Supplementary Methods (doc 60K)
advertisement
")
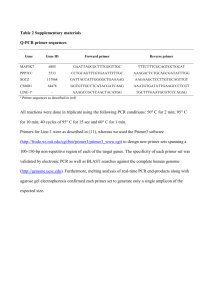
Supplementary Methods 16S rDNA 454-Pyrosequencing and data analyses. DNA extracts were normalized to 10 ng/l prior to PCR. Samples were amplified using a universal primer pair targeting the 16S rDNA V9 region. The forward primer was 515F (‘5-GTGCCAGCMGCCGCGGTAA-‘3) and the reverse primer was 907R (‘5CCGTCAATTCCTTTRAGTTT-‘3). The forward and reverse primers were downstream of the FLX-454 primer adapters and the reverse primer also contained a 12-bp barcode unique to a specific sample (Hamady et al. 2008). PCR reactions were performed using Takara Ex Taq polymerase (Takara, Madison, WI), with the following thermocycling parameters: initial denaturation at 95 C for 1 minute followed by 25 cycles of 95 C for 20 seconds, 30 seconds of annealing at 66 C and extension at 72 C for 1 minute. Final product extension was at 72 C for 10 minutes. Reaction primer dimmers were removed from the PCR products via SPRI bead purification according to the manufacturer’s protocol (AMPure XP, Beckman Coulter Genomics, Danvers, MA) before being checked for quality and quantity on a Bioanalyzer 2100 using a DNA 7500 chip (Agilent Technologies, Santa Clara, CA). Each PCR sample was normalized to 30 ng and pooled together for multiplex sequencing. Sequencing libraries were created using the SV emuPCR kit (Lib-A, Roche, Indianapolis, IN) and sequencing on a GS-FLX sequencer (Roche, Indianapolis, IN) at the Veterans Medical Research Foundation, La Jolla, CA. Reads between 200 and 1000 bp were used for downstream analysis in the Quantitative Insights Into Microbial Ecology (QIIME) pipeline (average = 426 bp) (Caporaso et al. 2010a. All sequences with a mean quality score below 25 and homopolymer run of 6 or more were removed. The total number of reads amounted to 56,182 reads with an average number of 6,208 sequences per sample (range: 1,90014,595). All reads were deposited at the European Bioinformatics Institute (EBI) with the accession number ERP003515. Sequences were clustered at 97% pairwise identity using the UCLUST (Edgar, 2010) reference-based OTU picking method, where the reference dataset was the GreenGenes 99% (GG99_12_10, Werner et al. 2012) data set. A representative sequence from each OTU was aligned to the GG99 data set using PyNAST (Caporaso et al., 2010b). The concatenated alignment of OTUs was filtered to remove gaps and hypervariable regions using the GreenGenes Lane mask (DeSantis et al. 2006). A phylogenetic tree was constructed from the filtered alignment using FastTree (Price et al., 2010). Taxonomic assignments were done with the naïve Bayesian algorithm (Wang et al., 2007) developed for the RDP classifier (Cole et al., 2009) using the GG99 taxonomy data set as training (Werner et al. 2012). Phylogenetic -diversity was quantified with a weighted Unifrac distance matrix which was constructed at a depth of coverage of 19,000 sequence (normalized coverage to the minimum number of sequences in the dataset). The resulting distance matrix was used to cluster samples using UPGMA hierarchical clustering algorithm. Support values for the UPGMA dendrograms were calculated using 100 jackknife permutations and nodes having at least 50% support were deemed to have high support. Supplementary References Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello, EK, Fierer N, Pena A.G, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Wildmann J, Yatsunenko T, Zaneveld J, Knight R. (2010a). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R, (2010b). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26: 266-267. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl Acids Res 37: D141-D145. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069-5072. Edgar RC. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460-2461. Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. (2008). Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5: 235-237. Price MN, Dehal PS, Arkin AP. (2010). FastTree2- approximately maximumlikelihood trees for large alignments. PloS One 5: e9490. Reeder J, Knight R. (2010). Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 7: 668-669. Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporasso JG, Angenent LT, Knight R, Ley RW. (2012). Impact of training sets on classifications of high-throughput bacterial 16S rRNA gene surveys. ISME J 6:94-103.