Chrom. I - UCSF Biochemistry & Biophysics
advertisement

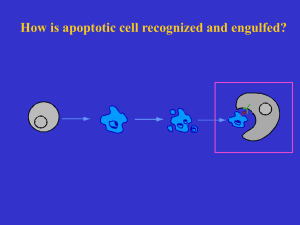
C. elegans problem set I. Basic concepts of diploid genetics and linkage You cross a dpy-5(e61) male (“dumpy” body shape phenotype, recessive mutation) with an unc-13(e51) hermaphrodite (“uncoordinated” movement phenotype, recessive mutation). a. What phenotype(s) do expect to see in F1 cross progeny? b. What assumptions are implicit in your answer? c. You single (i.e. move individual worms onto separate plates and let them self) several of the F1 cross progeny at a stage that you are certain they have not mated with their siblings. What genotypic and phenotypic ratios do you expect to see in the F2 if the two mutations are unlinked? (Draw out the dihybrid cross). How do these ratios change if the mutations are linked? d. At this point, let’s assume that the specific Dpy and Unc genes are linked. From the F2 progeny plate (see above) you single out several Dpy animals, 1 per plate (again, you are certain that they have not mated). What do you expect to see phenotypically in their progeny (F3 relative to the starting cross)? e. Of the 32 Dpy F2 animals that you single, 2 of the worms give rise to progeny (F3) that are ¼ DpyUnc. What is the genotype of these two F2 animals? Think about which chromosomes are parental, and which are recombination products. How many map units are Dpy and Unc apart? 2. Mapping using polymorphisms (SNPs) You have isolated a mutation that appears to be due to inactivation of a single locus. Your screen was started in the N2-Bristol background strain. To identify the mutated gene, you cross your mutant to the CB4856 Hawaiian mapping strain (polymorphic relative to starting strain). You pick F1 cross progeny, allow them to self, and from their progeny (F2), you pick animals that once again display the mutant phenotype. You pick 20 such animals. These animals are then placed individually on separate plates and allowed to lay eggs. About a week later, all 20 plates are filled with animals. From each plate you wash some of the animals away for DNA extraction and retain the plates (each plate will still have lots of animals). You know of SNIP-SNPs that inform on each of the chromosomes (polymorphisms that can be detected because they create/eliminate a restriction cutting site, hence SNIP; In each case, you PCR amplify ~0.5-1 kb region of the DNA in which the pre-determined SNIP-SNP is contained. Since the SNPs generate or destroy restriction sites, by subjecting the 1 fragments to specific restriction endonucleases and running gels, you can distinguish the presence or absence of a SNP). So, from the the collection of progeny of 10 of the mutant Fss that you have picked, you extract DNA, subject each DNA to PCR amplification for the six regions that contain the SNIP-SNPs that inform on the 6 chromosomes of the worm, perform restriction digest, and run a gel. Based on SNIPSNP cutting patterns you can score each SNP as CB4856 (CB), N2, or both. A gel showing the SNIP-SNP patterns for each chromosomal marker are shown. Lanes one and two show the band patterns for the N2 and CB strains as controls, and lanes 1-10 show the band patterns for each of 10 F2 clones that are tested. Chrom. I N2 CB _ _ 1 _ _ 2 _ _ 3 _ _ 4 _ _ 5 6 7 8 _ _ _ _ _ _ _ _ _ _ _ _ Chrom II N2 CB _ _ _ 1 _ _ _ 2 4 _ _ _ 5 6 7 8 _ _ 3 _ _ _ _ _ _ _ _ _ _ _ Chrom III N2 CB _ _ 1 _ _ 2 _ 3 _ 4 _ 5 7 _ 8 _ 6 _ _ 1 2 3 4 5 6 8 _ _ _ _ _ _ _ _ _ _ _ _ 7 _ 1 _ 2 _ 3 _ 4 _ 5 _ 6 _ 7 _ 1 _ _ _ 2 _ _ _ 3 _ _ _ 4 _ _ _ 5 6 _ _ _ 7 _ Chrom IV N2 CB _ _ _ Chrom V N2 CB _ _ _ Chrom X N2 CB _ _ _ _ _ 2 9 _ 10 _ _ _ 9 _ 10 _ _ _ 9 _ 10 _ 9 _ _ _ 10 _ _ _ 8 _ _ _ 9 _ 10 _ 8 _ _ _ 9 _ 10 _ _ _ _ _ _ _ _ a) Which chromosome does this mutation map to? Why? b) What is a rough estimate of map position of your mutation in relation to the SNP on that chromosome? Now, using SNPs along the chromosome you then get the following data (The SNPs are shown in proper genetic order, ie. SNP1 is on the left arm and SNP5 is on the right arm). Remember double recombination events on one chromosome occur but are rare): Starting strain CB4856 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 SNP1 N2 SNP2 N2 SNP3 N2 SNP4 N2 SNP5 N2 CB/CB CB/CB CB/CB CB/CB CB/CB N2/N2 N2/CB CB/CB CB/CB CB/CB N2/N2 N2/CB N2/CB N2/CB N2/CB N2/N2 N2/CB N2/N2 N2/N2 N2/N2 CB/CB N2/CB N2/CB N2/CB N2/N2 N2/N2 N2/CB N2/CB N2/CB CB/CB N2/N2 N2/CB N2/CB N2/CB N2/CB N2/N2 N2/CB N2/N2 N2/N2 N2/N2 N2/CB N2/CB N2/CB N2/CB N2/N2 N2/N2 N2/CB N2/N2 N2/N2 CB/CB N2/N2 N2/N2 N2/CB N2/CB N2/CB N2/N2 N2/CB N2/N2 N2/N2 N2/N2 N2/CB N2/N2 N2/CB N2/CB N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 CB/CB N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 N2/CB N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 N2/N2 N2/CB N2/CB N2/N2 N2/N2 N2/CB N2/CB N2/N2 N2/CB N2/N2 N2/N2 N2/N2 N2/N2 CB/CB N2/CB N2/CB N2/N2 N2/N2 N2/N2 N2/N2 N2/CB c) Which marker is most closely linked to the mutation? d) Which two markers does your mutation lie between? How do you know? e) Give one trivial explanation as to why worm #5 doesn't fit with the rest of the data. 3 3. Epistasis analysis and pathway building: During programmed cell death cells are killed off and the corpses are engulfed and cleared by neighboring cells. Regulatory relationships between pathway components may be inferred through epistasis. Consider the following results for ced (cell death defective) mutants (cell counts refer to final tally in adult hermaphrodites excluding the germ-line): Genotype Wild type ced-3(lf) ced-4(lf) ced-9(lf) ced-9(lf); ced-3(lf) ced-9(lf); ced-4(lf) live cells 959 1090 1090 0 1090 1090 dead cells 131 0 0 1090 0 0 A) What regulatory relationships can be inferred between ced-3, ced-4, ced-9 and their relationship to cell death? Corpse engulfment genes of programmed cell death are identified as mutations in which cell death still occurs leaving a visibly identifiable cell corpse but one that is not cleared away. Consider: Genotype ced-1(lf) ced-3(lf) ced-1(lf), ced-3(lf) phenotype cells die, but leave ugly corpses cells do not die cells do not die B) What is the epistatic relationship of ced-3 and ced-1? (initial identification of engulfment mutants (e.g. ced-1) allowed for identification of “killer” genes such as ced-3 since they emerged, as you can see, as suppressors of the ced-1 phenotype). It is quite difficult to count cell deaths through the lineage (tracing all 131). In practice, one can focus on one anatomical area of the worm and focus on death events there and, if needed, show that findings in that region are also mimicked by phenotypes seen in other cell types/anatomical regions. For instance, loss of function mutations in ced-1, ced-2, ced-7, and ced-10 were identified as engulfment genes as null mutations in each of these genes left, on average, one corpse per 4 pharynx (C. elegans feeding organ). However, more than one cell undergoes programmed cell death in the pharynx, implying that some corpse engulfment is still occurring in each of the single mutant backgrounds. The following results were obtained from mutant combinations (all cases are null alleles): ced-1; ced-2: average four corpses/pharynx ced-1; ced-7: average one corpse/pharynx ced-1. ced-10: average four corpses/pharynx ced-2; ced-7: average four corpses/pharynx ced-2; ced-10: average one corpse/pharynx ced-7; ced-10 average four corpses/pharynx C) What can you conclude about relationships between these four ced genes? 5