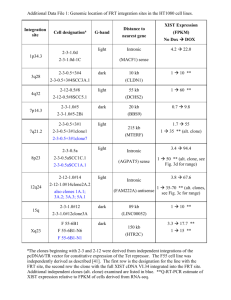
Murine spleen colony forming cell (CFU-S) assay
advertisement
 assay")
Supplemental Materials and Methods
Murine spleen colony forming cell (CFU-S) assay
2 x 106 bone marrow cells were pre-stimulated overnight in Dulbecco’s modified
essential medium supplemented with 20% fetal calf serum (Hyclone; Logan, Utah) and
recombinant 20ng/ml murine IL-3, 50ng/ml murine IL-6, 50ng/ml murine SCF (all
obtained from R & D Systems, Minneapolis, MN).
The following day, cells were
collected, pelleted, and resuspended in 1 ml of the above medium containing 2X
concentrations of the serum and cytokines, and 12 g/ml polybrene. An equivalent
volume of vector containing medium was added to the cell mixture to achieve an MOI of
5-50.
This mixture was placed into a RetroNectin (TAKARA Shuzo, Otsu, Shiga,
Japan)-coated (20ug/cm2) 6 well plate and incubated at 37° C in a humidified incubator
with 5% CO2. After 6 hours, the cells were collected and resuspended in PBS containing
2% FCS. Sublethally irradiated (900 cGy) C57Bl/6J mice (Jackson Laboratories, Bar
Harbor ME) were transplanted with either 5 x 104 cells or 1 x 105 by tail vein injection.
Thirteen days later the mice were euthanized and well-separated, discrete splenic colonies
were carefully dissected and a single cell suspension was prepared. Cells from each
colony were processed for genomic DNA and RNA. Additional globin vector transduced
CFU-S, originally derived in experiments previously reported{Hanawa, 2004 990 /id},
were obtained by transplanting BM cells from primary recipients that received transplants
of globin vector transduced -thalassemic cells, into secondary recipients.
The
microarray and qRT-PCR data in this report generated from those samples have not been
previously published.
1
Measuring transcript levels using Affymetrix microarrays
Gene Expression measurements for over 15,000 mouse transcripts were generated using
Affymetrix MOE-430A GeneChip arrays (Affymetrix, Santa Clara, CA) according to the
standard procedures of the Clinical Applications Core Technology Laborartory in the
Hartwell Center for Bioinformatics and Biotechnology (St. Jude Children’s Research
Hospital, Memphis, TN). RNA samples were processed using the Affymetrix small
sample version 2.0 protocol. First round cDNA synthesis was initiated using 200 ng of
total RNA annealed to a T7-oligo(dT) primer. After completion of second strand
synthesis (SuperScript II cDNA kit, Invitrogen, Carlsbad, CA), the purified cDNA was
used as template to generate cRNA (MEGAscript T7 in vitro transcription kit, Ambion,
Austin, TX). A second round of cDNA synthesis was initiated from 400 ng of cRNA
primed with random hexamers. The second round cDNA was used as template to
generate biotin-labeled cRNA using the T7 RNA polymerase Bioarray high-yield kit
(ENZO Diagnostics, Inc., Farmingdale, NY). The labeled cRNA (10 g) was fragmented,
added to a hybridization cocktail containing probe array controls and blocking agents,
and incubated overnight to a GeneChip at 45° C. Arrays were washed and stained with
streptavidin phycoerythrin (Invitrogen, Carlsbad, CA) using a GeneChip Fluidics Station
400, then scanned using the Affymetrix GeneChip Scanner 3000. Expression signals
were calculated using the Affymetrix GCOS software (version 1.2). Global scaling of
signals was applied to all arrays. The 2% trimmed mean signal was set to 500. Detection
calls for probesets were determined using the default settings in the GCOS software. The
microarray data will be made publicly available through a web site of the Hartwell Center
2
for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital
(Memphis, TN).
Statistical Analysis
We chose to evaluate the probesets contained within a 600 kb window, specifically 300
kb on either side of the vector insertion in a specific clone, for changes in signal values,
relative to both mock-transduced cells and to all other unrelated, transduced clones
containing different vector insertions. All signals were converted to log base 2 scale and
the value of each probeset for an individual vector-transduced clone was compared to the
distribution of those signal values in the mock-transduced clones and to that in the group
of unrelated, transduced clones. For gene activation events to be considered significant, a
present call for the probeset in the affected clone was required and the value had to be
increased at least 2-fold with statistical significance (p<0.05, see below), as compared to
both mock-transduced and unrelated, transduced clones. For decreased expression to be
considered significant, the same criteria were used with the additional requirement that
80% of mock-transduced clones had to have a present call.
Statistical significance was determined by the P value of the comparison of
the
expression level of a transcript near an insertion site to the expression level in the mock
samples or unrelated, transduced samples via the Student t-distribution as follows: Let X
denote the log-transformed expression level of the transcript near an insertion site, n
denote the number of mock samples or unrelated, transduced clone samples, A denote the
signal value of this transcript over the averaged log-transformed expression of the same
3
transcript in the mock samples, and S denote the standard deviation of log-transformed
expression among the mock samples. The t-statistic was computed as T = (A-X) / S. The
P value is then the two-sided tail probability beyond |T| and -|T| on the Student tdistribution with n-1 degrees of freedom. The expression of a gene was regarded
significantly affected by the insertion if at least one of the probesets (transcripts) for the
gene was significantly altered as compared to both the mock and the unrelated clones.
Changes in the signals of probesets representing transcripts fulfilling these requirements
were verified using quantitative real-time PCR (qRT-PCR) as described above.
As a further test to determine whether or not the observed effects on gene expression by
vector insertions in the transduced clones could be due to chance (completely random
variation), we generated 2000 Monte Carlo simulations of random insertions for both the
panel of 18 Globin clones and the 20 GFP clones. To generate a set of random insertion
locations, all the mouse chromosomes were concatenated into one long genome sequence
and a random position was selected for each clone. Affymetrix probesets falling within
300 kb on either side of the random positions were identified for evaluation of potential
changes, relative to the mock and transduced, unrelated clones. The t-statistic and P
value was calculated for each evaluation as above for the true insertions. If the random
position fell within the same chromosome as the true insertion for the particular clone, it
was rejected in order to exclude the possibility that effects from the true insertion might
influence the data from the randomly generated insertion. In the few cases where a clone
contained more than one insertion on different chromosomes, all chromosomes with the
mapped insertions were excluded from the randomization choices.
4
This process was
repeated 2000 times. The overall proportion of significantly affected genes and the
proportion of clones containing a random insertion that significantly affected at least one
gene in each simulation run were then compared with those proportions observed in the
original data to calculate statistical significance. The P value of a proportion calculated
from the original data was determined by the number of times that this proportion was
less than or equal to the same proportion calculated from simulated random insertions
divided by 2000.
5