Aplastic anemia – curs
advertisement

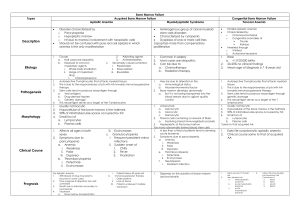
APLASTIC ANEMIA I. Background: Aplastic anemia is a bone marrow failure syndrome characterized by peripheral pancytopenia and marrow hypoplasia. Paul Ehrlich, introduced the concept of aplastic anemia in 1888 when he studied the case of a pregnant woman who died of bone marrow failure. However, it was not until 1904 that this disorder was termed aplastic anemia by Chauffard. Bone marrow failure is a term with a larger meaning, referring to disorders of the hematopoietic stem cell which involves either one cell line or all of the myeloid cell lines (ie, erythroid for red cells, myeloid for white blood cells, megakaryocytic for platelets). The lymphoid cells, which primarily are involved in lymphoproliferative disorders, usually are spared. II. Pathophysiology: The theoretical basis for marrow failure includes primary defects in or damage to the stem cell or the marrow microenvironment. The distinction between acquired and inherited disease may present a clinical challenge, but more than 80% of cases are acquired. In acquired aplastic anemia, clinical and laboratory observations suggest that this is an autoimmune disease. Bone marrow failure can be inherited or acquired. It can involve just 1 cell line or all 3 cell lines. The pathophysiology of these defects includes the following mechanisms of action: 1. decrease in or damage to the hematopoietic stem cells and their microenvironment, resulting in hypoplastic or aplastic bone marrow; 2. maturation defects - vitamin B-12 or folate deficiency; 3. differentiation defects - myelodysplasia. Generally, hematopoietic stem cells are damaged by a congenital defect or exposure to a noxious substance or factor. Pathophysiologic mechanisms are a. an acquired stem cell injury from viruses, toxins, and chemicals that leads to a quantitative or qualitative abnormality; b. abnormal humoral or cellular control of hematopoiesis; c. an abnormal or hostile marrow microenvironment; d. immunologic suppression of hematopoiesis (ie, mediated by antibodies, T cells, or lymphokines); e. mutations in genes, causing inherited bone marrow failure syndromes. The principal mechanism is implying the hematopoietic stem cell, because little evidence points to a defective microenvironment as a cause of aplastic anemia. In patients with severe aplastic anemia, the stromal cell function is normal, including growth factor production. The role of an immune dysfunction was suggested in 1970. Subsequently, numerous studies have shown that, in approximately 70% of patients with acquired aplastic anemia, immunosuppressive therapy improves marrow function. Suppression of hematopoiesis likely is mediated by an expanded population of the following cytotoxic T lymphocytes (CTLs): CD8 and HLA-DR+, which are detectable in both the blood and bone marrow of patients with aplastic anemia. These cells produce inhibitory cytokines, such as gamma interferon and tumor necrosis factor, which are capable of suppressing progenitor cell growth. III. Epidemiology The annual incidence of aplastic anemia in Europe is similar to that indicated by US data, at 2 cases per million population. Aplastic anemia is thought to be more common in Asia than in the West. The incidence determined was at 4 to 6 cases per million population Thailand and as high 14 cases per million population in Japan. The male-to-female ratio in acquired aplastic anemia is approximately 1:1, although some data suggest that a male preponderance may exist in the Far East. Aplastic anemia occurs in all age groups. A small peak in incidence in childhood is observed due to the inclusion of inherited marrow failure syndromes. The peak incidence of aplastic anemia is observed in people aged 20-25 years, and a subsequent peak in incidence is observed in people older than 60 years. IV. Causes: Congenital/inherited (20%) o Patients usually have dysmorphic features or physical stigmata. Occasionally, marrow failure may be the initial presenting feature. o Fanconi anemia o Dyskeratosis congenita o Shwachman-Diamond syndrome o Familial aplastic anemia Acquired (80%) o Idiopathic – 75% o Infectious causes such as hepatitis viruses, Ebstein-Barr virus (EBV), HIV, parvovirus, and mycobacterial infections o Toxic exposure to radiation and chemicals such as benzene o Drugs, such as chloramphenicol, phenylbutazone, and gold, may cause aplasia of the marrow. o Paroxysmal Noctural Hemoglobinuria is caused by an acquired genetic defect limited to the stem cell compartment affecting the PIGA gene. The PIGA gene mutations render cells of hematopoietic origin sensitive to increased complement lysis. Approximately 20% of patients with aplastic anemia have evidence of PNH at presentation as detected by flow cytometry. o Transfusional graft versus host disease o Orthotopic liver transplantation for fulminant hepatitis o Pregnancy o Eosinophilic fascitis V. History The clinical presentation of aplastic anemia includes symptoms related to the decrease in bone marrow production of hematopoietic cells. The onset is insidious, with the initial symptom relating to anemia or bleeding, but fever or infections often are noted at presentation. Anemia may manifest as pallor, headache, palpitations, dyspnea, fatigue, or foot swelling. Thrombocytopenia may present as mucosal and gingival bleeding or petechial rashes. Neutropenia may manifest as overt infections, recurrent infections, or mouth and pharyngeal ulcerations. Even though the search for an etiologic agent often is unproductive, an appropriately detailed work history, with emphasis on solvent and radiation exposure, and a family, environmental, travel, and infectious disease history should be obtained. - Significantly, in the absence of obvious phenotypic features, the presentation of an inherited marrow failure syndrome is subtle, and it may be suggested first by a thorough family history. - With regard to environmental agents, remember that much variation is observed in the time course of the occurrence of aplastic anemia and the exposure to the offending agent, and only rarely is an environmental etiology identified. VI. Physical exam The manifestations of bone marrow failure relate to the clinical effects of low blood counts. Patients with severe anemia may present with pallor and/or signs of congestive heart failure, such as shortness of breath. Bruising (eg, ecchymoses, petechiae) on the skin, gum bleeding, or nosebleeds frequently are associated with thrombocytopenia. Fever, cellulitis, pneumonia, or sepsis can be complications of severe neutropenia. A subset of patients with aplastic anemia present with jaundice and evidence of clinical hepatitis. Findings of adenopathy or organomegaly should suggest an alternative diagnosis. In any case of aplastic anemia, look for physical stigmata of inherited marrow failure syndromes such as skin pigmentation, short stature, microcephaly, hypogonadism, mental retardation, and skeletal anomalies. A careful examination of the oral pharynx, hands, and nailbeds should be preformed, looking for clues of dyskeratosis congenita. VII. Lab Studies: 1. CBC count and peripheral smear Anemia is common, and red cells appear morphologically normal. The reticulocyte count usually is less than 1%, indicating a lack of red cell production. Occasionally, the mean cell volume is elevated, with macrocytosis. Platelet counts are lower than normal, with a paucity of platelets in the blood smear. Platelet size is normal, but a low platelet count may cause greater heterogeneity in size. Agranulocytosis (ie, decrease in all granular white blood cells, including neutrophils, eosinophils, and basophils) and a decrease in monocytes are observed. A relative lymphocytosis occurs (ie, increased percentage) without an increase in numbers. The degree of cytopenia is useful in assessing the severity of aplastic anemia. The corrected reticulocyte count is uniformly low in aplastic anemia. The peripheral blood smear often is helpful in resolving aplasia from infiltrative and dysplastic causes. o The presence of teardrop poikilocytes and leucoerythroblastic changes is suggestive of an infiltrative process. o Patients with MDS often show certain characteristic abnormalities, including dyserythropoietic red blood cells and neutrophils with hypogranulation, hypolobulation, or apoptotic nuclei reaching to the edges of the cytoplasm. Monocytes are similarly hypogranular, and their nuclei may contain nucleoli. o A leukemic process may show evidence of blasts on the peripheral smear. 2. Peripheral blood Hemoglobin electrophoresis and blood group testing may show elevated fetal hemoglobin and red cell I antigen suggesting stress erythropoiesis, which is observed in both aplastic anemia and MDS and often is proportional to the macrocytosis. Biochemical profile, including evaluation of transaminases, bilirubin, lactic dehydrogenase, Coombs test, and kidney function, is useful in evaluating etiology and differential diagnosis. Serologic testing for hepatitis and other viral entities such as EBV, CMV, and HIV Autoimmune disease evaluation for evidence of collagen-vascular disease The Ham test or sucrose hemolysis test frequently is performed for excluding PNH. Histocompatibility testing should be conducted early to establish potential related donors, especially in younger patients. 3. Bone marrow aspiration and biopsy A bone marrow biopsy is performed in addition to the aspiration so that the cellularity may be assessed both qualitatively and quantitatively. In aplastic anemia, these specimens are hypocellular. Aspirations alone may appear hypocellular because of technical reasons (eg, dilution with peripheral blood), or they may appear hypercellular because of areas of focal residual hematopoiesis. A core biopsy provides a better idea of cellularity; the specimen is considered hypocellular if it is less than 30% cellular in individuals younger than 60 years or less than 20% in those older than 60 years. A proportion of marrow lymphocytes greater than 70% has been correlated with poor prognosis in aplastic anemia. Both are necessary to rull out a myelodisplasic syndrome (MDS), a leukemia or a metastatic cancers 4. Cytogenetic on bone marrow – is usefull to identify a MDS with trisomies of 8 and 21 and deletions of 5, 7, and 20 being most common. 5. Bone marrow culture - is useful in diagnosing mycobacterial and viral infections. 6. Imaging Studies Radiological studies generally are not needed to establish a diagnosis of aplastic anemia. A skeletal survey is especially useful for the inherited marrow failure syndromes, many of which show skeletal abnormalities. VIII. Differential Acute Myelogenous Leukemia Anemia Aplastic Anemia Hairy Cell Leukemia Paroxysmal Nocturnal Hemoglobinuria Immune pancytopenias in connective tissue disorders (eg, systemic lupus erythematosus, refractory anemia) All pancytopenias o Failure of production of blood cells a) bone marrow infiltration - acute leukemias - hairy cell leukemia - multiple myeloma - lymphoma - myelofibrosis - metastatic carcinoma b) aplastic anemia o Ineffective hematopoesis - myelodysplastic syndrome - vit.B12 and folate deficiency o Increased destruction of blood cells - hipersplenism - autoimmune disorders - paroxysmal nocturnal hemoglobinuria o Myelosuppression after irradiation or antiproliferative drugs IX. Staging Based on International Aplastic Anemia Study Group (Camitta, 1983) Blood o Neutrophils - Less than 0.5 X 109/L o Platelets - Less than 20 X 109/L o Reticulocytes - Less than 1% corrected (percentage of actual hematocrit [Hct] to normal Hct) Marrow o Severe hypocellularity o Moderate hypocellularity with hematopoietic cells representing less than 30% of residual cells Severe aplasia is defined by any 2 or 3 peripheral blood criteria and either marrow criterion. A further subclassification followed the recognition that individuals with neutrophils lower than 0.2 X 109/L constituted a very severe aplastic anemia (VSAA) group. This group is less likely to respond to immunosuppressive therapy. X. Treatment: X.1. Diet The diet for the patient with aplastic anemia who is neutropenic or on immunosuppressive therapy should be tailored carefully to exclude raw meats, dairy products, or fruits and vegetables that are likely to be colonized with bacteria, fungus, or molds. Further, a diet limiting salt is recommended during therapy with steroids or CSA. X.2. Activity: The patient should avoid the following: Any activity that increases the risk of trauma during periods of thrombocytopenia Risk of community-acquired infections during periods of neutropenia X.3. Medical treatment The goals of pharmacotherapy are to reduce morbidity, prevent complications, and eradicate malignancy. a. Supportive care : Supportive care is essential for patient survival. In patients with bone marrow failure, the resulting cytopenia can lead to life-threatening symptoms. If clinically indicated, initiate a blood transfusion using specific cells, such as packed red cells for anemia and platelets for thrombocytopenia. Supportive care gives only temporary relief of symptoms and does not treat the primary disease. Transfusions: Patients with aplastic anemia require transfusion support until the diagnosis is established and specific therapy can be instituted. For patients in whom marrow transplantation may be attempted, transfusions should be used judiciously because minimally transfused subjects have achieved superior therapeutic outcomes. Avoiding transfusions from family members is important because of possible sensitization against non-HLA tissue antigens of the donors. In considering blood bank support, attempt to minimize the risk of CMV infection. If possible, the blood products should undergo leuko-poor reduction to prevent alloimmunization and should be irradiated for prevention of third party graft versus host disease in BMT candidates. Judicious use of blood products is essential, and transfusion in conditions that are not life threatening should be performed in consultation with a physician experienced in the management of aplastic anemia. Anemia - can cause fatigue and can impair the patient's ability to function in daily activities. Transfuse packed red cells to maintain hemoglobin levels of 7-10 g/dL. Patients with coronary artery disease may need to be maintained at 10-12 g/dL if they are symptomatic at lower levels of hemoglobin. The benefits of this therapy are limited to 1 month because the life span of transfused red blood cells is limited to the average life span of collected cells. Also, each unit of transfused packed red cells introduces unwanted iron, which over time, accumulates in the patient. Although minimal, the risk of infection still is present (eg, HIV, hepatitis C). Bleeding/hemorrhage - resulting from thrombocytopenia is a major problem and may be life threatening if it occurs intracranially. Platelet transfusions are effective for stopping acute bleeding. Unfortunately, the platelet life span is short; the effects may last 2-4 days. This treatment temporarily stops bleeding, but it is not a practical maintenance therapy. Development of alloantibodies can make the patient refractory to platelet transfusions. Mucosal bleeding from the nose, gums, or teeth may be easily controlled by oral aminocaproic acid. Infections - Infections are a major cause of mortality in these patients. The risk factors include prolonged neutropenia and the indwelling catheters used for specific therapy. Fungal infections, especially Aspergillus, pose a major risk. After blood is drawn and other cultures are taken, broad-spectrum antibiotics should be started empirically in the presence of febrile neutropenia. Coverage for the most common gram-positive and gram-negative organisms should be considered. With the new broad-spectrum antibiotics, a single antibiotic generally is sufficient. The choice can be altered later, depending on the results of sensitivity tests from positive cultures. Specially consider including antipseudomonal coverage at initiation of treatment for patients with febrile neutropenia. The addition of antifungal agents should be considered in the presence of persistent fever despite adequate antibacterial coverage. Liposomal amphotericin B is indicated if renal dysfunction is present because of toxicity resulting from the drug in another form. Cytokine support with granulocyte colony stimulating factor (G-CSF) and granulocytemacrophage colony stimulating factor (GM-CSF) may be considered in refractory infections, although weighted against cost and efficacy. b. Bone marrow transplantation – is the treatment of choice for young patients (under 50-55 years) with a good potential for cure. The procedure is limited by the existence of a potential donor. HLA-matched sibling donor BMT is the treatment of choice for a young patient with severe aplastic anemia (controversial but generally accepted for age <60 y). The conditioning regimen most often used includes a combination of antithymocyte globulin (ATG), cyclosporine (CSA), and cyclophosphamide. The addition of ATG and CSA to the conditioning regimen has resulted in reduction of graft rejection. When radiation was used as part of the conditioning regimen, a higher incidence of chronic graft versus host disease and malignant disease was found. With current BMT regimens, most patients with severe aplastic anemia have a 60-70% long-term survival rate. Unrelated donor BMT probably only can be justified if the donor is a full match and the patient has failed immunosuppressive therapy or as part of a clinical trial. Using matched unrelated donors still is experimental (11-20% survival rates). Early referral to a transplant center at diagnosis is recommended in all younger patients, even if they lack a suitable related donor. c. Immunosuppressive therapy Immune suppression as a treatment for aplastic anemia is especially useful if a matched sibling donor for BMT is not available or if the patient is older than 60 years. The various options include combination therapy, including ATG, CSA, and methylprednisolone, with or without cytokine support. o Antithymocyte globulin, equine (Atgam) - inhibits cell-mediated immune response either by altering T cell function or eliminating antigen-reactive cells. The dose recommended : 10-20 mg/kg/day for 8-14 days. Attention to serum sickness. o Antithymocyte globulin, rabbit (Thymoglobulin) - may modify T-cell function and possibly eliminate antigen-reactive T lymphocytes in peripheral blood. The dose indicated : 0,75 mg/kg/day for 8 days. o Cyclosporine (Sandimmune, Neoral) - cyclic polypeptide that suppresses some humoral immunity and, to a greater extent, cell-mediated immune reactions for a variety of organs. For children and adults, base dosing on ideal body weight. Needs frequent drug level monitoring. To convert to PO dose, use a correction factor of 1:4 (IV:PO). The dose recommended : 1.5-2 mg/kg IV q12h, adjust to trough level of 500-800 ng/mL in initial 1 mo or so, then later adjust to trough level of 200 ng/mL. Evaluate renal and liver functions often by measuring BUN, serum creatinine, serum bilirubin, and liver enzymes; may increase risk of infection and lymphoma; reserve IV use only for those who cannot take PO o Methylprednisolone (Medrol, Solu-Medrol) - it is used in combination with antithymocyte globulin to decrease adverse effects (eg, allergic reactions, serum sickness). Further, it is an additional immunosuppressive agent. Higher doses or longer duration may be needed if serum sickness occurs with ATG. The dose recommende : 5 mg/kg IV on days 1-8; then tapered using PO 1 mg/kg on days 9-14; further tapering over days 15-29. Stop after 1 mo except in evidence of serum sickness. o Cyclophosphamide (Cytoxan) --Chemically related to nitrogen mustards. As an alkylating agent, the mechanism of action of the active metabolites may involve cross-linking of DNA, which may interfere with growth of normal and neoplastic cells. The dose recommended : 45 mg/kg/d IV for 4 d. Monitor carefully; only used on an investigational basis. The response, unlike other autoimmune diseases, is slow and usually takes at least 4-12 weeks to show early improvement, and the patient continues to improve only slowly thereafter. Most patients improve and become transfusion-independent, but many still have evidence of a hypoproliferative bone marrow. Even though the initial response rate is good, relapses are common and, often, continued immune suppression is needed. The best results seem to be obtained with triple association : globulines + cyclosporine + methylprednisolone. Preliminary data suggested that high-dose cyclophosphamide may result in durable remissions in some patients with aplastic anemia, but a recent report suggests that rates of fungal infections may practically limit this approach, and its use at present should be limited to clinical trials. d. Other treatments : Androgens : These agents push the resting hematopoietic stem cells into cycle, making them more responsive to differentiation by hematopoietic growth factors. They also stimulate endogenous secretion of erythropoietin. Androgens were used in the past, but most are masculinizing and poorly tolerated by females and children. Danazol is a nonmasculinizing androgen that may be useful. The response rate is limited to approximately 45%, and results may require 6-10 months of therapy. Hematopoietic growth factors - such as granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF), may be useful in patients with neutropenia who have infections, without requiring a WBC transfusion. X.4. Further Outpatient Care: Frequent outpatient follow-up is needed for monitoring blood counts and adverse effects of various drugs. Packed red blood cell and platelet transfusions also are administered on an outpatient basis. XI. Complications: Infections Bleeding Over time, the transfusion of packed red cells increases the total iron load to the patient. Measure iron stores in the form of ferritin. Increased levels of iron are toxic to various organs, including the heart. Iron toxicity can cause arrhythmia by blocking the bundle of His, diabetes by damaging the islets of Langerhans in the pancreas, liver cirrhosis, and bronze color in fair-skinned individuals. Administering a chelating agent is an effective method of removing excess iron. Chelating agents are composed of molecules that bind tightly with free iron and remove the iron by carrying it as the agent is excreted from the body. o Desferrioxamine is the iron chelator available in parenteral form. o Desferasirox is the iron chelator available orally. Complications of BMT o Graft versus host disease o Graft failure XII. Prognosis: The prognosis of bone marrow failure depends on the duration of the marrow function abnormality. Most inherited forms of bone marrow failure, such as Fanconi anemia, are associated with transformation into leukemia several years later. Viral causes, such as parvoviruses, usually are self-limiting. Acquired idiopathic aplastic anemia usually is permanent and life threatening. Half the patients die during the first 6 months.