nature01279
advertisement

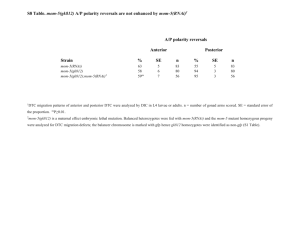
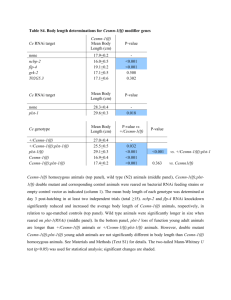
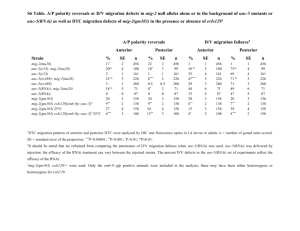
Supplementary Methods Strains Strains were maintained as described by Brenner27 at 25°C, except for when noted. OP50 E. coli was used for feeding. The wild type reference strain was N2 Bristol. The mutant strains used were: tub-1(nr2004)II15, and tph-1(mg280)II14. The following strains were grown at 15°C until the L2 stage then transferred to 25°C: daf-2(e1370)III, daf2(e1370)III;daf-3(mgDf90)X, daf-16 (mgDf47)I;daf-2(e1370)III, daf-2(e1370)III;daf-18 (mg198)IV, daf-16 (mgDf47)I, daf-18 (mg198)IV, daf-3 (mgDf90)X, daf-7(e1372)III, daf7(m62)III;daf-3(mgDf90)X. Many of these strains were provided by the Caenorhabditis Genetic Center. Fat staining a) Nile Red: Nile Red powder (N-1142 Molecular Probes) was dissolved in acetone at 500 g/ml, diluted in 1X phosphate buffered saline (PBS) and added on top of nematode growth media (NGM) plates already seeded with OP50 or RNAi bacteria, to a final concentration of 0.05 g/ml. Worms were placed on these plates as eggs or starved L1s. Their staining phenotypes were assessed prior to starvation at the L4 and the young adult stages. Fat content was monitored by fluorescence microscopy. b) BODIPY-labelled fatty acids: C1-BODIPY 500/510 C12 (4,4-difluoro-5-methyl-4-bora3a,4a-diaza-s-indacene-3-dodecanoic acid), and C8-BODIPY 500/510 C5 ( 4,4-difluoro-5octoyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoic acid) from Molecular Probes (D-3823 and D-3825) were dissolved in DMSO at 1 mg/ml. For staining animals, the stocks were diluted 1:10,000 in 1XPBS/20 M bovine serum albumin and added on top of plates already seeded with OP50. Worms were placed on plates as eggs or L1s. Incorporation of BODIPY-labelled fatty acids was assessed in L4 or non-starved young adults by fluorescence microscopy. When co-staining, Nile Red and BODIPY-labelled fats were added to the same NGM/E. coli plate at the indicated final concentrations. Fluorescence microscopy, image acquisition and intensity quantitation Nile Red and BODIPY -label fluorescence were visualized using a Zeiss SV11 M2-bio microscope equipped with rhodamine (emission 560-590 nm) and FITC/GFP (emission 500-515 nm) filters, respectively. Images were captured using a digital CCD camera (Hamamatsu C4742-95-12ER) attached to a Zeiss Axioplan II equipped with rhodamine and FITC/GFP filters. All Nile Red images were acquired using identical settings and exposure times. To quantitate pixel intensities and numbers, equal planes and regions of the worm body were selected. Using the Openlab software (Improvision Inc. Lexington, MA) total fluorescence for all Nile Red staining droplets within a region was calculated as the product of area multiplied by mean fluorescence. Lipid Analysis L1 synchronized worms were grown on four 6-cm plates at 15°C past the L2 stage then shifted to 25°C. Non-starved, young adult worms were washed off the plates with water, placed into 15 ml polypropylene screw-capped centrifuge tubes, spun at 1,000 x g for 1 min, and washed 5X with water. At least 100 mg of worm pellet was required. After the final wash, as much water as possible was removed, the sample was split into two equal portions, and each flash frozen in dry ice/ethanol. A portion of each sample was analysed for total soluble protein28 and the remaining pellet was used for lipid extraction. 5 ml of ice-cold chloroform/methanol/formic acid (10:10:1) was added to the worm pellet, vortexed for 2 min. and stored at –20°C overnight. 2.2 ml of H3PO4, 1 M KCL was added and lipids were recovered in the chloroform phase, dried under N2 and dissolved in 0.2 ml chloroform. After the chloroform/methanol extraction the remaining worm pellet was treated with 2.5% sulfuric acid in methanol at 80° to convert any remaining fatty acids to fatty acid methyl esters. The hexane extract of this reaction was analysed by gas chromatography and no fatty acids were detected. Therefore, the chloroform/methanol extraction is >98% efficient in extracting lipids from the nematodes. Lipids were spotted onto thin layer chromatography plates and developed in hexane-diethylether-acetic acid (80:20:20). Lipids were located by brief staining with I2. Silica gel from the triacylglyceride and phospholipid spots was transferred to screw capped tubes. 5 g of 15:0 was added as an internal standard and fatty acid methyl esters were prepared and analysed by gas chromatography as previously described29. Genome wide RNAi analysis Bacteria containing each RNAi clone were cultured in 300 l Luria Broth media containing 50 g/ml ampicillin for 6-14 hrs. 40 l of each culture was spotted in a single well of a 24-well plate containing NGM agar, 6 mM IPTG and 25 g/ml carbenicillin. After overnight incubation, Nile Red was added on top of each well to a final concentration of 0.05 g/ml. 10 synchronized L1 worms were placed per well and incubated at 20°C. Growth and Nile Red staining were assessed after 48, 72, and 96 hrs. by light phase and UV microscopy. For each batch of RNAi clones tested, L4440 (vector alone) and OP50 control wells were included. A phenotype was assigned to a well only if a majority of the animals displayed the phenotype. All phenotypes were confirmed by at least two additional rounds of testing on the selected clones. At the time of scoring the Nile Red phenotype, the identities of the target RNAi clones were unknown. Pathway analysis of the gene inactivations that cause reduced fat For each RNAi clone, aliquots from a single RNAi culture were simultaneously tested on wild type and all three increased fat mutant animals. Simultaneous testing of the cultures on multiple strains insured that each RNAi clone retained its fat reducing capacity on the wild type animals. All observations were confirmed by at least one additional round of testing.