Legend for Supplementary Figures online: (doc 35K)
advertisement
")
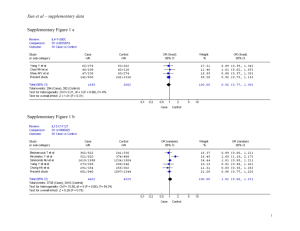
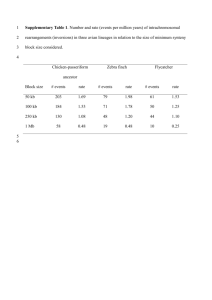
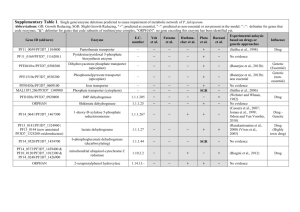
Legend for Supplementary Figures online: Supplementary Figure 1. Simplified life cycle of malaria parasites in human hosts. The parasite’s development within mosquito vectors is not represented. A few (5-20) haploid sporozoites are inoculated by bloodfeeding female Anopheles mosquitoes. After 30-60 min in the bloodstream, these uninucleate extracellular stages penetrate hepatocytes and start intense mitotic activity and nuclear division. The resulting mature, multinucleate liver-stage schizont bursts within 9-16 days and releases thousands of free merozoites, which are released into the bloodstream. Within 1-2 min of release, merozoites invade red blood cells, where they develop over 48 or 72 hours from early to late trophozoites and undergo a further phase of mitotic division, which generates erythrocytic-stage schizonts. When infected red blood cells rupture, each mature schizont releases 8-32 merozoites, each of which invades new erythrocytes. After a few cycles of asexual reproduction, some merozoites develop into sexual stages known as gametocytes. When gametocytes are taken up by feeding Anopheles mosquitoes, they mature into male and female gametes and unite to form a zygote in the midgut of the vector. The zygote is the sole diploid stage of malaria parasites; the only meiosis event during this life cycle occurs within a few hours of zygote formation. This process generates thousands of infective sporozoites, which migrate to the salivary glands of mosquitoes. Supplementary Figure 2. Comparison between nucleotide sequences of P. reichenowi Msp-6 (assembled from data generated by the P. reichenowi genome sequencing project) and the dimorphic lineages (K1 and 3D7) of P. falciparum Msp-6. Note that the alignment of repetitive domains between species is disturbed by the proliferation of different repeat units (see, for example, the GAA GAA AAA ACA motif [underlined] in P. reichenowi Msp-6), indicated by grey shading. Three blocks of sequence (15-, 24- and 129-bp long) present in K1-type and P. reichenowi alleles are absent from 3D7-type alleles (see also Figure 1 in the printed version of the paper). The following notation was used to highlight some features of the aligned sequences: (a) black shading shows nucleotide substitutions shared by P. reichenowi and one of the dimorphic alleles of P. falciparum, (b) boldface italicised letters indicate fixed differences between P. reichenowi and both P. falciparum alleles, (c) boldface non-italicised letters indicate polymorphic sites within P. falciparum lineages (nucleotides 295, 566 and 848), (d) lowercase letters indicate a position (nucleotide 639) where P. falciparum lineages and P. reichenowi all differ from each other, and (e) << and >> indicate repetitive regions. Supplementary Figure 3. Comparison of deduced amino acid sequences for P. reichenowi Msp-3 (AF252287) and four allelic lineages of P. falciparum, represented by the following isolates: (a) CSL2 (Thailand, U08852) and FCC1/HN (China, AF188190), (b) K1 (Thailand, U08851), (c) D10 clone of FC27 (Papua New Guinea, L07944) and 3D7 clone of NF54 (presumably from Africa, L28825). The following domains of MSP-3 (see also Figure 1 in the printed version of the paper) are highlighted: (a) three blocks of Ala-X-X-Ala-X-X-X heptad repeats (where Ala = alanine and X is any residue), grey shading (residues 95-129, 138-166 and 190-218), (b) the glutamic acid (E)-rich domain, black shading (residues 262-314), and (c) the putative leucine zipper-like motif, light grey shading (residues 340-382). The D10 allele is a mosaic of CSL2-type motifs (for example, residues 82-110) and 3D7-type motifs (for example, residues 111-160). Bold italicized letters indicate fixed differences between P. falciparum and P. reichenowi; codons with synonymous nucleotide substitutions are underlined. Supplementary Fig. 4. Critical values for the one-fourth rule. n is the number of individuals in the sample (or, in the case of balancing selection, the number of allelic classes), the mutation rate. For each pair of parameters, we performed 10,000 coalescent simulations. For each simulation, we calculated the ratio of the average genetic divergences 1) between pairs of individuals whose last common ancestor is more recent than the last common ancestor of the sample and 2) between pairs of individuals whose last common ancestor is the last common ancestor of the sample. which 95% of simulations yielded a higher ratio is given. The value for