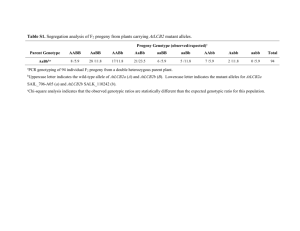
Quantitative trait loci mapping for the data simulated with selection
advertisement

QTLMDSS
Quantitative trait loci mapping for the data simulated with selection:
An appendix for the paper entitled “Selection bias in quantitative trait
loci mapping”
Chaeyoung Lee, Ph.D.
Consider a putative QTL flanked by adjacent makers A and B. The two
markers are linked with recombination rate r. The recombination rate of the QTL with
the marker A is r1, and that with the marker B is r2 (=r-r1). Marker means were
estimated using the following mixed model:
y 1 Zf uf Zmum e
where y was the vector of observations, was the vector of overall mean, 1 was
the summing vector whose elements were all unities (Searle, 1982). u f and um were
the vector of unknown random effects for full-sib litter and marker genotype, Zf and
Zm were the corresponding known design matrices relating the elements in y to those
in u f and um , respectively, and e was the vector of unknown random residuals.
Note that the full-sib litter effect was regarded as random because it reflected both
common environmental effects and possible genetic effects accumulated by various
mating histories. The random variables u f , um , and e were assumed to have Normal
distribution with zero means and variances equal to I f2 , I m2 , and I e2 where f2 ,
m2 , and e2 were full-sib litter, marker genotype, and residual variances. The
MIXED procedure of SAS software package (SAS Institute Inc., 1990) was used to
estimate um . Then, the estimates ( um ) of um were expressed as a linear function of
QTL means:
um L
where was the vector for QTL genotype effects, and L was the matrix of QTL
frequencies conditional on the flanking marker genotypes. A weighted least square
(WLS) analysis was applied to estimation of QTL genotype effects.
(L ' WL) L ' Wu m
where W was the diagonal matrix with diagonal element equal to the number of
corresponding observations in um .
Defining the matrix L was very demanding especially for a complex
pedigree with many loops. A simple way to define L was to divide a complex
pedigree into small full-sib families. These full-sib families were categorized as
progeny from backcross, intercross, or other types of mating.
For example, all the full-
sib litters simulated in the current study were classified into two groups of progeny
produced by backcrossing and by intercrossing. The conditional frequencies of QTL
genotypes given flanking marker genotypes were derived with their corresponding joint
frequencies of QTL and flanking marker genotypes using the mixed progeny. The
expected values for marker genotypes were also presented as following:
Marginal probabilities for nine flanking marker genotypes with mixed progeny
of backcross and F2 were:
2(1-r)n11 +(1-r)2 n 31
,
P(AABB)=
4(n11 +n 31 )
P(AABb)=
rn12 +r(1-r)n 32
,
2(n12 +n 32 )
P(AAbb)= 0.25r 2 ,
P(AaBB)=
rn14 +r(1-r)n 34
,
2(n14 +n 34 )
(1-r)(n15 +n 25 )+ {(1-r)2 +r 2 }n 35
P(AaBb)=
,
2(n15 +n 25 +n 35 )
P(Aabb)=
rn 26 +r(1-r)n 36
,
2(n 26 +n 36 )
P(aaBB)= 0.25r 2 ,
P(aaBb)=
rn 28 +r(1-r)n 38
, and
2(n 28 +n 38 )
2(1-r)n 29 +(1-r) 2 n 39
P(aabb)=
4(n 29 +n 39 )
where r was the recombination rate between the flanking markers; n1j , n 2j , and n 3j
were the sample sizes for marker genotype j corresponding to backcross to a parent with
QQ genotype, backcross to a parent with qq genotype, and intercross, respectively. The
probabilities were calculated under the assumption of no double crossover.
Joint probabilities for marker and QTL genotypes were:
2(1-r)n11 +(1-r) 2 n 31
,
P(AAQQBB)=
4(n11 +n 31 )
P(AAQqBB)= 0 , P(AAqqBB)= 0 ,
P(AAQQBb)=
r2 n12 +r2 (1-r)n 32
,
2(n12 +n 32 )
P(AAQqBb)=
r1n12 +r1 (1-r)n 32
, P(AAqqBb)= 0 ,
2(n12 +n 32 )
P(AAQQbb)= 0.25r22 , P(AAQqbb)= 0.5r1r2 , P(AAqqbb)= 0.25r12 ,
P(AaQQBB)=
r1n14 +r1 (1-r)n 34
,
2(n14 +n 34 )
P(AaQqBB)=
r2 n14 +r2 (1-r)n 34
, P(AaqqBB)= 0 ,
2(n14 +n 34 )
P(AaQQBb)= 0.5r1r2 ,
(1-r)(n15 +n 25 )+ {(1-r) 2 +r12 +r22 }n 35
P(AaQqBb)=
,
2(n15 +n 25 +n 35 )
P(AaqqBb)= 0.5r1r2 ,
P(AaQQbb)= 0 , P(AaQqbb)=
P(Aaqqbb)=
r2 n 26 +r2 (1-r)n 36
,
2(n 26 +n 36 )
r1n 26 +r1 (1-r)n 36
,
2(n 26 +n 36 )
P(aaQQBB)= 0.25r12 , P(aaQqBB)= 0.5r1r2 , P(aaqqBB)= 0.25r22 ,
P(aaQQBb)= 0 , P(aaQqBb)=
P(aaqqBb)=
r2 n 28 +r2 (1-r)n 38
,
2(n 28 +n 38 )
r1n 28 +r1 (1-r)n 38
,
2(n 28 +n 38 )
P(aaQQbb)= 0 , P(aaQqbb)= 0 , and
2(1-r)n 29 +(1-r) 2 n 39
P(aaqqbb)=
4(n 29 +n 39 )
where r1 and r2 were the recombination frequencies between the QTL and markers.
Their corresponding conditional probabilities of QTL genotypes given flanking marker
genotypes were derived as the joint probabilities divided by the marginal probabilities.
P(QQ AABB)= 1 , P( Qq AABB)= 0 , P( qq AABB)= 0 ,
P( QQ AABb)= 2 , P(Qq AABb)= 1 , P(qq AABb)= 0 ,
P(QQ AAbb)= 22 , P( Qq AAbb)= 21 2 , P( qq AAbb)= 12 ,
P( QQ AaBB)= 1 , P(Qq AaBB)= 2 , P( qq AaBB)= 0 ,
(n15 +n 25 +n 35 )r 2 1 2
,
P( QQ AaBb)=
(n15 +n 25 )(1-r)+ n 35{(1-r)2 +r 2 }
(n15 +n 25 )(1-r)+ n 35{(1-r) 2 +r12 +r22 }
P( Qq AaBb)=
,
(n15 +n 25 )(1-r)+ n 35{(1-r) 2 +r 2 }
(n15 +n 25 +n 35 )r 2 1 2
P( qq AaBb)=
,
(n15 +n 25 )(1-r)+ n 35{(1-r) 2 +r 2 }
P(QQ Aabb)= 0 , P(Qq Aabb)= 2 , P( qq Aabb)= 1 ,
P(QQ aaBB)= 12 , P( Qq aaBB)= 21 2 , P( qq aaBB)= 22 ,
P( QQ aaBb)= 0 , P(Qq aaBb)= 1 , P( qq aaBb)= 2 ,
P(QQ aabb)= 0 , P(Qq aabb)= 0 , and P(qq aabb)= 1
where 1
r1
r
and 2 2 1 1 .
r
r
Therefore, the trait expected values for the marker genotypes were:
E(yAABB )=P(QQ AABB)QQ +P(Qq AABB)Qq +P(qq AABB)qq QQ ,
E(y AABb )= 2 QQ 1Qq ,
E(yAAbb )=22 QQ 2 1 2 Qq 12 qq ,
E(y AaBB )=1QQ 2 Qq ,
E(y AaBb )=
(n15 +n 25 +n 35 )( QQ qq )r 2 1 2 [(n15 +n 25 )(1-r)+ n 35{(1-r) 2 +r12 +r22 }]Qq
(n15 +n 25 )(1-r)+ n 35{(1-r) 2 +r 2 }
E(y Aabb )= 2 Qq 1 qq ,
E(yaaBB )=12 QQ 2 1 2 Qq 22 qq ,
E(yaaBb )=1Qq 2 qq , and
E(yaabb )=qq .
,
The method above was applied to data simulated with selection. This method
was devised, first to estimate marker genotype means using a mixed model that
accounted for full-sib litter effects and then to estimate the QTL effects using a
weighted least square analysis based on the conditional frequencies of QTL given
marker genotypes. Extension of this method to QTL analysis with various types of
progeny from multiple generations was straightforward. This approach would be
utilized for QTL mapping with complex pedigree, along with the methods by George et
al. (2000) and Yi and Xu (2001). Furthermore, the principles of the method for QTL
mapping in this study can be incorporated with other QTL analyses with a few
modifications. For example, the current method can be extended to composite interval
mapping (Zeng, 1993) by adding a few other well-chosen markers to the framework
shown in this study.
George AW, Visscher PM, and Haley CS, 2000. Mapping quantitative trait loci in
complex pedigree: a two-step variance component approach. Genetics 156:2081-2092.
Searle SR, 1982. Matrix Algebra Useful for Statistics. Johm Wiley & Sons, New York,
NY.
Yi N and Xu S, 2001. Bayesian mapping of quantitative trait loci under complicated
mating designs. Genetics 157:1759-1771.
Zeng ZB, 1993. Theoretical basis for separation of multiple linked gene effects in
mapping quantitative trait loci. Proc Natl Acad Sci USA 90:10972-10976.