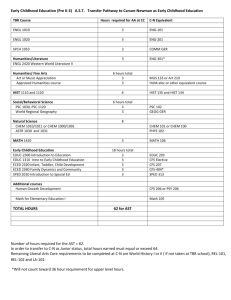
Woodchuck whole transcriptome sequencing reveals mechanisms
advertisement

Serum anti-WHs (—) Serum WHsAg (—) Serum WHV DNA (—) Innate immunity Adaptive immunity Lifetime HCC risk Chronic = 60-75% 100% Resolved = 25-40% 15-20% 0 WHV7P1 (WHV7-11) 4 8 12 16 t (post inoculation)/wk 20 24 88 Median time of sampling Supporting Figure 1. Neonatal woodchuck model of experimental WHV infection. Schematic serological profiles for WHV DNA, WHV surface antigen (WHsAg), and virus-neutralizing antibody to WHsAg (anti-WHs) in chronic and resolved WHV infections in the neonatal woodchuck model. Woodchucks born to WHV-negative dams during early spring were subcutaneously infected at 3 days of age with 5 x 106 infectious doses of the same WHV7P1 inoculum containing WHV strain WHV7-11. The proportions of chronic and resolved WHV infection outcomes usually range between 6075% and 25-40%, respectively. Chronic WHV infection is characterized by high blood levels of virus and viral antigens without seroconversion to antiWHs antibody. Resolved WHV infection is characterized by a substantial clearance of virus and viral antigens from the blood and seroconversion to antiWHs antibody. The lifetime risk for the development of HCC in established chronic and resolved WHV infections is 100% and 15-20%, respectively. HCC in uninfected, WHV-negative woodchucks is not observed. The time point for tissue (liver, spleen and kidney) sampling, at a median age of 88 weeks = 22 months (range 15-28 months), is indicated. Approximate time intervals for the development of innate and adaptive immunity are shown. Liver Carrier Liver Resolved Liver Negative PBMC Carrier PBMC Negative All reads (5,741,102) combined (100%) Map to WHV7-11 genome (85% id, 80% cov) Unmapped (99.6%) Map to Human Refseq (85% id, 80% cov) Mapped (22.7%) Chimeric, Repeat, or Unmapped (76.1%) 23,618 Contigs Map to Human Genome (85% id, 80% cov) (avg: 749, N50: 1,005) Unmapped (53.7%) Mapped (3.3%) Chimeric (12.6%) Contigs De-novo assembly 10,037 Isotigs (avg:1,022; N50:1,203) De-novo assembly 25,615 Singletons Merge to contigs (48,933) 61,039 Contigs, Singletons and WHV sequences submitted for chip design 33,554 Isotigs (avg:1,039; N50:1,340) 571,556 Singletons Singletons longer than 540 bp excluding repeats (12,101) 5 WHV transcript regions Supporting Figure 2. Workflow of the woodchuck transcriptome assembly. All reads from the five sequenced samples (i.e., liver carrier, liver resolved, liver negative, PBMC carrier, and PBMC negative) were combined and mapped against the WHV7-11 genome (NC_004107) to filter out WHV reads (0.4%). The reads were then mapped to the human Refseq database and to the human reference genome (hg19) to obtain transcript contigs conserved in human. The reads chimerically mapped to the human Refseq or reference genome were likely novel transcript forms and were therefore assembled together using Newbler (454 Life Sciences, Branford, CT), while other unmapped reads were assembled separately. Finally, all assembled contigs were merged into 48,933 contigs using Phrap (Phil Green, Genome Sciences, University of Washington). These contigs, together with 12,101 non-repeat singletons longer than 540 bp, and 5 WHV transcript regions (WHV polymerase, mature and signal peptide regions of WHsAg, WHx and WHcAg), were used for microarray design. ID: identified, cov: coverage, avg: average. PC #2 (12.7%) U R C-N C-H PC #3 (8.4%) Principal Component (PC) #1 (18.4%) Supporting Figure 3. Persistent infection and HCC induce extensive changes in the liver transcriptome. Principal component analysis of normalized liver expression data for U (n=10), R (n=11), and paired C-N and C-H (n=13) samples. Technical repeats (n=3) for each sample are linked. Leukocyte extravasation p=0.0001 Cell cycle control of chromosomal replication p<0.0001 Antigen presentation p<0.0001 Bile acid and steroid hormone metabolism p<0.0001 C-N -2 0 R U 2 Supporting Figure 4. Persistent WHV infection markedly alters the liver transcriptome. Unsupervised hierarchical clustering of the top differentially expressed intrahepatic genes for C-N, R and U samples. All genes had an absolute fold-change >1.5 with a Benjamini-Hochberg corrected FDR<0.05. Heatmap columns represent samples from individual animals, and rows represent different genes. Red and blue coloring of cells represents high and low expression levels, respectively, as indicated by the scale bars for normalized values. Functional annotation of gene clusters was performed by Ingenuity Pathway Analysis, with the top canonical pathway for each cluster being displayed. Pathway enrichment was calculated with the Fisher’s exact test with multiple testing correction by the Benjamini and Hochberg method. Gene clusters that were not significantly enriched for a pathway were not functionally annotated. p450 metabolism p=0.0001 Antigen presentation p<0.0001 GPCR signaling p=0.0468 TRAIL signaling FDR=0.0391* MYC pathway FDR=0.0447* C-N -2 0 C-H 2 4 Supporting Figure 5. WHV-induced HCC extensively modulates intrahepatic gene expression. Unsupervised hierarchical clustering of the top differentially expressed intrahepatic genes for paired C-H and C-N samples. All genes had an absolute fold-change >1.5 with a Benjamini-Hochberg corrected FDR<0.05. Functional annotation of gene clusters was performed by Ingenuity Pathway Analysis, with the top canonical pathway for each cluster being displayed. Pathway enrichment was calculated with the Fisher’s exact test with multiple testing correction by the Benjamini and Hochberg method. In cases where Ingenuity did not identify significant pathways, the annotation was performed by GSEA (indicated by asterisks). Gene clusters that were not significantly enriched for a pathway were not functionally annotated. Supporting Figure 6. MYC intrahepatic transcriptional signature in WHV-induced HCC. Network (“protein synthesis”) created by Ingenuity Pathway Analysis. Color intensity indicates magnitude of differential expression in C-H relative to C-N. All genes had an absolute fold-change > 1.5 with a BenjaminiHochberg corrected FDR<0.05. a p<0.001 p<0.001 20 15 10 p>0.05 5 p<0.001 6 LYZ expression MPO expression 25 5 p=0.001 4 3 p>0.05 2 1 0 0 C-N R U C-N R (n=11) (n=11) (n=10) (n=11) (n=11) U (n=10) b 8 p<0.001 IGFBP3 expression IGF2 expression 7 1.5 6 5 4 3 2 1 0 p<0.001 1.0 0.5 0.0 C-N C-H C-N C-H (n=11) (n=11) (n=11) (n=11) Supporting Figure 7. qRT-PCR verification of select intrahepatic genes. qRT-PCR data expressed as foldchange relative to (a) the mean of U, and (b) the mean of C-N. The bar height indicates the mean of each group, and the errors bars represent the standard error of the mean.





![Major Change to a Course or Pathway [DOCX 31.06KB]](http://s3.studylib.net/store/data/006879957_1-7d46b1f6b93d0bf5c854352080131369-300x300.png)