2. the phase field model for multicomponent alloys
advertisement

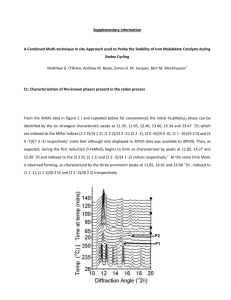
NUMERICAL SIMULATION FOR TRANSFORMAION IN Fe-Mn-C SYSTEM BY PHASE-FIELD MODEL Dong-Hee Yeon, Pil-Ryung Cha, Jong-Kyu Yoon School of Materials Science and Engineering, Seoul National University, Seoul 151-742, Korea ABSTRACT In Fe-Mn-C system, there is a large difference in mobility of solute Mn and C. Substitutional element Mn diffuses much slower than interstitial element C in Fe. By definition, paraequilibrium means that only the mobile elements are equilibrated while the sluggish ones behave as a single element. The transformation may be under local equilibrium, paraequilibrium, or lying between these two limits. The evolution mode could be determined by the diffusion velocity of solute elements in the matrix in front of the moving interface and the interface migration. Phase-field model could be applicable to simulate these phenomena without any constraints or boundary conditions at the interface. The objective of this study is to find out evolution mode for paraequilibrium or full thermodynamic equilibrium by the phase-field model of ternary system. 1. INTRODUCTION Many researches have been carried out for the transformation of Fe-Mn-C system both theoretically and experimentally [1-5]. In transformation of Fe-Mn-C system, there is a regime that can be characterized by the absence of partitioning of the substitutional alloying element Mn. This type of transformation is called paraequilibrium. Under paraequilibrium, carbon diffuses at an appreciable rate but the alloying element Mn is almost immobile relative to iron, the growing phase inherits the alloy content form the parent phase. While the mobile element carbon is in the local equilibrium at the phase interface, the chemical potentials of alloying substitutional element have no physical meaning and thus these substituional elements behave thermodynamically as if they were only a single element [4]. Many works have been done mainly about the limit in which the transformation under paraequilibrium can be possible thermodynamically and about the transition from fully local equilibrium to paraequilibrium. But, both fully local equilibrium and 375 paraequilibrium states are thermodynamically unique states and the reaction of course may choose any one of infinite numbers of undefined nonequilibrium states lying between the equilibrium and paraequilibrium limits. Whether nature in fact attains such states between two limits is a very subtle problem in kinetics. So the model is needed that can select the state with considering the thermodynamics and kinetics of the system correctly. The Phase field model can be recommended as the unified model that is applied to any systems under fully local equilibrium, paraequilibrium and nonequilibrium states lying between two limits. It has been reported that phase field model is able to describe successfully the interface kinetics such as solute trapping effect when it is applied to solidification in binary system [6,7]. In order to describe the paraequilbrium state, the phase field model for multicomponent alloy is needed and it can distinguish the interstitial alloy elements from the substitutional alloy elements because the large difference in atomic mobility is induced from the different diffusion mechanism. However, the existing phase field models for alloys are restricted to binary alloys. And, although these models are applicable to only binary system that contains only substitutional alloy element, they are often applied to the system that has interstitial alloy element such as carbon. [8,9] In this study, the transformation in Fe-Mn-C system is simulated by the phase field model for multicomponent alloys. The result is compared with the simulation by the model that does not distinguish between intestitial and substituiuional alloy elements. The phase field model is certified by the comparison with the results of the system that is composed of only substituional alloy elements, so the difference of atomic mobility is small. 2. THE PHASE FIELD MODEL FOR MULTICOMPONENT ALLOYS When the phase field model is applied to the multicomponent alloy system, the phase field evolution and diffusion equation could be established from a LandauGinzberg free energy functional F(c1,c2,). For this purpose, (x,t) is postulated to characterize the phase of the system. The phase field is defined as a continuous variable between =1 at one phase and =0 at the other phase. In the interfacial region that has the finite width, (x,t) has the value between 0 and 1 and varies steeply but smoothly. In multicompnent systems, the free energy functional F(c1,c2,) is the functional of the phase field variable with solute concentration. [10] 2 2 F f , c1 , c2 , , , cn dV V 2 376 (1) Here, f(c1,c2,) is the free energy density (free energy per unit volume) and it may be written in the form, f , c1 , c2 , , cn h f c1 , c2 , , cn 1 h f c1 , c2 , , cn wg , (2) where f and f are the free energy densities of each phase, respectively and functions of solute concentration ci. It is assumed that the phases in the interfacial region are the mixture of phases that are composed of same concentration (WBM Model) [6]. In equation (2), we choose h(), g() in the following forms. h 3 6 2 15 10 (3) g 1 2 2 (4) where g() is the double-well potential associated with phase change. The evolution equation of phase field and diffusion equations are driven from gradients of the above functional. F (5) M t n 1 J k M ki i 1 F ci (6) In the equations (5) and (6), M is the phase field mobility and Mki is the mobility matrix. From equations (5) and (6), it is possible to get governing equations. M 2 2 f t (7) 2 2 M h f f g ck n1 M ki f ci t i 1 In the above equations, f and fCi mean (8) f f , respectively. Since equation (8) c i should be the same form with traditional diffusion equation in one phase region, we n 1 can determine Mki using Dkj M ki f ci c j . The parameter in equation (7) and (8) i 1 should be related to the interfacial properties, the interfacial energy and interface thickness 2 respectively. The parameters are determined at the sharp interface limit when we define interfacial region as the region 3 2 where changes from 0.1 to 0.9 at - to . The phase field mobility can be V determined at the sharp interface limit as F , where F is the driving M 2 condition, , 2 2.2 2 force per unit volume and V is the interface velocity. The phase field mobility can be 377 calculated with the relationship between driving force and interface velocity V M i F where Mi is interface mobility. [6,10] 3. CONSIDERATION OF INTERSTITIAL ALLOY ELEMENT The existing phase field models cannot be applied to the system that contains interstitial alloy element because these models use the Cahn-Hilliard type diffusion equation and they usually use atomic fraction of solute atoms as the concentration variables. These models cannot afford to distinguish the different diffusion mechanisms of substitutional alloy elements and interstitial alloy elements. The diffusion of substitutional solute atoms always accompanies with the diffusion of solvent atoms while the diffusion of interstitial solute atoms does not. If one uses the atomic fraction of solute atoms as the concentration variables, the increase of concentration of interstitial alloy elements means the decrease of the concentration of solvent atoms. In order to consider the interstitial alloy element in the diffusional transformation, the site fraction is used as the independent variables [3,11]. However, the definition of the site fraction in the mixture phases is a delicate problem. So, we use moles per unit volume as the concentration variables. It is assumed that the solvent atoms and the substitutional elements contribute to the total molar volume of the alloy and there is no contribution from the interstitial elements. There is no constriction on the movement of the interstitial atoms and total moles of substitutional atoms are conserved. The two constraints can be written as follows. 1 ci const . (9) VS iS c iI i cVa const. (10) VS is the molar volume of substitutional alloy elements, cVa and ci is the mole numbers of vacancies per unit volume in the interstitial sites and S, I denote substitutional element and interstitial element respectively. It is assumed that all substitutional elements have the same molar volume and interstitial elements have no volume. We did not consider the vacancies in the substitutional sites. From the above assumptions, we can evaluate correct driving force of solute diffusion. The free energy per unit volume can be written as the following, n f ci i (11) i 1 From equation (8), if we do not consider the off diagonal term of atomic mobility matrix, the driving force of solute diffusion per unit volume is f ci . For the case of 378 substitutional elements, the driving force per unit volume is i Fe and for interstitial elements, f ci i . 4. NUMERICAL SIMULATION OF TRANSFORMAION The isothermal transformation in Fe-Mn-C system is simulated by numerical method. The overall compositions of each element are Mn 1.0% and C 1.0% and the temperature of the system is 873K. In this system, the carbon diffuses 107~109 times faster than Mn. So the ferrite grows under paraequilibrium. The simulation is performed that the ferrite, which occupies initially 10% of whole system with the same composition in austenite, grows into austenite phase. The result is shown in Fig. 1. Figure 2 is the same result in which the concentration is expressed as a function of the atomic fraction. 1450 Mn (Mole per unit Volume) 12000 1440 C (Mole per unit Volume) 10000 Initial profile 0.5E-4sec 1.0E-4sec 1.5E-4sec 2.0E-4sec 1.2sec 8000 6000 4000 2000 1430 0.0 1.0x10 -2 2.0x10 -2 3.0x10 -2 4.0x10 0 -2 0.0 1.0x10 Position( m) -8 -8 2.0x10 3.0x10 Position( m) -8 4.0x10 -8 Fig. 1. Concentration Profile as a Function of Mole per Unit Volume 12 Initial profile 0.5E-4sec 1.0E-4sec 1.5E-4sec 2.0E-4sec 1.2sec 1.1 8 1.0 C (at %) Mn(at %) 10 0.9 6 4 2 0 0.8 0.0 -2 -2 1.0x10 2.0x10 3.0x10 Position( m) -2 4.0x10 -2 -2 0.0 1.0x10 -2 -2 2.0x10 3.0x10 Position( m) -2 4.0x10 -2 Fig. 2. Concentration Profile as a Function of Atomic Fraction From the above results, we can see that the phase-field model describes the phase transformation under paraequilibrium. The result without considering interstitial element is shown in Fig. 3. Comparing with Fig. 2, there is no change of the atomic 379 fraction of Mn and only concentration of carbon changes. This means that there is a change of the atomic fraction of Fe and only Fe atoms move without movement of Mn atoms. During the growth of ferrite, the difference of chemical potentials of carbon is very small as compared with that of Mn across the interface. Kirkaldy et. al said that under paraequilibrium as the mobile atom is fully local equilibrium state at the interface and the chemical potential of immobile atom is discrete [2]. The small difference in chemical potential of carbon might be kinetic effect. 1.02 8 7 Initial Profile 1.25E-5 2.50E-5 3.75E-5 Final Profile (8.6sec) 5 C (at %) Mn (at % ) 6 1.00 4 3 2 1 0.98 0 0.0 1.0x10 -2 2.0x10 -2 3.0x10 -2 4.0x10 -2 0.0 Position ( m) 1.0x10 -2 -2 2.0x10 3.0x10 Position ( m) -2 4.0x10 -2 Fig. 3. Concentration Profile as a Function of Atomic Fraction during Transformation of Fe-Mn-C System Without Considering the Interstial Atom In order to compare with the above calculation, another Transformation in the Fe-Cr-Ni system, which contains only substitutional elements, is studied. The overall compositions of each element are Cr 23.0% and Ni 9.04% and the temperature of the system is 1373K. This system was verified thermodynamically and kinetically by the concept of local equilibrium [12,13]. In this system, Cr diffuses only 10~102 times faster than Ni. So Both Cr and Ni atoms are partitioned at the interface and the transformation is controlled by diffusion of Ni atoms because Ni moves slower. The simulation is performed that the austenite, which occupies initially 10% of whole system with the same composition with ferrite, grows into ferrite phase. Figure. 4 show that the transformation is governed by Ni diffusion. 32 20 initial profile 0.02sec 0.08sec final profile (1.24sec) 28 26 Ni( at % ) Cr ( at % ) 30 24 22 20 10 18 16 0.0 1.0x10 -2 2.0x10 -2 3.0x10 Position ( m) -2 4.0x10 0 -2 0.0 1.0x10 -2 2.0x10 -2 3.0x10 Position ( m) -2 4.0x10 -2 Fig. 4. Concentration Profile as a Function of Atomic in Fe-Mn-C System 380 When the system reaches equilibrium state, the equilibrium volume fractions of ferrite and austenite are 20.11% and 79.89% respectively and equilibrium concentrations of Cr and Ni in ferrite are 29.26at% and 5.69at% respectively and those in austenite are 21.42at% and 9.88at% [13]. The result performed by the phase field model is that the equilibrium volume fractions of ferrite and austenite are 20.44% and 79.56% respectively and equilibrium concentrations of Cr and Ni in ferrite are 29.22at% and 5.70at% respectively and those in austenite are 21.43at% and 9.90at% respectively. From this result, the phase field model reflects thermodynamics and solute conservation properly. For Fe-Mn-C system, it takes too long to reach the equilibrium state. So, we calculate the equilibrium volume fraction of each phase and their compositions by increasing the diffusivities of Mn hypothetically in both phases. The result predicted by the thermodynamic calculation is that the ferrite (Fe-0.511at% Mn-0.077at%C) occupies 86.6% of total volume and the austenite (Fe-4.172at%Mn6.984at%C) occupies 13.4% at equilibrium state. The result by phase field model is that the compositions of Mn and C in ferrite is Fe-0.507at% Mn-0.0737 at %C and that in austenite is Fe-4.169at%Mn-6.965at%C respectively and volume fractions of ferrite and austenite are 86.5% and 13.5% respectively. From the results presented, we can say that the transformation mode between fully local equilibrium and paraequilibrium can be described properly using the phase field model. The selection of transformation mode depends on the relationship between the interface mobility and the diffusivities of the solute atoms. 5. CONCLUSION Using the phase field model, the transformation of Fe-Mn-C system is simulated. It is shown that the phase field model can describe not only thermodynamics effects of the phase transformation but also the kinetics such as partitioning of solute atoms, so it is possible to simulate various modes of partitioning behavior at the interface without extra boundary condition for moving interface. By consideration of interstitial elements, we can simulate the transformation under paraequilibrium properly. ACKNOWLEDGEMENTS The authors appreciate Prof. Kyu-Hwan Oh in Seoul National University for his help in thermodynamic evaluation Authors are grateful to the financial support of Brain Korea 21 program supported by Ministry of Education, Korea. 381 REFERENCES 4. Gilmour J. B., Purdy G. R. and Kirkaldy J. S.: ‘Thermodynamics controlling the proeutectoid ferrite transformation in Fe-C-Mn Alloys’ Metall. Trans. 1972 3 1455-64 Speich G. R., Demarest V. A. and Miller R. L.: ‘Formation of austenite dufing intercritical annealing of dual-phase steels’. Metall. Trans. A 1981 12A 1419-28 Agren J.: ‘Computer simulations of the austenite/ferrite diffusional transformations in low alloyed steels’, Acta. metal. 1982 30 841-51 Liu Z. and Agren J.: ‘On the transition from local equilibrium to 5. paraequilibrium during the growth of ferrite in Fe-Mn-C Austenite’, Acta. metal. 1989 37(12) 3157-63 Enomoto M., Sonoyama T. and Yada H.: ‘Kinetics of austenite to ferrite 1. 2. 3. 6. 7. transformation in 3 mass % Mn low carbon steels’ Mater. Trans. JIM. 1998 39(1) 189-95 Wheeler A. A., Boettinger W. J. and McFadden G. B.: ‘Phase-field model for isothermal phase transitions in binary alloys’ Phys. Rev. A, 1992 45(10) 7424-39 Ahmad N. A. and Wheeler A. A.: ‘Solute trapping and solute drag in a phse-field model of rapid solidification’ Phys. Rev. E 1998 58(3) 3436- 50 8. Lee J. S. and Suzuki T.: ‘Numerical Simulation of Isothermal Dendritic Growth by Phase-field Model’ ISIJ int. 1999 39(3) 246-52 9. Ode M., Lee J. S., Kim S. G., Kim W. T., Suzuki T: ‘Numerical simulation of the critical velocity for particle pushing/engulfment transition in Fe-C alloys using a phase-field model’ ISIJ. Int. 2000 40 (2) 153-60 10. Kim S. G., Kim W. T., Suzuki T.: ‘Interfacial compositions of solid and liquid in a phase-field model with finite interface thickness for isothermal solidification in binary alloys’ Phys. Rev. E, 1998 58(3) 3316-23 11. Agren J.: ‘Diffusion in phases with weveral components and sublattices’ J. Phys. Chem. Solids 1982 43(5) 421-30 12. Lee B. J.: ‘A Thermodynamic Evaluation of the Fe-Cr-Ni System’ J. of the Korean Inst. Of Met. & Mater. 1993 31(4) 480-489 13. Lee B. J. and Oh K. H.: ‘Numerical Treatment of the Moving Interface in Diffusional Reactions’ Z. Metallkd. 1996 87(3) 195-204 382