Data flow and distribution
advertisement

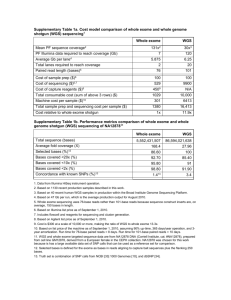
DEEP: Data Standards and Policies Introduction The German Epigenome Program DEEP is a consortium targeted at interdisciplinary epigenome research in Germany associated with IHEC initiative. It comprises 31 research centers, currently. The aim of the project is to generate and analyze more than 70 human epigenomes of 13 tissue types in the context of metabolic, inflammatory and neurodegenerative diseases. ChIP-seq, DNase-seq, RNA-seq and bisulfite sequencing data will be processed along with genomic sequencing. Among those experiments, due to high sequencing depth (more than 30X), processing of bisulfite sequencing data will be particularly demanding with respect to resources. The substantial volume and diversity of generated data requires adoption of standardized data formats and experiment descriptions. Consistent quality standards are crucial for downstream data integration. This document describes data standards and policies used by the DEEP consortium to exchange data between producers and the Data Coordination Center (DCC) and make the data available to the whole consortium. We will propose both detailed data exchange policies and standard data formats for data distribution. Data flow and distribution The six data providers in the DEEP project will submit the raw sequence data to the DCC at the DKFZ. Metadata describing sequencing experiments as well as quality values will be transferred to the DCC together with sequencing results. The DCC at the DKFZ will provide access to this data for the whole consortium. The sequencing data will be transferred (on request/automatically) to the Data Analysis Center (DAC) in Saarbrücken via an Aspera interface. Metadata describing biological samples will be transferred from the sample providers to the DCC by data producer in parallel with or previous to sequencing results. Primary Analysis results will be accessible from the DCC, if feasible. Initially, this will be restricted to the Consortium members. Public access will be provided according to the IHEC policies (http://ihec-epigenomes.net/about/policies-andguidelines/). We plan to follow BLUEPRINT recommendation and make our high level analysis results (the ChIP-seq, DNAse-seq, methylation signals and RNA-seq expression analysis) visible via a common web portal. The Biomart approach already being realized for ICGC was given a first attempt by the Spanish colleagues of BLUEPRINT (groups of Ivo Gut from Barcelona Supercomputing Center (BSC) and Alfonso Valencia from Spanish National Cancer research Center (CNIO)). Biomart failed as it did not scale up to epigenetics approaches. Consequently, the BLUEPRINT colleagues are currently developing a new database using MongoDB (a NoSQL technology). There was agreement that DEEP will wait for the availability of the MongoDB realization provided by BLUEPRINT’s Spanish colleagues. Afterwards, we will continue the discussion with the BLUEPRINT team to harmonize the web presentation of high level analysis results between DEEP and BLUEPRINT. For access to the raw data from outside the DEEP consortium, the sequencing data will be additionally transferred (automated, on request?) to the European Genomephenome Archive (EGA). The EGA will provide access to the wider public under the review of DEEP Data Access Compliance Office (DACO) (which still has to be established). Data access rules will follow the general guidelines from ICGC and IHEC. Sample laboratory Sample meta-data Sequencing groups Reads Sequencing meta data Consortium members Data Coordination Center (DCC) Data Analysis Center (DAC) Reads EGA meta data Approved researchers DEEP DACO Data Access Compliance Office EGA Sample meta-data Reads Sequencing meta-data Biomart RNA-seq analysis (Kiel) Figure 1 The Data Coordination Center has a central role in the project, providing a platform for storage and reception of raw data Data Formats Large-scale projects like DEEP need to specify their data formats and data standards to ensure interoperability and maximum usability of the data. DEEP is supporting community driven data standards for all its data types. Each kind of data transfer (each vertical arrow in figure 1) requires a clearly defined data format specification. Metadata for sequencing raw data (transfer from sequencing groups to DCC): o Format will be used according to ICGC specifications. Metadata for sequencing experimental data (transfer from sequencing groups to DCC): o Format will be used according to IHEC recommendations (NIH Roadmap). Metadata for primary cell lines (transfer from sample provider to DCC): o Have to be determined by sample provider. o Format will be used according to NIH roadmap for IHEC specification. o In agreement with BLUEPRINT we propose to use EBI or NIH Biosample database as a guideline. http://www.ncbi.nlm.nih.gov/biosample http://www.ebi.ac.uk/biosamples/index.html Metadata for patient information (transfer from sample provider to DCC): o Have to be defined by sample provider. o Sample naming will be defined together by the DAC and the DCC. Metadata for raw data to EGA (for submission from DCC to EGA at EBI): o According to EGA requirements. It’s similar to ICGC operation. o A Data Access Compliance Office (DACO) is required Data exchange from DCC to DAC o DAC obtains access to all the data gathered by DCC Data (high-level analysis) exchange from DAC to DCC o Based on BLUEPRINT precedent. High level analysis results (for submission from DCC to the BLUEPRINT MongoDB): o Based on BLUEPRINT precedent. o Naming conventions for high-level analysis files will be defined together by DCC and DAC. Primary data formats The primary data formats produced by DEEP are described in Table 1 Experiment Type Data Type All Alignment RNASeq Expression Levels BiSulphite, DNase and Regions ChipSeq BiSulphite, DNase and Signal ChipSeq Data Format Sam/Bam GTF BED WIG Table 1: File Formats used to store different data types produced by DEEP Quality Metrics The DCC will record some standard metrics to assist with quality control of both the incoming data and primary analysis results. All DEEP data will be submitted to three main types of quality control. First, all sequence data will be checked for quality and contamination. Second, all alignments and corresponding quality statistics will be calculated and distributed by DCC together with certain quality statistics and distributed for QC purposes. Finally, there will be data type specific metrics for RNA-Seq, Chip-Seq, DNase-Seq and BS-Seq sequencing which are also described here. It is expected that each sequencing center will perform internal quality control prior to submission to the DCC. Once sequence reads are stored by the DCC group, they are assessed for quality with specialized software. Sequence Quality and Contamination FastQC assesses multiple aspects of sequence quality and library diversity. These include the per-base quality across reads and the degree of sequence duplication and overrepresented sequence. (http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen) FastQ Screen is used to check the data for contamination and ensure that the reads map to the expected genome. Reads are mapped against the following: Human genome & transcriptome E. coli and yeast genomes UniVec (http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html) Common contaminants & PhiX (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) The output from both programs is available via DCC web portal, allowing transparent comparison of sequencing quality by all interested consortium members. Alignment Quality Metrics All our alignment metrics are collected using programs from the Picard toolkit (http://picard.sourceforge.net/) for BAM files. The alignment quality metrics are collected using AlignmentSummaryMetrics and DuplicationSummaryMetrics. These provide statistics like the number of reads mapped, mismatch rates, read aligned in pairs and duplication rates. (Table 2a and 2b) These statistics will be presented in a tab-delimited format DCC web page alongside any defined data freezes. RNA-Seq Quality Metrics Picard also has an RNA-Seq statistics utility called RnaSeqMetrics which also collects information such as Number of Ribosomal Bases, Number of coding bases and other coverage statistics for the RNA-Seq data set against a specific transcriptome (Table 3). ChIP-Seq and DNase-Seq Quality Metrics For these data sets measures including: percentage of reads mapped to enriched regions, mean and median region length and region length variance will be calculated. The ENCODE Project also defined a number of ChIP-Seq metrics that we will also measure like the Irreproducible Discovery Rate (IDR), Normalised Strand Crosscorrelation coefficient (NSC) and Uniquely Mappable Reads. These statistics will be distributed in tab-delimited files alongside the region and signal files. (http://genome.ucsc.edu/ENCODE/qualityMetrics.html#chipSeq) (Table 4) BS-Seq Quality Metrics The primary metric measured for BS-Seq is the conversion efficient. This is estimated using reads that uniquely map to the phage lambda reference; these should be derived from the spiked in unmethylated lambda DNA. Conversion efficiency λ is estimated from the formula: λ=1-(nC1+nG2)/(nG1+nC2), where nXy is the number of observed X bases in read y. This metric will be distributed along the BS-Seq data. A Metric #Reads #Noise Reads #Reads Aligned #Aligned_Bases #High-Quality Aligned Reads #High-Quality Aligned Bases #High-Quality Aligned Q20 Bases Median Mismatches Mismatch Rate In-del Rate Mean Read Length Reads Aligned in Pairs %Reads Aligned in Pairs #Bad Cycles Strand Balance %Chimeras %Adapter Description Total Number of Reads Number of Noise Reads Number of Reads Aligned Number of Aligned Bases Number of Aligned Reads with Mapping Quality of 20 or higher Number of Aligned Bases with Mapping Quality of 20 or higher Number of High Quality Aligned bases with Base Quality of 20 of higher Median Number of Mismatch High Quality Reads Rate of Mismatches in all reads Number of Indels per 100bp Average Read Length Number of Read Pairs where both reads aligned to the reference Percentage of Read Pairs where both reads aligned Number of Cycles where 80% of the bases were no calls Ratio of Number of Reads Aligned to the Positive Strand to the Number of Reads Aligned to the Whole Genome Number of Read Pairs where the reads map outside the maximum insert size or on different chromosomes Number of Reads which failed to map to the genome but do map to a known adaptor sequence B Metric #Unpaired_Reads #Read Pairs #Unmapped Reads #Unpaired Read Duplicates #Read Pair Duplicates #Read Pair Optical Duplicates %Duplication Estimated Library Size Description Total Number of unpaired Reads Total Number of Read Pairs Total Number of Unmapped Reads Number of Unpaired Reads which are Duplicated Number of Read Pairs which are Duplicated Number of Read Pairs which are Optical Duplicates Percentage of Duplication Estimated Number of Unique Molecules in the Library Table 2 Alignment Quality Metrics Collected by Picard AlignmentSummary and DuplicationSummary Metrics Metric #Bases #Aligned Bases #Ribosomal Bases #Coding Bases #UTR Bases #Intronic Bases #Intergenic Bases %Ribosomal Bases %Coding Bases %UTR Bases %Intronic Bases %Intergenic Bases %mRna Bases (#UTR Bases + #Coding bases / #Aligned Bases) Description Number of Bases Number of Aligned Bases Number of Ribosomal Bases Number of Coding Bases Number of UTR Bases Number of Intronic Bases Number of Intergenic Bases Percentage of Ribosomal Bases Percentage of Coding Bases Percentage of UTR Bases Percentage of Intronic Bases Percentage of Intergenic Bases Percentage of mRNA Bases %Usable Bases (#UTR Bases + #Coding bases / #Bases) Percentage of Useable Bases Median CV Coverage (Median Coefficient of Variation of Coefficient of Variation coverage of the 1000 most highly expressed transcripts) Coverage Median 5' Bias Median 5' bias Median 3' Bias Median 3' Bias Median 5' to 3' Bias Median 5' to 3' Bias Table 3 RNA-Seq Quality Metrics Collected by Picard RNASeqMetrics Metric Region Enrichment Median Region Length Mean Region Length Region Length Variance Region Length Standard Deviation Description Percentage of reads maapping to enriched regions Median Region Length Mean Region Length Table 4 ChIP-Seq and DNase-Seq Quality Metrics of DACO (Data Access Compliance Office): Public dissemination For users to access the raw data files the correct authentication and authorization is required. The EGA does not grant access to the data; data access must be applied for from the IHEC Data Access Compliance Office (DEEP-DACO). The EGA provides a personal account with access permissions for each successful applicant. Applicants must also sign the Data Access Agreement with the DEEP-DACO that provides instruction on how data can be stored, used and transferred once it has been download from our system. Conclusions This report describes coherent set of standards and policies for the DEEP consortium to follow. As the project progresses and data types evolve, the DCC will seek to find appropriate new measures to ensure the data DEEP produces continues to represent a high quality epigenomic resource. Scheduling issues Metadata for sequencing raw data (transfer from sequencing groups to DCC): o First test submission of raw data from sequencing centers to DCC will be initiated Time: after the first runs have been performed (expected in summer 2013) Metadata for sequencing experimental data (transfer from sequencing groups to DCC): o The extract of the current BLUEPRINT metadata is available and will be distributed by the DAC to the sequencing centers and sample labs. Time: will be discussed at the Saarbrücken conference (28.6.) Metadata for patient information (transfer from sample provider to DCC): o Has been initiated by the DAC. o Time: waiting for answer from sample provider Metadata for raw data to EGA (for submission from DCC to EGA at EBI): o According to EGA requirements. It’s similar to ICGC operation. o A Data Access Compliance Office (DACO) is required. Initiation will be done by DEEP coordinator. o Time: waiting for answer of coordinator