DOC
advertisement

BCB 444/544 Fall 07 Oct 29
BCB 444/544
Homework 5
Due Mon Nov 5
HW 5
Name _________________________________________
(please bring to class or deliver to MBB 106)
Objectives:
1. Learn to use Fitch’s algorithm for computing the parsimony score of a phylogenetic tree
2. Complete a computer exercise to align a set of sequences and build some phylogenetic trees
Note: you may work together on these problems, but each student must submit answers in his/her own words.
Introduction:
In lecture we will discuss using parsimony methods for phylogenetic trees. There are two parsimony
problems considered, the large parsimony problem and the small parsimony problem. The large parsimony problem
is: find the tree that explains the observed data with the fewest number of evolutionary changes. This problem is
rather difficult since it requires exploring all possible trees. The small parsimony problem is: given a tree, what is
the parsimony score? The small parsimony problem is quite a bit easier to solve. One method for solving the small
parsimony problem is Fitch’s algorithm. It is based on set operations, and all evolutionary events (mutations) are
treated equally (i.e. a mutation from an A to a C is scored the same as a mutation from an A to a G, and so on). I
will now give you the precise notation for Fitch’s algorithm.
v in a tree has a set X v .
If v is a leaf, X v is the nucleotide (or amino acid for protein based trees) observed at v .
If v is a node with descendants u and w :
o Let Y X u X w
o If Y make X v Y
o If Y make X v X u X w and count one evolutionary step.
Each node
If this notation is not clear, perhaps an example of the algorithm in action will help.
Consider the following tree:
A
T
G
C
A
G
A
T
BCB 444/544 Fall 07 Oct 29 HW 5
We will start at the leaf nodes (the tips of the tree) and work our way down to the root. Look at the first internal
node, the branch point to the A and T on the left side of the tree. The intersection of the sets of the descendant
nodes is the empty set. ( A T ). Therefore, we take the union of the sets and count one evolutionary
step. I have filled in the sets for the first level of internal nodes and marked the ones where we took an
evolutionary step with an *.
A
T
G
C
{A,T}*
A
G
A
{A,C}*
T
{A,T}*
We can now repeat the process with the next level of internal nodes. Here is the next level marked:
A
T
{A,T}*
G
C
A
{A,C}*
{A,C,G}*
G
A
T
{A,T}*
{A,G,T}*
So far, we have taken the union of the sets at each node and counted an evolutionary step each time. The next
node will be different:
BCB 444/544 Fall 07 Oct 29
A
T
G
C
{A,T}*
A
G
A
{A,C}*
HW 5
T
{A,T}*
{A,C,G}*
{A,G,T}*
{A,G}
Ah, now that’s more interesting. We finally had an intersection of two sets that was not empty. Now, for the
conclusion, let's find out what possible states our root ancestor had at this position:
A
T
G
C
{A,T}*
A
{A,C}*
{A,C,G}*
G
A
T
{A,T}*
{A,G,T}*
{A,G}
{A}
Our root ancestor could only have had an A at this position. Also, if we simply count our sets marked with an *, we
will get the number of evolutionary steps it takes to explain our observed data. We have 5 evolutionary steps, and
because all steps are treated equally by the Fitch algorithm, the parsimony score for this tree is 5.
BCB 444/544 Fall 07 Oct 29
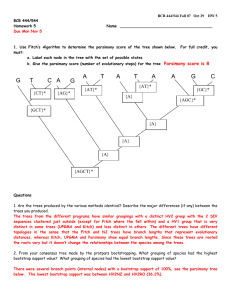
1. Use Fitch’s Algorithm to determine the parsimony score of the tree shown below.
must:
a. Label each node in the tree with the set of possible states
b. Give the parsimony score (number of evolutionary steps) for the tree
G
T
C A
G
A
T
A
T
A
A
HW 5
For full credit, you
G
C
2. This exercise follows one from the Appendix of the Xiong textbook. The exercise starts on page 308 of
the book and requires the sequences at:
http://www.cup.cam.ac.uk/catalogue/catalogue.asp?isbn=9780521600828&ss=res
A. The file we need for this exercise is Sequences for Exercises – gp120. Follow the instructions in the textbook
for “Constructing and Refining a Multiple Sequence Alignment.” The textbook includes references to nedit, but any
text editor will work for these steps.
B. Unfortunately, after the first section, some of the servers mentioned in the textbook no longer allow access, so
we cannot follow all of the instructions. We will use the alignment generated in step A as input to some programs
for building phylogenetic trees.
All of the phylogenetic programs we will run in this homework can be found at:
http://bioweb.pasteur.fr/seqanal/phylogeny/intro-uk.html
If this server is busy, slow, down, etc. This server has the PHYLIP programs with the same input forms:
BCB 444/544 Fall 07 Oct 29
HW 5
http://bioinfo.hku.hk/phylipinterface.html
We will first use the PHYLIP program protpars to build a tree based on maximum parsimony.
Click on PHYLIP at the top of the page, select the advanced form for protpars.
Enter your email address and your alignment in PHYLIP format.
Scroll down the page to the Bootstrap options and click on the checkbox for “Perform a boostrap before analysis”
Enter an odd number in the random number seed box
Click on the checkbox for compute a consensus tree
Then scroll down and click Run protpars.
The output of the program will be emailed to you along with a URL for viewing all of the results. Browse through
the results to see what information the program returns to you. You’re probably wondering how to get a pretty
looking tree out of all that text-based output. Go to the URL that contains all of the protpars results that was
emailed to you.
Near the top of the page, there is a button that says “Run the selected program on outtree.” Choose either
drawtree or drawgram and click the button to get a nice looking version of your tree. Drawtree produces unrooted
trees and drawgram produces rooted trees.
The output of these two programs is a postscript file. If your computer cannot display postscript files, save the
file to your computer, and try out PS2PDF (http://silver.ps2pdf.com/convert.htm) to convert the postscript file to
a PDF.
Go back to the main phylogenies page at
http://bioweb.pasteur.fr/seqanal/phylogeny/intro-uk.html
The next set of programs we will run require a distance matrix as input.
Use the PHYLIP program protdist to compute the distance matrix. Note that the input needed for protdist is your
alignment.
Use the following programs to build a tree from your distance matrix:
PHYLIP neighbor using NJ
PHYLIP neighbor using UPGMA
PHYLIP fitch
PHYLIP kitsch
The input for each of these programs is your distance matrix. Each of these programs has an input page similar to
the one for protpars and produces similar output. Save your trees from each program.
BCB 444/544 Fall 07 Oct 29
HW 5
Questions
1. Are the trees produced by the various methods identical? Describe the major differences (if any) between the
trees you produced.
2. From your consensus tree made by the protpars bootstrapping, What grouping of species had the highest
bootstrap support value? What grouping of species had the lowest bootstrap support value?