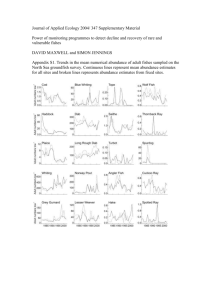
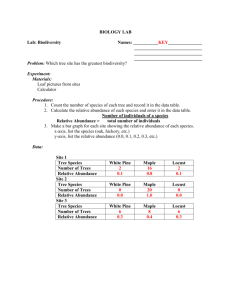
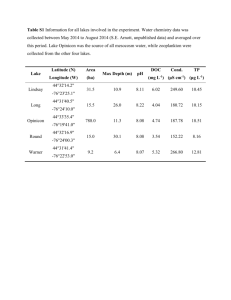
Electronic Supporting Information Multistage Reactive Transmission
advertisement

Electronic Supporting Information Multistage Reactive Transmission-Mode Desorption Electrospray Ionization Mass Spectrometry Kevin C. Peters, Troy J. Comi, and Richard H. Perry* TM2-DESI Characterization Propranolol Intensity 1.3×105 c) Parameters Liquid Flow Rate = 10 mL/min [Propranolol] = 5.0 ×10-5 M 1.0×103 0 N2 Threshold 100 2.5×104 Propranolol Intensity a) 200 [Propranolol] (M) 5.0×10-6 M 1.0×10-5 M 5.0×10-5 M 1.0×10-4 M 5.0×10-4 M 0 300 [Propranolol] (M) b) d) 0 1 2 3 Inter-Mesh Distance (mm) 7.0×104 Propranolol Intensity Propranolol Intensity 100 Relative N2 Velocity 2.0×105 0 6.0×10-4 0 Nitrogen Line Pressure (PSI) 0 Parameters N2 Flow Rate = 200 PSI Liquid Flow Rate = 10 mL/min y = 2336.2x – 8546.8 R² = 0.9828 1.0×103 0 4 Parameters N2 Flow Rate = 200 PSI [Propranolol] = 5.0×10-5 M 10 20 Liquid Flow Rate (µL/min) 30 Figure S1. The performance characteristics of TM2-DESI were evaluated by monitoring the intensity of [2+H]+ from M2 as a function of various experiment parameters. (a) N2 line pressure. (b) Inter-mesh distance (200 psi N2 pressure; 15 μL/min flow rate; 5 μL of 5 x 10-5 M 2 deposited). Inset shows COMSOL Multiphysics simulation of N2 flow through M1 and M2 with 1.0 mm separation. (c) Concentration of 2 deposited on M2 (solvent is 4:1 CH2Cl2:CH3OH for all trials). (d) Liquid flow rate. TPFPP Ligand Oxidation by Fe-TPFPP Relative Abundance a) 100 973.051 x10 No Catalyst 989 : 973 (x1000) = 2.5 ± 0.2 989.044 0 974 976 978 980 982 984 986 988 Relative Abundance b) 100 973.051 x10 + Fe-TPFPP 989 : 973 (x1000) = 23 ± 5 989.044 0 974 976 978 980 982 m/z 984 986 988 Figure S2. (a) Negative mode TM1-DESI mass spectrum of free TPFPP ligand (5 μL of 5 × 10-4 M) with 5 × 10-4 M mCPBA in 4:1 CH2Cl2:CH3OH as spray solvent (10 μL/min, 200 psi N2). (b) Same experiment performed as in part (a), with 5 μL of 5 x 10-4 M FeTPFPP added to a new TPFPP sample spot; n = 3 for each experiment. Tandem MS Characterization of Propranolol Hydroxylation (Aromatic and Aliphatic) and Screening of Various Fe-porphyrin Catalysts TM2-DESI-MS screening of five catalysts’ reactivities towards hydroxylation of 2 was carried out by depositing five spots of [2+HCl] (5 μL of 5 × 10-5 M in CH2Cl2) on M2, and each spot was outlined with red permanent marker for reference. Then, M1 was added in front of M2, and 5 mL of each catalyst (5 × 10-5 M in CH2Cl2) was deposited directly over each deposited spot of 2. The meshes were sufficiently spaced to prevent M1-M2 contact. In the TM2-DESI experiment, the mesh holders were positioned on each of the five pairs of spots to initiate reaction (triplicate measurements were acquired for each). Aliphatic hydroxylation product 2a yielded characteristic fragments at m/z 114, 183, 198, and 232 (Fig. S3c). Aromatic hydroxylation gives characteristic fragment ions at m/z 98, 116, 173, and 199 in TM2DESI-MS/MS spectra (Fig. S3d), which all have O-atom addition on the ring evidenced by a shift of 16 Da on fragments containing the naphthyl ring (Fig S3e). The aliphatic to aromatic oxidation ratio (Fig. 2a) was estimated by multiplying the summed intensities of the aromatic or aliphatic hydroxylation fragment ions in MS/MS spectra (viz. the fragment ions mentioned above; Fig. S3) by the 276/260 intensity ratio. Relative Abundance a) 260.165 100 276.160 260 262 264 266 268 b) 270 272 183.1 Relative Abundance 100 260.2 157.0 98.0 218.1 100 120 140 160 180 200 242.2 220 240 c) Relative Abundance 276 116.0 80 100 114.1 132.1 157.1 120 140 160 200 116.0 100 280 300 276.2 198.1 180 260 258.2 183.1 d) Relative Abundance 274 234.1 220 240 260 280 300 258.2 199.1 276.2 173.0 98.0 80 100 234.1 120 140 160 180 200 m/z 220 240 260 280 300 e) m/z 98.1 m/z 116.0 m/z 242.2 m/z 157.0 m/z 218.1 m/z 183.1 Figure S3. (a) Mass spectrum from spraying a pre-mixed solution of 2.5 × 10-6 M 2 and 2.5 × 10-5 M mCPBA in a sonic spray ionization (SSI) experiment (30 μL/min, 200 psi N 2). (b) MS/MS spectrum of [2+H]+ (m/z 260) from (a). (c) Tandem MS of [2a+H]+ at m/z 276 from (a), which was generated in the absence of a catalyst. (d) Tandem MS of [2b+H]+ generated by Fe-TDFPP in TM2-DESI experiment (5 μL of 10-5 M catalyst on first mesh; 5 μL of 5 × 10-5 M propranolol on second mesh; 5 × 10-5 M mCPBA sprayed at 30 μL/min with 200 psi N2, and 1.0 cm sprayer-to-inlet distance). (e) Possible structures for some of the fragments observed in (b) – (d) (other isomers are possible). Fe-TPFPP Hydroxylation of 1-Naphthyl-3-Pyrrolidinyl Ether by TM2-DESI-MS 18x 90x 214.121 Relative Abundance 100 230.120 0 220 230 m/z Figure S4. Positive mode TM2-DESI-MS showing the oxidation of 1-naphthyl-3-pyrrolidinyl ether ([M+H]+ = m/z 214.12) by mCPBA (5 × 10-5 M in 4:1 CH2Cl2:MeOH used as the spray solvent; 10 μL/min and 200 PSI N2 line pressure) and Fe-TPFPP (5 μL of 5 × 10-4 M deposited on M1; 2 mm between meshes; Emitterto-inlet distance was 1.25 cm). Other Substrates Investigated by TM2-DESI-MS Figure S5. Structures for various substrates that were hydroxylated using Fe-TPFPP in TM2-DESI-MS experiments. Product to substrate intensity ratios (P:S) are shown below each structure (all P:S ratios are × 1000). Tandem Mass Spectrometry of Rhodamine B Tandem MS of Rhodamine B (m/z 443.2) shows loss of C2H4 and C3H8 (m/z 415.2 and m/z 399.2, respectively) from the alkyl chains (Fig. S5a). MS/MS of oxidized Rhodamine B (m/z 459.2) also shows a primary fragment at m/z 415.2, indicating that the molecule is oxidized on the alkyl chain that undergoes dissociation (Fig. S5b). Further evidence for this assignment is found at m/z 441.2 (loss of H2O), as dehydration from an aromatic ring is unlikely. Another peak is observed at m/z 430.3, corresponding to C2H5 loss. All of these results indicate that Rhodamine is oxidized on the aliphatic substituents rather than the aromatic ring. a) 443.2 Relative Abundance 100 399.2 415.2 395 b) 415 435 455 415.2 Relative Abundance 100 441.2 459.2 Figure S6. (a) Positive mode TM1-DESI MS/MS of Rhodamine B (m/z 443.2). Structure shown represents the m/z 399.2 fragment ion. (b) Positive mode TM2-DESIMS/MS of in-situ-oxidized Rhodamine B (m/z 459.2). Structure shown represents the m/z 415.2 fragment ion. (Parameters: collision energy = 30%; activation time =30 ms; Solvent = acetonitrile). 430.2 395 415 435 455 m/z Tandem Mass Spectrometry of Rhodamine 6G MS/MS of Rhodamine 6G (m/z 443.2) also shows a significant fragment at m/z 415.2, indicating C2H4 loss (Fig. S6a). Oxidized Rhodamine 6G (m/z 459.2) shows a prominent fragment ion at m/z 415.2 (Fig. S6b), indicating alkyl chain oxidation. H2O loss is also observed (m/z 441.22), as well as combined [C2H4 + C2H4O] loss (m/z 387.17), all of which indicate alkyl chain, rather than aromatic, hydroxylation. The only peak that does not definitively suggest aliphatic hydroxylation is m/z 431.20, which contains the ethyl substituent and an oxygen atom. a) 443.2 Relative Abundance 100 443.2 415.2 385 405 425 445 459.2 415.2 100 Relative Abundance b) 431.2 441.2 459.2 387.2 385 405 m/z 425 445 Figure S7. (a) Positive mode TM1-DESI MS/MS of Rhodamine 6G (m/z 443.2). Structure shown represents the m/z 415.2 fragment ion. (b) Positive mode TM2DESI MS/MS of in-situ-oxidized Rhodamine 6G (m/z 459.2). Structures shown represent m/z 387.2 (left) and m/z 431.2 (right) fragments respectively. (Parameters: collision energy = 30%; activation time =30 ms; Solvent = acetonitrile). Tandem MS of Fluorescein Tandem MS of fluorescein (m/z 331.1) produce fragment ions at m/z 287.1 and m/z 286.1, which corresponds to a CO2 and -CO2H loss, respectively (Fig. S7a). Upon oxidation, there is a 16 Da mass unit shift (Fig. S7b), which confirms hydroxylation. a) Relative Abundance 100 287.1 286.1 285 331.1 295 305 b) 31 5 325 335 345 303.1 Relative Abundance 100 347.1 302.1 285 295 Figure S8. (a) Negative mode TM1-DESI MS/MS of fluorescein (m/z 331.1). Structure shown represents m/z 286.1 fragment. (b) Negative mode TM2-DESI MS/MS of in-situ-oxidized fluorescein (m/z 347.1). Structure shown represents m/z 302.1 fragment. Reaction performed in 2:1 dichloromethane:acetonitrile. 305 315 m/z 32 5 335 345 Tandem MS of Lauric Acid Finally, tandem MS of lauric acid (m/z 199.17) shows a single fragment at m/z 181.2, which corresponds to loss of H2O from the carboxylic acid group (Fig. S8a). Tandem MS of oxidized lauric acid (m/z 215.2, Fig. S8b), however, shows a peak corresponding to loss of water and hydroxyethene at m/z 153.1. This result suggests that Fe-catalyzed oxidation of lauric acid occurs primarily on one of the two carbon atoms furthest from the carboxylic acid group. 100 181.2 155 100 Relative Abundance b) 199.2 199.2 Relative Abundance a) 165 175 185 195 215.2 a 197.1 153.1 155 165 175 m/z 185 195 Figure S9. (a) Negative mode TM1-DESI MS/MS of lauric acid (m/z 199.17). (b) Negative mode TM2-DESI MS/MS of insitu-oxidized lauric acid (m/z 215.17). Reaction performed in acetonitrile. Comparison of Signal Intensities at 0 kV and 5 kV (Positive and Negative Modes) Relative Abundance (a) 100 1027.980 +0 kV NL: 8.9E4 1043.975 0 1025 1050 Relative Abundance (b) 100 1027.976 +5 kV NL: 1.9E5 1043.971 0 1025 1050 Relative Abundance (c) -0 kV NL: 9.0E4 100 1337.957 1198.962 1217.936 1097.914 0 1100 1200 1300 (d) -5 kV NL: 1.2E5 100 1097.914 1217.935 1198.961 1100 1200 m/z 1337.956 0 1300 Figure S10. TM1-DESI-MS of Fe-TPFPP (deposited 5 mL of 10-4 M Fe-TPFPP in CH2Cl2; DESI micrdroplet spray contains 10-5 M mCPBA in 4:1 CH2Cl2:CH3OH) using 0 kV ((a) and (c)) and 5 kV ((b) and (d)) in positive and negative modes. Comparison of Signal Intensities at Different Inlet Capillary Temperatures (a) Relative Abundance -0 kV; 225 oC NL: 5.6E4 1337.958 100 1198.963 1097.915 1217.936 0 1100 1200 1300 (b) Relative Abundance -0 kV; 325 oC NL: 4.2E4 1337.959 100 1198.963 1097.916 1217.938 0 1100 1200 1300 m/z Figure S11. TM1-DESI-MS of Fe-TPFPP (deposited 5 mL of 10-5 M Fe-TPFPP in CH2Cl2; DESI micrdroplet spray contains 10-5 M mCPBA in 4:1 CH2Cl2:CH3OH) using -0 kV spray voltage, as well as 225 oC (a) and 325 (b) oC capillary temperatures. Calculation for Estimating the Mean Lifetime of the Iron Peroxo Intermediate An Arrhenius plot was constructed using previously reported data [1] describing the temperature dependence of Fe-TMP peroxo (Fe-OOR) O-O bond cleavage. This Arrhenius plot is described by where k is the forward rate constant for this reaction and T is the temperature (in Kelvin). Using this relationship, the estimated activation energy (Ea, obtained by multiplying the slope by -R) is about 17 kJ/mol, which agrees with the previously reported value of about 17 kJ/mol. Substituting room temperature (298 K) for T gives an estimated rate constant (k298) of 5.4 × 104 s-1, which corresponds to a mean lifetime (τ298) of approximately 19 μs (assuming first order kinetics with respect to Fe-OOR). Comparison of Time Scales of Ambient Mass Spectrometric Methods The timescales of ESI-MS, TM1-DESI-MS, and DESI-MS can be compared by deriving mathematical expressions for the reaction time (t) as a function of ion intensities in acquired mass spectra. The integrated rate law for the second order reaction A + B P that applies to the ESI-MS experiment is: where kf is the forward rate constant (an unknown value), [4a] and [4a]0 refer to the concentrations of final and initial concentrations of reduced DCIP, respectively, and [AA] and [AA]0 refer to initial and final concentrations of ascorbic acid, respectively. The variables [4a]0 and [AA]0 are known, but the final concentrations of DCIP and ascorbic acid must be estimated from ion intensities corresponding to oxidized and reduced DCIP (I4a and I4b, respectively). [4a] is relatively simple to calculate, since any 4a consumed turns into 4b. So, by multiplying [4a]0 by the fraction of unreacted 4a observed in the mass spectrum yields [4a]. Because 4a and 4b have nearly equal ionization efficiencies, the fraction of unreacted 4a is simply given by the intensity of 4a divided by the total DCIP intensity (I4a + I4b). Thus, the concentration of 4a can be found by: The final concentration of ascorbic acid can be found by assuming that all ascorbic acid consumed in this reaction is consumed by DCIP reduction. Thus, the final ascorbic acid concentration is simply the difference between the initial concentration and the amount of reduced DCIP ([4b]), which can again be measured directly from the mass spectrum. Analogous to the calculation of [4a], [4b] can be given as: So, substituting this term into the calculation of ascorbic acid remaining at the end of the reaction, the equation becomes: Substitution of these terms for [AA] and [4a] and solving for t yields the equation given in the main text: Calculation of the reaction time of TM1-DESI-MS is made difficult by the fact that the starting concentration of DCIP is not known, since it is desorbed from a mesh by the DESI spray. To perform this calculation, we can make the reasonable assumption of pseudo-first-order kinetics due to an excess of ascorbic acid in the spray relative to the small amount of DCIP that is desorbed from the mesh. This assumes that the concentration of ascorbic acid is not significantly affected by its reaction with the small amount of DCIP. The integrated rate law is then given by: The ratio in the logarithm can be calculated from the MS data by the following: If it is assumed that [AA]0 is the initial concentration placed in the DESI spray solvent, then substitution yields: An identical analysis can be performed for reactive DESI as well. These “time“ values can then be compared to compute factor differences in apparent timescale for the three MS techniques. The only shortcoming of this model is the fact that in both TM1-DESI and DESI, rapid microdroplet evaporation increases the actual concentrations of both reagents in the droplets as the reaction proceeds [2]. Comparison of TM1-DESI-MS to Other MS Techniques in the Reduction of DCIP Four separate sets of conditions were used to explore the reduction of DCIP by each of the three MS methods, generated by varying both ascorbic acid concentration and the distance from the solvent emitter to the MS inlet. The equations outlined above were used to estimate the timescales for continuous-flow ESI-MS, DESI-MS, and TM1-DESI-MS. Table S1 shows a summary of the results for all of these experiments. All eight experiments show shorter effective reaction time for TM1-DESI compared to the other techniques. Table S1. Comparison of TM1-DESI to ESI and DESI-MS for the reduction of DCIP under various experimental conditions. For TM1-DESI and DESI reactions between DCIP and L-AA, 5 μL of 5 x 10-3 M DCIP in CH3OH was deposited. For all continuous-flow ESI-MS experiments, the concentration of DCIP was decreased by one order of magnitude to compensate for desorption of DCIP from the surface. In all experiments, the calculated reaction yield, calculated as I4b/(I4a + I4b), was statistically different (p ≤ 0.05) between the two techniques under study in a give experiment. tTM refers to the calculated timescale of TM 1-DESI; Same for tESI and trDESI. [L-AA] (M) Emitter-Inlet Distance (cm) tESI tTM trDESI tTM 4 × 10-3 4 × 10-3 2 × 10-3 2 × 10-3 0.6 1.0 0.6 1.0 48 47 244 200 3.5 1.6 3.0 1.6 Relative Abundance Reduction of Dichlorophenolindophenol by Reactive Desorption Electrospray Ionization 267.993 100 270.008 0 268.0 268.2 268.4 268.6 268.8 269.0 m/z 269.2 269.4 269.6 269.8 270.0 Figure S11. Reactive DESI mass spectrum of DCIP reduction by ascorbic acid. L-AA (2 × 10-3 M in 1:1 CH3OH: H2O) was sprayed (5 μL/min; 200 PSI nebulizing gas; 0.6 cm emitter-to-inlet distance; +3 kV applied) towards a paper surface bearing DCIP (5 μL of 5 x 10-3 M in CH3OH). Schematic of Continuous-Flow Electrospray Ionization Mass Spectrometry Figure S11 shows a schematic of DCIP reduction studied by continuous-flow ESI-MS. A short capillary connects the mixing tee to the ESI emitter, resulting in a reaction time of only ~7 seconds at the liquid flow rate studied. L-AA Mixing Tee Mass Spectrometer Inlet N2 In Electrospray Emitter Flow Direction Reaction Mixture Out: Vol = 574 nL Fused Silica Capillaries DCIP Figure S12. Schematic of the continuous-flow ESI-MS experiment. Estimation of Microdroplet Velocities Previous studies by Cooks and co-workers demonstrated that primary droplets had velocities of 120 m/s at 2 mm distance from the DESI emitter tip [3]. COMSOL simulations show that the N2 velocity in TMn-DESI is 35% slower between M1 and M2 and 20% slower after M2, which corresponds to a velocity of ~9 m/s and a maximum microdroplet flight time of ~450 ms before entering the mass spectrometer, which further supports an estimated tTM range of ~350 ms – 500 ms. Since the velocity of the microdroplets desorbed from M2 is approximately 2-3 times faster compared to DESI (4 m/s after leaving the surface), we hypothesise that the droplet velocity is a significant factor leading to the ability to access shorter times. However, As a result, the tTM range of ~350 ms – 500 ms represents the experimentally ideal case, and that TMn-DESI has the potential to access reaction time scales on the order of hundreds of microseconds. References 1. 2. 3. Groves, J. T. and Watanabe, Y.: Reactive Iron Porphyrin Derivatives Related to the Catalytic Cycles of Cytochrome-P-450 and Peroxidase - Studies of the Mechanism of Oxygen Activation. J. Am. Chem. Soc. 110(8443-8452 (1988) Girod, M., Moyano, E., Campbell, D. I. and Cooks, R. G.: Accelerated Bimolecular Reactions in Microdroplets Studied by Desorption Electrospray Ionization Mass Spectrometry. Chem Sci. 2(3), 501-510 (2011) Venter, A., Sojka, P. E. and Cooks, R. G.: Droplet Dynamics and Ionization Mechanisms in Desorption Electrospray Ionization Mass Spectrometry. Anal. Chem. 78(24), 8549-8555 (2006)