Mitochondria and Epilepsy
advertisement

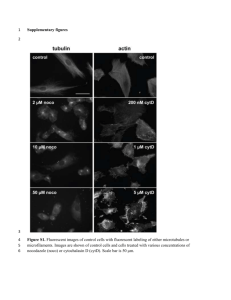
Mitochondria and Epilepsy Major Research interests: Dysfunction of the mitochondrial respiratory chain associated with epilepsy Mitochondrial oxidative phosphorylation provides the major source of ATP in neurons. It consists of five multienzyme complexes located in the mitochondrial inner membrane (Figure). The complexes I to IV are oxidoreductases which participate in the transfer of reducing equivalents from NADH and FADH2 to oxygen and create an electrochemical proton gradient across the mitochondrial inner membrane. Complex V – the F0F1-ATPase uses this proton gradient for the synthesis of ATP. Defects of oxidative phosphorylation in the CNS are the characteristic sign of mitochondrial encephalopathies. In a broad variety of these diseases epileptic seizures have been observed. An overview of the most common mitochondrial disorders presenting with an epileptic phenotype is given in the Table. Most of them are associated with mutations in the autonomous mitochondrial DNA, consisting of 13 polypeptide genes, a 16S and a 12S rRNA gene and 22 tRNA genes. A well known mitochondrial disorder with an epileptic phenotype is the MERRF (myoclonus epilepsy with ’ragged red fibers’) syndrome which has been associated with mutations in the mitochondrial tRNALys gene. However, as shown in the Table, other mitochondrial DNA mutations predominantly localised in the mitochondrial tRNA genes for lysin, serin, leucin, isoleucin or cystein have been also associated with epileptic phenotypes. These mutations affect the protein biosynthesis of all mitochondrial-encoded subunits of the following complexes of the mitochondrial oxidative phosphorylation pathway: complex I (NADH:CoQ oxidoreductase, containing 7 mitochondrial-encoded subunits), complex III (CoQH2:cytochrome c oxidoreductase, containing 1 mitochondrial-encoded subunit), complex IV (cytochrome c oxidase, containing 3 mitochondrial-encoded subunits) and complex V (F0F1-ATPase, containing 2 mitochondrial-encoded subunits). Quite rarely, also mutations in polypeptidecoding mitochondrial genes have been reported in patients with epilepsy – in the ATPase 6 gene, in the CO III gene and in the ND 1 gene. We have recently identified a novel mutation in the CO I gene associated with Epilepsia patialis continua. The large variation in the clinical phenotype, even for a given mutation, is a well known feature of mitochondrial diseases. It is therefore very likely that the distribution of the mitochondrial defect within the CNS is the responsible factor which determines the association of a certain mutation with epilepsy. Thus KSS is almost a white matter disorder affecting preferentially brainstem tegmentum, white matter of cerebellum and cerebrum, cervical spinal cord, basal ganglia, and diencephalon. On the other hand, MELAS is characterised by foci of necrosis, which are predominantly localised in the cerebral cortex and also in the hippocampus – a highly epileptogenic area, whereas MERRF involves preferentially the inferior olivary nucleus, the cerebellar dentate nucleus, the red necleus and the pontine tegmentum – structures being implicated in the genesis of myoclonus. In contrast to the relatively rare mitochondrial encephalopathies being associated with mtDNA mutations epilepsy is a frequent neurological disorder usually well controlled by presently available drugs. However, 20 to 30% of patients do not experience seizure control with available medication. The majority of these patients suffer from focal epilepsies which frequently develop subsequently to brain trauma, complicated febrile convulsions, status epilepticus, ischemic lesions and brain tumours. The areas of epileptogenesis in these cases are usually characterised by cell loss. It is well documented that during seizures both nerve cells and glia undergo necrotic and apoptotic cell death. Neuropathological investigations have repeatedly pointed to a similarity between ischemic and seizure related alterations of neurons characterised by swollen and often disrupted mitochondria. In patients with Ammon’s horn sclerosis mitochondrial ultrastructural pathology was described as characteristic feature of hilar neurons. In this context is noteworthy to mention, that in addition to the pathological abnormalities also functional defects of mitochondria have been reported in the areas of epileptogenesis. Thus, we observed a severe impairment of respiratory chain complex I activity for CA3 neurons in the hippocampus from patients with Ammon’s horn sclerosis and in the parahippocampal gyrus of patients with parahippocampal lesions. In these reports the mitochondrial abnormalities have been observed only close to or directly in the epileptic focus, while the investigated surrounding brain tissue (e.g. the parahippocampal gyrus of patients with a clearly pronounced hippocampal pathology and a hippocampal seizure focus) showed now mitochondrial pathology. Mitochondria as potential trigger for neuronal cell death observed in epilepsy Prolonged seizures (status epilepticus) induced in experimental models by kainic acid or pilocarpine are known to activate programmed cell death mechanisms. This cell death observed also in human epilepsy is one of the most important aspects of epileptogenesis. In the hippocampus the loss of CA1, CA3 and CA4 pyramidal neurons, with relative sparing of the granular neurons of the dentate gyrus and some types of interneurones, is the histopathological hallmark of Ammon’s horn sclerosis. The mechanism that underlies this regional selectivity remains to be elucidated, some data point to differential expression of proaportotic and antiapoptotic genes. The probably most important factor preceding neuronal cell death after status epilepticus is the increased level of reactive oxygen species observed in various models of experimental epilepsy – kainate-induced hippocampal damage, pilocarpine treatment and low Mg2+-induced epileptiform activity in brain slices and slice cultures. Mitochondria are known to be the most important source of production of reactive oxygen species. Moreover, increased production of reactive oxygen species is a feature of partially respiratory chain-inhibited mitochondria and it is noteworthy to mention in this context that a severe impairment of respiratory chain complex I activity is present in the areas of epileptogenesis - in CA3 neurons of the hippocampus from patients with Ammon’s horn sclerosis and in the parahippocampal gyrus of patients with parahippocampal lesions. Similar observations we made recently in the vulnerable CA1 and CA3 hippocampal subfields of pilocarpine-treated chronic epileptic rats. As potential cause of the detected respiratory chain impairment we could delineate a decrease of the mtDNA copy number. This finding indicates a possible substantial role of oxygen radicals in causing neuronal mtDNA damage occurring selectively in the areas of epileptogenesis. How mitochondrial dysfunction can alter neuronal excitability ? Beside alterations of mitochondrial substrate oxidation and ATP synthesis due to diseaseassociated mutations discussed in detail before, also the direct partial inhibition of enzymes of mitochondrial respiratory chain – of cytochrome c oxidase by cyanide, and of succinate dehydrogenase by 3-nitropropionic acid – evoke seizures. Potential direct links between the observed impairment of mitochondrial function and the increased neuronal excitability causing epileptiform activity are (i) decreased intracellular ATP levels and (ii) alterations of neuronal calcium homeostasis. A relatively high impact of neuronal ATP levels can be postulated since epileptic seizures are observed in Leigh syndrome patients harbouring the mutations T8993G and T8993C in the ATPase 6 gene. Under these conditions mitochondria still have a high membrane potential enabling normal ion transport. Therefore, for cybrids with the T8993G NARP mutation normal mitochondrial calcium handling properties at decreased cellular ATP levels were observed. It has to be mentioned that mitochondrial oxidative phosphorylation provides the major source of ATP in neurons and adequate ATP levels are essential to maintain the neuronal plasma membrane potential via the sodiumpotassium ATPase which consumes about 40% of the energy. Therefore, the decreased neuronal plasma membrane potential is most likely responsible for epileptic seizures observed in Leigh syndrome patients harbouring ATPase 6 gene mutations. On the other hand it is well established that mitochondria are an important intracellular Ca2+ sequestration system. Especially due to this feature, mitochondria are believed to modulate neuronal excitability and synaptic transmission which is altered in epilepsy. In agreement with this concept in kainate-treated chronic epileptic rats impaired oxidative phosphorylation due to Ca2+ cycling at the inner membrane of hippocampal mitochondria has been demonstrated by us. Similarly, impaired cellular Ca2+ homeostasis due to substantial alterations of mitochondrial Ca2+ handling was the predominant feature of cybrid cells harbouring the mitochondrial T8356C mutation being associated with MERRF. Brain energy metabolism as potential target of neuroprotective strategies For a certain number of neurodegenerative diseases with established mitochondrial pathology, like amyotrophic lateral sclerosis and Chorea Huntington neuroprotective strategies have been suggested. The proposed treatment is buffering of neuronal energy levels by systemic creatine administration. The supplemented creatine passes the blood-brain barrier and increases the total pool of phosphocreatine / creatine available for buffering of the neuronal ATP levels by creatine kinase. Creatine supplementation has been shown to protect motor neurons in a transgenic animal model of amyotrophic lateral sclerosis and striatal neurons in an animal model of Huntingston’s disease. Our NMR spectroscopic data on patients with amyotrophic lateral sclerosis showed upon creatine supplementation increased N-acetyl aspartate levels in the motor cortex of these patients, suggesting at a potential improvement of neuronal energy levels. Interestingly, the buffering of brain energy levels with creatine seems to be effective not only in the mentioned neurodegenerative disorders but also in hypoxia-induced or traumatic brain injury. Moreover, 3 g/kg creatine were observed to reduce hypoxia-induced seizures in rat and rabbit pups. The neuroprotective effect of the creatine treatment in human pathology remains, however, still to be shown. Selected recent publications: 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. Kunz, W.S., Kuznetsov, A.V., Clark, J.F., Tracey I. & Elger, C.E. (1999) Metabolic consequences of the cytochrome c oxidase deficiency in brain of copper-deficient Movbr mice. J. Neurochem. 72, 1580-1585 Kunz, W.S., Goussakov, I.V., Beck, H. & Elger, C.E. (1999) Altered mitochondrial oxidative phosphorylation in hippokampal slices of kainate-treated rats. Brain Res. 826, 236-242 Kudin, A., Vielhaber, S., Beck, H., Elger, C.E. & Kunz, W.S. (1999) Quantitative investigation of mitochondrial function in single hippocampal slices: A novel application of high-resolution respirometry and laser-excited fluorescence spectroscopy. Brain Res. Prot. 4, 93-98 Vielhaber, S., Kunz, D., Winkler, K., Wiedemann, F.R., Kirches, E., Feistner, H., Heinze, H.-J., Elger, C.E., Schubert, W. & Kunz, W.S. (2000) Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 123, 1339-1348 Schröder, R., Vielhaber, S., Wiedemann, F.R., Kornblum, C., Papassotiropoulos, A., Broich, P., Zierz, S., Elger, C.E., Reichmann, H., Seibel, P., Klockgether, T. & Kunz, W.S. (2000) New insights in the metabolic consequences of large scale mtDNA deletions: A quantitative analysis of biochemical, morphological and genetic findings in human skeletal muscle. J. Neuropath. Exp. Neurol. 59, 353-360 Kunz, W.S., Kudin, A.P., Vielhaber, S., Blümcke, I., Zuschratter, W.,Schramm, J., Beck, H. & Elger, C.E. (2000) Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 48, 766-773 Kunz, W.S., Kudin, A., Vielhaber, S., Elger, C.E., Attardi, G. & Villani, G. (2000) Flux control of cytochrome c oxidase in human skeletal muscle. J. Biol. Chem. 275, 2774127745 Kunz, W.S. (2001) Control of oxidative phosphorylation in skeletal muscle. Biochim. Biophys. Acta (Bioenergetics) 1504, 12-20 Debska,G., May, R., Kicinska, A., Szewczyk, A., Elger, C.E. & Kunz, W.S. (2001) Potassium channel openers depolarize hippocampal mitochondria. Brain Res. 892, 4250 Vielhaber, S., Schröder, R., Winkler, K., Weis, S., Sailer, M., Feistner, H., Heinze, H.J., Schröder, J.M.& Kunz, W.S. (2001) Defective mitochondrial oxidative phosphorylation in myopathies with tubular aggregates originating from sarcoplasmic reticulum. J. Neuropath. Exp. Neurol. 60, 1032-1040 Vielhaber, S., Kaufmann, J., Kanowski, M., Sailer, M., Feistner, H., Tempelmann, C., Elger, C.E., Heinze, H.-J. & Kunz, W.S. (2001) Effect of creatine supplementation on metabolite levels in ALS motor cortices. Exp. Neurol. 172, 377-382 12. 13. 14. 15. 16. 17. Kunz, W.S. (2002) The role of mitochondria in epileptogenesis. Curr. Opin. Neurol. 15, 179-184 Kunz , D., Winkler, K.,Elger, C.E. & Kunz, W.S. (2002) Functional imaging of mitochondrial redox state. Method. Enzymol. 352, 135-150 Kudin, A.P., Kudina, T.A.,Seyfried, J., Vielhaber, S., Beck, H., Elger, C.E. & Kunz, W.S. (2002) Seizure-dependent modulation of mitochondrial oxidative phosphorylation in rat hippocampus. Eur. J. Neurosci. 15, 1105-1114 Varlamov, D.A., Kudin, A.P., Vielhaber, S., Schröder, R., Sassen, R., Becker, A., Kunz, D., Haug, K., Rebstock, J., Heils, A., Elger, C.E. & Kunz, W.S. (2002) Metabolic consequences of a novel missense mutation of the mtDNA COI gene. Hum. Mol. Genet. 11, 1797-1805 Kudin, A.P., Vielhaber, S., Elger, C.E. & Kunz, W.S. (2002) Differences in flux control and reserve capacity of cytochrome c oxidase (COX) in human skeletal muscle and brain suggest different metabolic effects of mild COX deficiencies. Biocomplexity 1, in press Vielhaber, S., Varlamov, D.A., Kudina, T.A., Schröder, R., Kappes-Horn, K., Elger, C.E., Seibel, M., Seibel, P. & Kunz, W.S. (2002) Expression pattern of mitochondrial respiratory chain enzymes in skeletal muscle of patients harbouring the A3243G point mutation or large scale deletions of mitochondrial DNA. J. Neuropath. Exp. Neurol., in press