jwsPROTv59.4.655.figs
advertisement

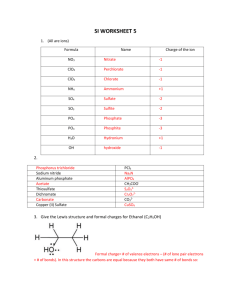
Supplementary Data The Nmr Data Model Package The link between Resonance and Atom objects is shown in Supplementary Figure 1. A Resonance is linked to the relevant Atom objects via two other objects: the ResonanceSet and the AtomSet. The AtomSet groups together atoms that are, for solution NMR, in fast exchange (e.g. protons in methyl groups), while the ResonanceSet handles the ambiguity of which resonance or resonances might be linked to which set of equivalent atoms. Supplementary Figure 1 illustrates this with an example for the methyl groups of a leucine, both in the stereospecific and nonstereospecific assignment case. Here, the AtomSet groups together the three protons of the methyl group. These atoms have the correct covalent and stereochemical IUPAC definition. This means that if a Resonance is stereospecifically assigned, the links to the relevant AtomSet go directly via one ResonanceSet and are unambiguous. In the non-stereospecific case this is not possible: two Resonances for the side chain methyl groups (whether they have the same chemical shift or not) are linked via one shared ResonanceSet to the two relevant AtomSets. This exactly describes the ambiguity of the assignment; the Resonances are known to exist for the stereospecific Atom Sets – it is the precise connections that are not known. Resonance ResonanceSet AtomSet res 1 resSet 1 atomSet 1 Atom H11 H12 H13 Leucine HD groups (stereospecific) H21 res 2 resSet 2 atomSet 2 H22 H23 H11 res 1 atomSet 1 H12 H13 Leucine HD groups (not stereospecific) resSet 1 H21 res 2 atomSet 2 H22 H23 Supplementary Figure 1. Resonance to Atom link. An AtomSet has multiple atoms when they are in fast exchange (in this case the atoms of a methyl group), and a ResonanceSet describes the ambiguity for the Resonance-Atom link. For a stereospecific assignment each resonance is linked to a specific AtomSet, for a nonstereospecific assignment both resonances are linked via one resonance set, so describing their ambiguity. NMR measurements are mostly handled through the generic MeasurementList object (Supplementary Figure 2A). A MeasurementList has Measurements, which are linked to one or more Resonance objects depending on the measurement type. The setup for most types of measurements is derived from this generic description. For example, a ShiftList has Shift objects, each of which has to be linked to one and only one Resonance. A Resonance on the other hand can be linked to several Shift objects, so that different shifts observed for the same atom in different spectra or under different conditions are all still linked to the same object. This also illustrates the importance of the Resonance object: if in a range of spectra at different conditions a particular peak is clearly discernible all its shifts in one dimension can already be assigned to a Resonance before the atom it belongs to is actually known. As soon as the assignment of the Resonance is known and set in the Data Model all the shift information is then also linked to the relevant atom(s). Data that is derived indirectly from measurements (e.g. PkaLists) are handled in a very similar way. Measurement MeasurementList Shift ShiftList (A) DihedralConstraintItem DihedralConstraint DihedralConstraintList ConstraintItem Constraint ConstraintList DistanceConstraintItem DistanceConstraint DistanceConstraintList (B) Resonance PeakDimContrib PeakContrib (C) PeakDim Peak PeakList Supplementary Figure 2. Simplified description of measurements, constraints and peaks in the data model. Grey arrows indicate subclass relations, diamond arrows indicate parent/child relationships, plain lines indicate normal links. Constraint lists are, similar to measurement lists, handled via a generic ConstraintList object (see Supplementary Figure 2B, which gives a DistanceConstraintList as a concrete example). A ConstraintList object has Constraints, which have ConstraintItems that are linked to one or more Resonance objects. This set-up describes the ambiguity at the level of the constraint separately from the assignment of the Resonance to the atom. In this way the stereospecific assignment (e.g. this is either a tyrosine H2 or H3 atom) is separate from the constraint ambiguity (e.g. this is a constraint between an H atom and an H or an H* atom). An exception DihedralConstraintList. to In this the generic case the constraintList resonances setup is the are linked to the DihedralConstraint, while the DihedralConstraintItem describes an angle range of the dihedral angle so that multiple angle regions can be allowed (e.g. between –60° and -20° or between 40° and 80°). This flexibility in describing differences in very similar systems from a generic model (in this case constraint lists) is inherent in the Data Model. This principle also applies to the PeakList object. In Supplementary Figure 2C only the system for handling normal peaks is described, but in the full Data Model this whole set-up is mirrored for sub-peaks that are used for peak splittings (in this way the complete information from, for example, DQF-COSY or E-COSY type peaks can be handled). The normal description allows the creation of Peaks, each with a PeakDim object for each of the dimensions involved. Each PeakDim has PeakDimContribs that, similarly to the ConstraintItems, describe the assignment ambiguity at the peak level (e.g. this peak in this dimension can be assigned to either a non-stereospecifically assigned leucine H methyl group or to an alanine H methyl group). These PeakDimContribs can be combined using PeakContrib objects: with this method combinations of assignments can be grouped together (e.g. either residue 3 H to H or residue 7 H to H). Also crucial to the Data Model for NMR is the description of an NMR experiment (Supplementary Figure 3). The main object here is the Experiment, which has dimensions ExpDim, each of which has one or more ExpDimRefs. The ExpDimRef object describes multiple references for a particular dimension (e.g. for a combined 3D 15N/13C NOESY HSQC the ExpDim corresponding to the hetero nuclei will have an ExpDimRef for the 15 N and an ExpDimRef for the 13 C). An Experiment has DataSource objects, which handle the original time (or other) domain data, and transforms of that data. For a typical experiment, a DataSource exists which describes the main characteristics of the raw data (the type of data file, the location of the data file, etc.). In this case it has FidDataDims corresponding to each of the ExpDims. Each FidDataDim describes the number of recorded points, the number of valid points, etc. for that dimension. Another DataSource would be created for the processed data: in this case it has FreqDataDims. Each FreqDataDim holds the number of points used for the Fourier transform in its dimension, the phase settings, etc. Furthermore, a FreqDataDim can have DataDimRef objects, which are linked to an ExpDimRef described above. Each of these DataDimRef objects describes a particular referencing for that dimension: this again allows multiple references to exist within the same dimension (e.g. in the case of a combined 3D 15 N/13C NOESY HSQC). Also note that the PeakDim discussed previously is linked to a DataDimRef. This allows multiple references to exist within the same peak list. Finally, the Experiment itself is also linked to objects describing the physical setup, such as NmrSpectrometer, Sample, etc. (not shown in Figure 3). Experiment DataSource ExpDim DataDim FreqDataDim ExpDimRef FidDataDim DataDimRef PeakDim Supplementary Figure 3. Simplified description of the NMR experiment setup in the data model. Grey arrows indicate subclass relations, diamond arrows indicate parent/child relationships, and plain lines indicate normal links. This concise overview of the crucial areas of the Data Model for NMR does not describe many of the objects and subtleties that are contained within it – more detail can be found at http://www.ccpn.ac.uk/. For example, there are a number of objects that allow the description of intermediate assignment data (e.g. a grouping of Resonances that belong to the same residue, etc.). Also, there is scope for describing multiple conformational (or other) states of molecules in the sample, and there are many convenient and informative but non-crucial links between related objects (e.g. a ConstraintList can be linked to multiple Experiments). CcpNmr Analysis The assignment of NMR spectra proceeds via the Resonance object and is made in two steps; the connection of the Resonance to a dimension of a peak (PeakDim) and the connection of the Resonance to an atom or atoms (AtomSet). The connection of the Resonance to AtomSets can be made once sufficient information is gleaned, but prior to that a partial assignment serves as a useful point of reference to connect data. For example, a peak in a 15N HSQC experiment can have a resonance assigned for the 1 H and 15 N dimensions and these can be grouped together into a spin system (ResonanceGroup). Groups of peaks in other spectra that correspond to this spin system are then easily assigned to the same Resonances, even though the atomic identity is undetermined. Once the sequential assignment of the chain is made and the identity of the spin system determined, the atomic assignment, albeit only physically specified for one peak, is immediately transferred to all peaks that represent the same spin system because they are linked to the same Resonances. In the final stages of assignment, for example when deriving structural information from an NOE experiment, most Resonances have been identified and the assignment step mainly involves choosing existing resonances from a curated, ranked list. The Resonances in such a list can be ranked by the closeness of chemical shift match or the spatial distance between assigned AtomSets, given a draft or intermediate structure for the molecule. The screen shots in Supplementary Figures 4-8 illustrate some additional features of the CcpNmr Analysis program. Supplementary Figure 4. Reference chemical shift information from the BioMagResBank can be viewed within Analysis for any of the represented atoms and residues. Supplementary Figure 5. The Edit Spectrum window with the name of an experiment being edited; an example of one of the many editable tables found within Analysis which allow parameters and NMR objects to be readily changed without the need for configuration files. Supplementary Figure 6: Peak selection and assignment within analysis. The assignment possibilities (top panel) are shown for all peak dimensions and can be ranked according to chemical shift closeness and atomic distance, given a preliminary structural model. Peaks selected in spectrum windows (bottom panel) can be edited within the Analysis tables and may be assigned independently of the spectrum contour windows. Supplementary Figure 7. The Calculate Heteronulear NOE window. This an example of Analysis performing some of the more complex data manipulations without the need for specialist scripting. Supplementary Figure 8. An example of a Python macro written for Analysis. This script assigns spin systems per peak. By importing functions from Analysis the user can create powerful high-level functionality without having to be concerned with the fine details of the NMR Data Model. CcpNmr FormatConverter Application data Often not all information can be transferred into a specific attribute or class inside the Data Model because it is application specific (e.g. force constant information). In these cases the information is stored in ApplicationData classes and can still be used for export to the specific format. Information stored at this level, or information that is not contained within a specific format, is always lost in a transfer between formats, e.g. the nmrView ‘shape’ information for peaks cannot be transferred to an XEasy peak list. This problem is due to the definitions of the formats and cannot be avoided. Resonance-Atom link Most data formats lack the concept of Resonances, and therefore assign NMR parameters directly to atoms using their own particular naming system. A crucial task in conversion is thus to map these data format names to the IUPAC naming used in the Data Model. In the set of scripts that make up the format conversion software, only the linkResonances module deals with linking Resonances to Atoms. During import, no assumptions are made about which atoms are referred to by the names read in from the data format; the Resonance concept allows linking of all NMR data from the data format file to a shared object within the Data Model, as long as the naming is consistent within the data format file(s). After all relevant NMR data is read in, linkResonances is executed to allow the user to define which atom(s) correspond to which name. The Data Model contains reference data on which atoms are prochiral and/or NMR equivalent, as well as several common naming conventions for these atoms (e.g. XPLOR, DIANA, etc.). The script first checks how well the available naming systems fit the data format names that were read in. The user can then decide which naming convention should be used to interpret the atom names. All names that have a matching name in that naming convention will then be automatically linked to the correct atom(s). For unknown names or cases where a resonance already exists for that name, user interaction is necessary and pop-ups appear that allow the user to define exactly what the name should mean. The atom name mapping can be propagated at this stage to residues/residue types within the molecular system, molecule or chain. In the next step the information is reorganized in order to group resonances that should be treated together (e.g. a threonine H22 atom should be treated together with the H2 methyl group). Finally, the Resonance objects are linked to atoms in the correct way. Although options are available to treat all assignments as stereospecific (or not stereospecific), the default option will prompt the user with a choice if the information is ambiguous. Other ambiguities are also dealt with at this stage. For example, if only one atom of a prochiral center has been assigned to a Resonance, this can mean that the original name was in fact referring to both atoms, and a new Resonance that is linked to the same information as the first one has to be created. This is necessary because it is possible that even though these atoms have the same chemical shift under a particular set of circumstances, they could be resolved under a different condition. At the end of this process the user has unambiguously defined what atom(s) the NMR information derives from. During export the link between the Resonance and the atom(s) is used to derive the meaning of the Resonance. The actual name of the atom for export is arbitrary. As far as the Data Model is concerned any supported naming system can be used. Since all atom information in a file format will again have to be contained within a number of strings, however, the information content is inevitably reduced after export. It is possible to save the names that were used for export to a format so that the files can later be read in again without having to use linkResonances, but it is essential that this is done only if no changes were made in the names or their meaning in the exported files. At any time after the linking process the writeMappingFile script can produce a mapping file, which relates the original format names to the atom(s) within the Data Model.