nph12434-sup-0002-MethodsS1
advertisement

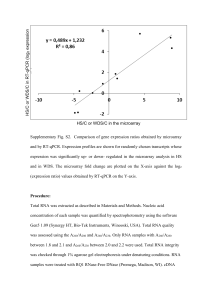
Supporting Information Methods S1 Supplemental Materials and Methods MiRNA microarray MiRNA samples isolated from the young leaves and roots of control or stress-treated S. bicolor plants were used for miRNA microarray hybridization. All samples were assessed for the presence of enriched miRNA using an Experion Bioanalyzer (Bio-Rad, Hercules, CA, USA). The plant GeneChip® miRNA 3.0 Array (Affymetrix, Santa Clara, CA, USA) was used in this study, and contained 3741 well-characterized plant miRNAs from Sorghum bicolor, Arabidopsis thaliana, Zea mays, Oryza sativa, Glycine max, and other species as noted in the miRBase v17 coverage and various controls (http://www.mirbase.org/). The miRNAs were labeled using the FlashTag Biotin RNA labeling kit (Genisphere, Hatfield, PA, USA) according to the provided protocol and then hybridized to Affymetrix GeneChip miRNA 3.0 microarrays. Three replicates each for leaf and for root, using a total of 12 arrays, were applied to compare the control and Zn deficiency conditions. The Affymetrix GeneChip 3000 TG microarray hybridization was performed with the assistance of Biochip Corporation (Tianjin, China). Array hybridization, washing, and staining were performed per the manufacturer's instructions, and arrays were scanned with a GeneChip Scanner 7G 4C. Only plant miRNAs were considered in our analysis. The Affymetrix miRNA QC Tool software (Affymetrix, Inc.) was used for data summarization, normalization, and quality control. The miRNAs with P < 0.05 and fold change > 2.0 compared to the control were defined as differentially expressed. 5’ RACE of miRNA cleavage For mapping the internal cleavage site in the predicted target genes of the miRNAs, RLM-RACE was performed using the GeneRacer Kit (Invitrogen, USA). A modified procedure for RLM-RACE was performed following the GeneRacer Kit instructions as described previously (Song et al. 2009). Total RNA was extracted from the leaves and roots of S. bicolor seedlings using TRIzol reagent (Invitrogen, USA). The RNA quality and integrity were checked before the cDNA was synthesized using a Bio-Rad Experion RNA StdSens analysis kit (Bio-Rad, Hercules, CA, USA). Poly(A)+ mRNA was purified from the pooled RNA from all tissues using a PolyA kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The GeneRacer RNA Oligo adapter was directly ligated to mRNA without calf intestinal phosphatase and tobacco acid pyrophosphatase treatment. The GeneRacer Oligo dT primer was then used to synthesize first-strand cDNA. This cDNA was subjected to an amplification procedure with the GeneRacer 5’ Primer and the GeneRacer 3’ Primer to generate a pool of non-gene-specific 5’RACE products. Gene-specific 5’RACE reactions were performed with the GeneRacer 5’Nested Primer and gene-specific primers (Table S1). In each case, a unique gene-specific DNA fragment was amplified. After amplification, 5’RACE products were gel-purified and cloned into the pMD 19-T vector (TaKaRa, Otsu, Japan), and at least 10 independent clones were randomly chosen and sequenced. RT-qPCR analysis of gene expression The RNA was extracted from the young leaves, mature leaves, and roots of control or stress-treated S. bicolor plants using TRIzol (Gibco/BRL, Life Technologies). The RNA quality and integrity were checked before the cDNA was synthesized using the Bio-Rad Experion RNA StdSens analysis kit (Bio-Rad, Hercules, CA, USA). For the quantitative RT-PCR (RT-qPCR), we first accurately quantified the RNA concentration using spectrophotometry. The cDNA was synthesized from the DNase-treated total RNA (1 µg) using a Reverse Transcription System Kit (Promega) and oligo (dT) primers. The cDNA produced was diluted 1:15, and 3 μl was used in the RT-qPCR reaction in a 7500 Real-Time System (Applied Biosystems) using Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen, USA). Based on semi-quantitative RT-PCR analysis and the Genevestigators’ RefGenes tool V3 (Hruz et al., 2008), we selected ACT (Sb01g003250), GADPH (Sb04g025120), UBI3 (Sb01g030340), UBQ (Sb04g031060), TuA2 (Sb01g009570), and EF-1alpha (Sb02g036420) as the candidate reference genes for RT-qPCR normalization. The geNorm software (http://medgen.ugent.be/~jvdesomp/genorm/) was used to choose the most suitable reference genes. The qRT-PCR for each gene was done on three biological replicates with duplicates for each biological replicate. The relative transcript level was determined for each sample and averaged over the six replicates. Specific primers for each gene are listed in Table S1. All primer pairs produced only one peak in DNA melting curves indicating high specificity of the primers. To verify the gene expression patterns of target genes, the RT-qPCR analysis was performed for selected genes. We selected ACT, GADPH, UBI3, UBQ, TuA2, and EF-1alpha as the candidate reference genes for RT-qPCR normalization. The expression of ACT was the most stable in Sorghum, and the pairwise variation V value of ACT and TuA2 was 0.11 (Fig. S1), which suggested that the two genes were suitable reference genes for RT-qPCR normalization.