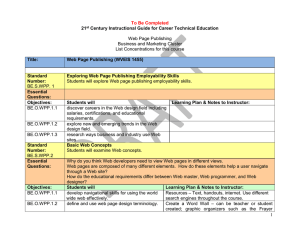
X-ray crystal structure of the bis(dihydrogen) complex RuH2(H2)2
advertisement
 complex RuH2(H2)2")
This journal is © The Royal Society of Chemistry 2000 X-Ray crystal structure of the bis(dihydrogen) complex RuH2(H2)2(PCy3)2 Andrzej F. Borowski, Bruno Donnadieu, Jean-Claude Daran, Sylviane Sabo-Etienne and Bruno Chaudret Following the publication of this paper, the attention of the authors was drawn by a number of colleagues to the likely centrosymmetry of this structure. Although all the statistical tests indicate a noncentrosymmetric system, it is obvious from the examination of the structure that it is very close to centrosymmetry, if not really centrosymmetrical. The resolution and refinement of the structure in space group P1 had been previously carried out but not discussed in the published paper. So, the authors wish to present here the results concerning the refinement in the centrosymmetric space group P1.† In this case, the Ru atom is located on an inversion center and only half of the molecule has to be refined. The heavy atoms were located with the help of SIR97 and refined by full matrix least squares using SHELXL97. After including, using a riding model, the hydrogen of the cyclohexyl groups, a difference Fourier map in the equatorial plane of the Ru atom showed two large peaks (four by reason of symmetry) with an electron density of ca. 1.5 e Å-3. These two positions could be refined as H atoms without any restraints leading to the results showed in Table 1. Table 1 Comparison of refinement between the non-disordered model and the disordered one Model with no disorder Disordered model Data/restraints/parameters Goodness-of-fit on F2 3084/0/187 3084/12/197 1.045 1.045 Final R indices [I>2σ(I)] R1 = 0.0321, wR2 = 0.0709 R1 = 0.0314, wR2 = 0.0676 R indices (all data) R1 = 0.0373, wR2 = 0.0730 R1 = 0.0366, wR2 = 0.0696 Largest diff. peak and hole/e Å-3 0.491 and -0.362 0.372 and -0.355 This journal is © The Royal Society of Chemistry 2000 The Ru-H distances are 1.52(2) and 1.60(3) Å, respectively, and the H-Ru-H angle is 98.2(1.4)°. However, owing to the fact that there is good evidence that this compound contains hydride and dihydrogen ligands, it was attempted to resolve these large electron density peaks. Introducing only one half of hydrogen on each site and calculating a new Fourier difference map resulted in the appearance of two residual densities around each site. This result led us to consider a disordered distribution of hydride and dihydrogen ligands on the two sites. The refinement of this new model was carried out with the help of soft restraints (Ru-H and H-H distances) to maintain, as far as possible, a reasonable geometry. The result of the refinement is given in Table 1 and a view of the molecule is shown in Fig. 1. It is obvious that the model shown in this figure does not lead to an unambiguous attribution for the structure of RuH2(H2)2(PCy3)2, but is in agreement with the proposed structure established on a spectroscopic basis.1 Fig. 1 In conclusion, the main question remains, is the structure centrosymmetric or noncentrosymmetric? Owing to the statistical test, we were inclined to consider the possibility of a twin by This journal is © The Royal Society of Chemistry 2000 inversion (racemic twin) and to refine the structure in the P1 space group to eliminate the disordered model. It is clear that from a pure crystallographic approach, the refinement in P1 is more satisfactory with a better set of C-C distances within the cyclohexyl groups. A comparison of the Ru-P, Ru-H and HH distances between the two hypotheses is given in Table 2. Finally note that the structure of the very similar complex RuH2(H2)2(PPri3)2 has recently been reported.2 Table 2 Comparison of bond distances (Å) and bond angles (º) between the P1 and the disordered P1 refinement Ru(1) - P(1) Ru(1) – P(2) Ru(1) - H(5) Ru(1) - H(6) Ru(1) - H(1) Ru(1) - H(2) Ru(1) - H(3) Ru(1) - H(4) H(1) - H(2) H(3) - H(4) H(2) - H(3) H(1) - Ru(1) - H(2) H(2) - Ru(1) - H(3) H(3) - Ru(1) - H(4) H(4) - Ru(1) - H(5) H(5) - Ru(1) - H(6)' H(6) - Ru(1) - H(1)' P1 2.330(2) 2.347(2) 1.71(4) 1.69(4) 1.54(3) 1.55(3) 1.52(3) 1.53(3) 0.85(4) 0.86(4) 1.41(7) 31.8(1.4) 54.6(2.5) 33.0(1.5) 79.8(2.3) 78.9(2.4) 83.5(2.5) P1 2.3384(7) 1.73(4) 1.74(5) 1.48(3) 1.49(3) 1.49(3) 1.49(3) 0.87(4) 0.86(4) 1.48(6) 34.1(1.7) 59.7(2.8) 33.3(1.7) 68.6(2.4) 83.1(2.3) 68.7(2.5) The authors wish to acknowledge Professors G. Parkin and A. Rheingold for interesting comments on the reported structure. Notes and references †Crystal data for C 1 8 H 3 6 PRu 0 . 5 , M = 333.97, triclinic, space group P1, a = 8.1781(13), b = 9.9339(17), c = 11.7055(19) Å, = 76.18(2), = 84.45(2), = 69.39(2)°, Z = 2, V = 864.2(2) Å 3 , D c = 1.283 gcm - 3 , Mo-K radiation ( = 71073 Å), = 5.69 cm - 1 , crystal dimensions 0.5 x 0.2 x 0.1 mm, F(000) = 362. Stoe IPDS diffractometer, T = 160(2) K. Numerical absorption corrections were applied. From This journal is © The Royal Society of Chemistry 2000 8239 reflections, 3084 were unique ( R i n t = 0.0447). Data/restraints/parameters ratio 3084/12/197, R = 0.0314, wR2 = 0.676 [2735 reflections with F > 4(Fo)], R = 0.0366, wR2 = 0.0696 (all data), S = 1.030. CCDC 182/1678. See http://www.rsc.org/suppdata/cc/a9/a908567j/addition.htm for revised crystallographic files in .cif format. 1 V. Rodriguez, S. Sabo-Etienne, B. Chaudret, J. Thoburn, S. Ulrich, H.-H. Limbach, J. Eckert, J.-C. Barthelat, K. Hussein and C. J. Marsden, Inorg. Chem., 1998, 37, 3475. 2 K. Abdur-Rashid, D. G. Gusev, A. J. Lough and R. H. Morris Organometallics, 2000, 19, 1652.