 1. Method interferences may be caused by contaminants in solvents, reagents, glassware and other sample processing apparatus that lead to discrete artifacts or elevated baselines in chromatograms. All reagents and apparatus must be routinely demonstrated to be free from interferences under the conditions of the analysis by analyzing method blanks. Subtracting blank values from sample results is not permitted. 2. The use of high purity reagents and solvents helps to minimize interference problems. Solvent blanks should be analyzed for each new bottle of solvent before use. An interference free solvent is a solvent containing no peaks yielding data at > MDL and at the retention times of the analytes of interest. 3. Interfering contamination may occur when a sample containing low concentrations of analytes is analyzed immediately following a sample containing relatively high concentrations of analytes. Routine between-sample rinsing of the sample syringe and associated equipment with MtBE can minimize sample cross-contamination. After analysis of a sample containing high concentrations of analytes, one or more injections of MtBE should be made to ensure that accurate values are obtained for the next sample. 4. Matrix interferences may be caused by contaminants that are co-extracted from the sample. The extent of matrix interferences will vary considerably from source to source, depending upon the water sampled. Page 12 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT DEFINITIONS 1. SURROGATE ANALYTE -- A compound which is added to a sample aliquot in known amount(s) before extraction or other processing and is measured with the same procedure used to measure other sample components. The purpose of the surrogate is to monitor method performance with each sample. 2. LABORATORY DUPLICATES -- Two aliquots of the same sample designated as such in the laboratory. Each aliquot is extracted, derivatized and analyzed separately using identical procedures. Analysis of these duplicates indicates the precision associated with laboratory procedures, but not with sample collection, preservation or storage procedures. 3. FIELD DUPLICATES -- Two separate samples collected at the same time and place under identical circumstances and treated exactly the same throughout field and laboratory procedures. Analysis of these duplicates gives a measure of the precision associated with sample collection, preservation and storage, as well as with laboratory procedures. 4. METHOD BLANK (MB) -- An aliquot of reagent water or other blank matrix that is treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, internal standards, and surrogates that are used with other samples. The MB is used to determine if method analytes or other interferences are present in the laboratory environment, the reagents, or the apparatus. 5. MATRIX SPIKE (MS) -- An aliquot of an environmental sample to which known quantities of the method analytes are added in the laboratory. The MS is analyzed exactly like a sample, and its purpose is to determine whether the sample matrix contributes bias to the analytical results. The background concentrations of the analytes in the sample matrix must be determined in a separate aliquot and the measured values in the MS corrected for background concentrations. 6. STOCK STANDARD – Neat material or a commercially purchased concentrated solution used in the calibration process 7. PRIMARY DILUTION STANDARD -- A solution of the target analyte(s) prepared in the laboratory from stock standard solutions and diluted as needed to prepare secondary standards. 8. SECONDARY STANDARD -- A solution prepared from the primary dilution standard and used to calibrate the instrument response with respect to analyte concentration. 9. SECOND SOURCE STANDARD – A solution of method analytes of known concentration which is used to fortify an aliquot of reagent water. It is obtained from a source different from that of the standards used to calibrate and is used as a check on the accuracy of standards being used to calibrate the analytical instrumentation. Page 13 CENTER FOR DRINKING WATER OPTIMIZATION 08/08/01 DRAFT 10. METHOD DETECTION LIMIT (MDL) -- The minimum concentration of an analyte that can be identified, measured and reported with 99% confidence that the analyte concentration is greater than zero. 11. MATERIAL SAFETY DATA SHEET (MSDS) -- Written information provided by vendors concerning a chemical's toxicity, health hazards, physical properties, fire and reactivity data including storage, spill, and handling precautions. XI INSTRUMENT SOFTWARE AND OPERATION The Agilent GC Chemstation software (Rev. A.08.03 [847]) is used for data acquisition. Method files are named as follows: Sequence files are named as follows: Data files are named as follows: HAAMMDD.M MMDDA.S MMDDA00X.D where MM = Month, DD = Day, X represents consecutive numbering REFERENCES 1 APHA, AWWA and WEF (1998) Standard Methods for the Examination of Water and Wastewater 20th Edition, Washington, D.C. 2 USEPA (1996) DBP/ICR Analytical Methods Manual, Office of Water, Cincinnati, OH. 3 USEPA (1995) Method 552.2: Determination of haloacetic acids and dalapon in drinking water by liquid-liquid extraction, derivatization and gas chromatography with electron capture detection, In Methods for the Determination of Organic Compounds in Drinking Water-Supplement III, EPA/600/R-95/131. Cincinnati, Ohio: National Exposure Research Laboratory, USEPA Office of Research and Development. Page 14