Supplementary Materials (doc 44K)
advertisement
")
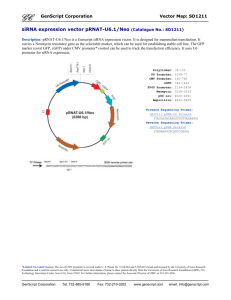
Supplementary Materials Western Blot Analysis. 25 µg of fractionated or immunoprecipitated proteins were resolved in 4-20% SDS-polyacrylamide gels, and electrotransferred onto nitrocellulose membranes. The blots were then blocked for 1 h at 25°C in PBS containing 5% nonfat dry milk and 0.05% Tween. Thereafter, the blots were incubated either with Sp1 antibody (Upstate, Inc, Lake Placid, NY) anti-CTCF (Abcam, Cambridge, England), or anti-C-myc (Invitrogen, Carlsbad, CA) for 1 h at room temperature or 4oC overnight, washed three times with PBS containing 0.05% Tween, and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The blots were washed five to six times in PBS containing 0.05% Tween, and the immunocomplexes were detected using the Super Signal chemiluminescent substrate (Pierce). Immunoprecipitation. The concentrations of nuclear extracts were adjusted to 2 µg/µl protein with chilled NET buffer (150 mM NaCl, 0.1% Nonidet P-40, and 50 mM Tris-HCl, pH 7.5) containing protease inhibitor cocktails. Nuclear lysates were precleared by incubation with 50 µl of protein G agarose (Amersham Biosciences, Piscataway, NJ) for 2 h at 4 °C followed by centrifugation. For immunoprecipitation experiments, nuclear proteins were incubated with 2 µg of antibodies recognizing Sp1 (Upstate), BORIS (Abcam), or CTCF (Abcam) overnight at 4 °C. After addition of 50 µl of protein G agarose, the suspension was incubated for another 2 h at 4 °C. The beads were pelleted by centrifugation, washed three times with cold NET buffer, and resuspended in 50 µl of 2x SDS sample buffer. After the suspension was heated to 95 °C for 10 min, 20-µl samples were resolved in denaturing SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and probed for the presence of specific proteins using western blot techniques. Mutagenesis of Sp1 recognition site on NY-ESO-1 promoter DNA. Mutations of the Sp1 binding site within the NY-ESO-1 promoter were performed with reference to the results from gel shifting experiments. Two mutated versions of plasmid pGL3/NY762 containing point mutations within the Sp1 recognition site were constructed using the QuikChangeTM mutagenesis 1 kit (Stratagene, La Jolla, CA) with the following forward primers and its antisense oligonucleotide as a reverse primer 5'-GGGGAGGGGCATGGTTGGTGAGAACCGGTC-3' and GGGGAGAAGCGGGGTTGGTGAGAACCGGTC, and designated as pGL3/NY762M2 and pGL3/NY762M3, respectively. The base substitutions are underlined and mutagenesis was confirmed by DNA sequencing. Six truncated versions of plasmid pGL3/ESO-762 were prepared by cloning serially shortened PCR products into Mlu-Xho I sites within pGL3 basic vector, each with a deletion of approximately 100 bp from the previous one at the 5’ end of the 762 bp fragment. The reverse primer 5’-TGGGCAGCCCGGGATCC-3’ was shared by all six PCR reactions; the six forward primers as well as the designation of corresponding plasmid construct (in parenthesis) were: 5’-TGGTGGGAGGCCACCCGCCAAC-3’ (pGL3/ESO-662), 5’-TAGGAGTGAGATCCGGCCCCG-3’ (pGL3/ESO-562), 5’ACATGCAGTTCCAGCTACCC-3’ (pGL3/ESO-462), 5’-TCCCCCAAGGACGGACAGGG-3’ (pGL3/ESO-362), 5’-ACAGGCTGGGCCCCGCAGTG-3’ (pGL3/ESO-262), 5’TGAGAACCGGTCACGTGCTC-3’ (pGL3/ESO-162), respectively. Similarly, a truncated plasmid construct designated pGL3/ESO/deltaSp1 bearing deletion of the proximal Sp1 binding site, was made using primer pairs (forward) 5’-TACTGCCGAACCCAGGAGCC-3’ and (reverse) 5’-GCCGGAAGGTGGCCGCAATG –3’. Transfections and Reporter Assays. All transfections were performed using LipofectAMINE 2000CD (Invitrogen), and luciferase assays were carried out using the Dual-Luc Reporter Assay System (Roche, Nutley, NJ) according to the respective vendor instructions. Briefly, cells were plated at a density of 2.5 x 106 cells/6-cm dish in RPMI 1640 medium (10% fetal bovine serum, with antibiotics omitted) and transfected using Lipofectamine Plus reagent (Invitrogen). For all transfection experiments except for those otherwise specified, 15 µg of plasmid test DNA and 2 µg of Renase control plasmid were co-transfected. Transfected cells were harvested and rinsed in 100 µl cold PBS. Cells were lysed by three rounds of freezing and thawing, and centrifuged at 12,000 x g. The resulting supernatants were assayed immediately, or stored at -800C for further use. For all transfection assays, four to five independent experiments were performed, and luciferase values were normalized for transfection efficiency against Renilla Luciferase activity. Nuclear Protein Extraction. Subconfluent cells were scraped from 100-mm dishes with icecold PBS and centrifuged for 5 min at 240 x g. The cells were resuspended in 1 mL of lysis 2 buffer containing 10 mM HEPES, pH 7.6, 1 mM EDTA pH 8.0, 60 mM KCl, 1 mM dithiothreitol, 0.5% Nonidet P-40, 1 µM phenylmethylsulfonyl fluoride, 10 µM leupeptin, and 0.2 U/mL aprotinin. Intact nuclei were isolated by centrifugation, and resuspended in nuclear suspension buffer containing 250 mM Tris-HCl pH 7.8, 1 mM dithiothreitol, 1 µM phenylmethylsulfonyl fluoride, 10 µM leupeptin, and 0.2 U/mL aprotinin. Nuclei were then lysed by freezing and thawing, and the protein concentration in the resulting nuclear lysate was determined by the Bradford Kit (Pierce). siRNA Construction and Transfection. Cells were grown to 60% confluence in 6-well plates on the day of transfection. 15 µl of RNAiFect siRNA transfection reagent was added to100 µl of normal medium, mixed by vortexing and incubated at room temperature for 20 minutes. 5 ug of siRNA was added to the diluted transfection reagent, mixed gently by pipetting, and incubated at room temperature for 20 minutes to form the transfection complexes and then diluted with 2 ml of medium. The culture media from the cells were aspirated and immediately replaced with the diluted transfection mixture. The siRNA complexes were evenly dispersed by gently rocking the plate. Electrophoretic Mobility Shift Assay (EMSA). Wild type and four oligonucleotide probes containing base substitutions in the Sp1 consensus sequence were designed. The forward strands are as follows: WT, 5’CAAGCTGGCATTGCGGCCACCTTCCGGCCCGGGCTCTCTTGGGGAGGGGCGGGGTTG GTGAGAACCGGTCACGTGCTCCGGGGCTCACTCGGGGTCTCCC-3’; M1, 5’CAAGCTGGCATTGCGGCCACCTTCCGGCCCGGGCTCTCTTGGGGAGGTTCGAAGTTG GTGAGAACCGGTCACGTGCTCCGGGGCTCACTCGGGGTCTCCC-3’; M2, 5’CAAGCTGGCATTGCGGCCACCTTCCGGCCCGGGCTCTCTTGGGGAGGGGCATGGTTG GTGAGAACCGGTCACGTGCTCCGGGGCTCACTCGGGGTCTCCC-3’; M3, 5’CAAGCTGGCATTGCGGCCACCTTCCGGCCCGGGCTCTCTTGGGGAGAAGCGGGGTTG GTGAGAACCGGTCACGTGCTCCGGGGCTCACTCGGGGTCTCCC-3’; M4, 5’CAAGCTGGCATTGCGGCCACCTTCCGGCCCGGGCTCTCTTGGGGAGTTACGGTATTTG GTGAGAACCGGTCACGTGCTCCGGGGCTCACTCGGGGTCTCCC-3’ (Sp1 consensus was italicized and base substitutions are underlined.). Probe annealing was performed by heating 100 µM of each complementary strand of the oligonulecotide to 95°C for 2 min, and then cooling 3 gradually to 25°C over a period of 45 min in a thermal cycler. The annealed oligonucleotides were DIG-labeled using DIG Gel Shift Kit (Roche) per manufacturer’s instructions. The labeled probes were incubated with 4 µg of the nuclear extracts in gel shift binding buffer [20% glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM dithiothreitol, 250 mM NaCl, 50 mM Tris-HCl, pH = 7.5 and 0.25 mg/ml poly (dI-dC)·poly (dI-dC)] for 15 min in a total reaction volume of 20 µl. Then, labeled probes were added, and the reactions were allowed to continue at room temperature for 30 min, following which the samples were resolved in a 10-cm 5% native PAGE gel at 4°C with a 20 mA current for 1.5 h. In competition assays, the unlabeled competitor oligonucleotides were pre-incubated with the nuclear extract prior to the addition of labeled probe. In the antibodymediated supershift assay, approximately 2 µg of Sp1-specific antiserum (SC-420 X, Santa Cruz Biotechnology, USA) was incubated with 8 µg of nuclear extract in gel shift binding buffer for 1 h on ice, and then incubated further with the labeled probes for another 30 min before resolving the reaction products in gels. The gels were then blotted onto nitrocellulose filter, and processed according to vendor’s instructions (DIG Gel Shift Kit, Roche). Thereafter the blots were exposed to x-ray film. Chromatin immunoprecipitation (ChIP). 1.2 x 106 NHBE, H1299, or Calu-6 cells were prepared using a ChIP Assay Kit from Upstate (Charlottesville, VA) according to the manufacturer’s recommendations using anti-Sp1(Upstate) or AcK9H3 (Abcam) antibodies. The following primers were designed to amplify a 198-bp region that included a putative Sp1 binding site within the NY-ESO-1 promoter: forward 5’-CTCCTCGATGTGAGGGAGAC-3’ and reverse 5’-GAGCACGTGACCGGTTCT-3’. PCR was done using Advantage GC 2 Polymerase Mix (Clontech, Mountain View, CA) with 1 M of GC Melt and the following variables: 94˚C x 3 minutes followed by (94˚C x 30 seconds, 61˚C x 1 minute, 68˚C x 1 minute) x 34 cycles and an extension of 68˚C x 3 minutes. PCR products were analyzed by 1.5% agarose gel electrophoresis. Bisulfite sequencing of the NY-ESO-1 promoter. The NY-ESO-1 promoter sequence was obtained from the University of California Santa Cruz Genome Browser (http://genome.ucsc.edu/cgi-bin/hgBlat) and was analyzed using the CpGPlot website (http://www.ebi.ac.uk/emboss/cpgplot/index.html). Genomic DNA from cultured cells 4 was subjected to bisulfite modification using the CpGenome™ DNA Modification Kit (Millipore, Billerica, MA). A 135-bp region of the NY-ESO-1 gene (–207 to –73) was amplified using the primer sequences: forward 5'-GTGAGGGGTTTAAGTTGGTATTG3', reverse 5'-AAACAAAAAAACTACAAAAAATTCC-3', Clontech Advantage GC 2 polymerase (Clontech, Palo Alto, CA), and the following thermal cycle variables: 95°C for 2 minutes followed by (95°C for 45 seconds, 56°C for 30 seconds, 68°C for 1 minute) x 35 cycles then 68°C for 3 minutes. PCR fragments were ligated into the TOPO TA cloning vector (Invitrogen, Carlsbad, CA). Following transformation, plasmids from individual bacterial colonies were isolated, and the NY-ESO-1 promoter fragments were sequenced using an ABI 310 prism apparatus (Applied Biosystems, Carlsbad, CA). 5