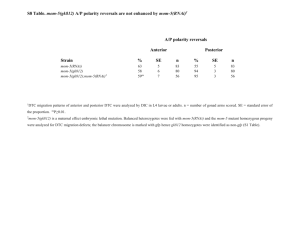
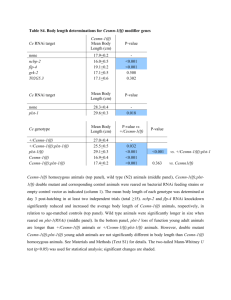
Complementation of lethal mutations
advertisement

Fly Genetics (fall 2011) Pat O’Farrell ofarrell@cgl.ucsf.edu - 6-4707 Lecture 3 How genes function in space and time & Intro to the “Genetic tool box” Genetics concepts new to this section: P-element tranformation – the fly version of transformation. The GAL4/UAS two component expression system. Using FLP mediated site specific transformation at FRTs – clonal lose and gain of function. Positive marking of clones. RNAi – the fly version Developmental Principle: Cells whose fate is determined produce clones that are limited to specific tissues/areas. Reciprocally, the distribution of daughter cells reveals the developmental potential of an earlier cell. General reading: This is very lucid description of key developmental concepts highlighted by clonal analysis in Drosophila. The pdf is on line and it is recommended reading. Crick FH, Lawrence PA. (1975) Compartments and polyclones in insect development. Science 189, 340-7 Development of P element mediated transformation: Rubin GM and Spradling AC (1982) Genetic Transformation of Drosophila with Transposable Element Vectors. Science 218, 348-353 The single hop insertational mutagenesis strategy: Cooley, L., Kelley, R., and Spradling A. (1988) Insertional mutatgenesis of the Drosophila genome with single P elements. Science 239, 1121- 8 This now dated review still provides a background on many of the diverse tools that have been developed on the basis of regulated expression of transgenes by the GAL4 transcription factor. Duffy, JB (2002) GAL4 System in Drosophila: A Fly Geneticist;s Swiss Army Knife. Genesis 34, 115 A technical paper that gives an excellent description of the process of producing positively marked clones, which can also be used for ectopic expression of genes in specific cells and for RNAi knockdown in specific cells. Wu, JS and Luo L (2006) A protocol for mosaic analysis with a repressible cell marker (MARCM) in Drosophila. Nature Protocols 1, 2583 Hybrid dysgenesis/Some strange biology http://www.math.princeton.edu/~jgevertz/public_html_Pelement/indexpage.html http://engels.genetics.wisc.edu/Pelements/Pt.html For a few decades bizarre phenomena were reported when laboratory Drosophila melanogaster were crossed to flies from the wild. If wild male flies were crossed to laboratory females, the progeny had shriveled gonads, reduced fertility, significant recombination in males and increased mutation. Infection by Transposition Element Remarkable detective work led to the realization that the wild flies where carrying a transposition element and that laboratory flies had been sheltered from a world wide sweep of transmission of this transposition element. The dysgenesis was a reflection of the infection strategies of the element. Transposition elements: Pieces of DNA equipped with mechanisms that lead to their movement from one DNA sequence to another. They are considered selfish pieces of DNA that parasitize other replicating molecules. There are three modes of transposition, conservative (or cut and paste), replicative and retrotransposition. The P element uses cut-andpaste. The P element encodes an enzyme called transposase that cuts out P elements by recognizing a 31-bp inverted terminal repeat sequence at the ends of the element. The excision of the Pelement leaves a double strand break that needs to be repaired. If excision occurs in G2, the break is likely to be repaired from the sister, which still has its copy of the P element. Repair using the homologous sequences on the sister restores the original P element by gene conversion and leaves the excised copy to find a new home. The transposase cuts a target sequence with a staggered cut and end to end joining inserts the P and fill-in of the staggers creates a short direct repeat at the site of insertion. Regulation – the transposase includes an intron whose excision is suppressed in somatic cells so that transposition is effectively limited to the germline. It is the high level germline transposition activity that is thought to contribute to dysgenesis. However, dysgenesis is down regulated once the P has successfully taken up residence in a strain. This appears to be due to repression of transposase by a product that accumulates in the infested strains. The repressor appears to be generated as the result of an entirely different category of RNA mechanism called PIWI interacting RNA (or piRNA) but there may also be an involvement of RNAi. P element as a Transgenesis Tool Transposase source: To control transposition the transposase that catalyzes the event has been separated from the transposing sequences. A mutated derivative of a P element inserted at 99B can no longer transpose acts as simple stable transposase source. It is called delta 2-3 in reference to it lack of an intron. Importantly, it can be followed by its association a dominant third chromosome marker, Kinked (Ki). P element vectors: Plasmids that replicate in E. coli (thin line) but with the Cis sites for transposition — 31 bp sequences defining the two ends of transposing sequence bracketing a marker gene detectable in fly plus other useful stuff. Generally, the marker is a w+ gene called mini-white or just white (w). It is a streamlined version of the endogenous white gene. It behaves like a weak white gene complementing w- mutants. The intensity of eye color (orange to red) is a convenient indicator of the number of P elements in a strain. Special vectors give great flexibility. Insertion of DNA: Injection of w+ marked P element constructs into embryos with 2-3 results in transposition from the plasmid to the genome. This occurs in a few cells, so the flies arising from these injected embryos, called G0 flies, are usually still white eyed. However, if the marked P element inserts into a germ cell precursor, some of the progeny will carry the insert and have red eyes (w+). Select w+ progeny that lack Ki — these will lack the chromosome with the transposase (2-3) so that the newly transposed element will be stable. Cross w+ progeny to Balancers to find out what chromosome it is on. There are many applications. For example, the upstream regulatory region of gene with tissue specific, or position specific pattern of expression can be positioned up stream of a reporter such as GFP and the regulatory capacity of the upstream sequences assayed. Mobilizing a P – to mutagenize/trap etc. It is useful to hop insertional elements all over the genome. For this, use a chromosome that has any dominant marker (Dom) and the [w+] P element you want to hop (ammunition chromosome). Cross w-; Dom [w+] P flies to w-; 2-3 Ki flies and select males that are w-; Dom [w+] P; 2-3 Ki — transposition occurs in these males. Cross these to any w- stock and search for progeny flies with reddish eyes (w+) that lack Dom and lack Ki. Since there is no recombination in males, Dom and [ w+] P will not separate and Ki and 2-3 will not separate. Thus, the selected progeny will lack the ammunition chromosome and the w+ must have relocalized/transposed. Furthermore, the selected flies will lack transposase (2-3) so the new insertions are stable. P element mutagenesis – a gene knockout collection: Insertional gives mutations that can quickly be characterized molecularly. Elements are mobilized as above and if they insert in a gene they will inactivate the gene partially or completely. Several labs worked together to knock out all of the genes in the fly. They mobilized P elements or other transposition elements (e.g. minos. Hops were mapped to the genome sequence by inverse PCR and instertions that interrupted coding sequences are held in a collection available to everyone. Most fly genes have been knocked out or at least tagged with a nearby insertion element. Genes and insertions in 100 kb of genome (see FlyBase) . Enhancer trapping: When a reporter gene is under the control of a minimal promoter, it is usually inactive, but if the element inserts near endogenous enhancers, it “traps” them, so that they direct expression of the reporter. Many of the endogenous enhancers have refined developmental programs of expression, and the reporter now recapitulates these programs. This reveals the programs and provides fantastic markers for cells, tissues etc, which can be used in additional genetic analyses – e.g. screens for alterations in the development of a cell or tissue type. An enhancer trap marks dentridritic projections of specific sensory cells in the epidermis of the larvae (right). The image to the left shows the projections of one cell. Two component systems e.g. The GAL4 System The GAL4 transcription factor of S. cerevisiae works in other species where it will specifically induce transcription of promoters associated with the appropriate enhancer UASGAL4. When used in flies UASGAL4 is usually referred to simply as UAS. If a gene is under the control of a UAS associated minimal promoter, it can be introduced into Drosophila on a P element, but it usually will not be expressed in the absence of GAL4. If a second strain is made that carries a P element expressing GAL4, and the two are crossed together, the UAS associated promoter will be activated. GAL4 Drivers: P elements in which GAL4 was expressed from a minimal promoter were constructed. When introduced into flies these often were not expressed, but just as described for enhancer trapping, if these elements are mobilized by 2-3 expression, they will move to new sites and at some of these sites they will trap enhancers. A cross to a UAS-GFP line (responder/reporter) will reveal cases in which GAL4 is now expressed and will also reveal the spatial and temporal pattern of the expression. Lines that express GAL4 in patterns are called drivers, because they can drive any UAS constructs to be expressed in the same pattern. As we will see this has allowed targeted gene expression in specific cells, targeted recombination, and targeted loss of gene function. Thousands of GAL4 drivers have been made, either by the described trapping procedure or by an alternative process in which known enhancers are placed upstream of GAL4 to design a driver with known behavior. This bipartite GAL4/UAS expression system has proved extraordinarily powerful. Adding temporal control to the GAL4 system: Several strategies have been created to control the timing of activation of UAS transgenes, but the most flexible is based on GAL80. GAL80 binds to GAL4 and inhibits transcriptional activation. Its widespread expression using the tubulin promoter blocks GAL4 dependent transcription. One of its uses stems from the availability of ts allele of GAL80. In a three element system in which a GAL4 dependent transcription of a UAS transgene is restrained due to the presence of a PtubGAL80ts, expression will be extinguished at low temperatures (18 -23 C) where the GAL80ts is active and will induced at high temperatures where GAL80 repression is inactivated. This allows control of gene expression in space (using the GAL4 driver specificity) and time (using temperature shifts) to control expression of transgenes. FLP/FRT System The yeast FLP recombinase acts at FRT sites to induce recombination. This activity has been put to great use in flies. FLP based mitotic recombination to produce clones: There were numerous disadvantages of using X-rays to induce mitotic recombination. For example, there is significant cell lethality due to X-rays, the position of the induced recombination event is random and frequency is low. Now days, recombination is induced by FLP. Chromosomes have been produced with FRT sites inserted at the base of each major chromosome arm (near the centromere). The figure shows an example in which an FRT (blue box) is at the 'base" of the X chromosome and FLP mediated recombination produces a twin spot with two marked cells – one marked with body color marker yellow (y) and one marked with a twisted bristle marker singed (sn). This is used to make marked clones, but it can also be used in mutant screens. Numerous genes that are required for development of the eye are also required for earlier processes such as embryogenesis. How can you genetically dissect the involvement of such genes in eye development? One solution is to make clones of the mutant later in development. If the mutation is not “a cell lethal mutation” you should be able to get viable cells. If the gene were absolutely required for cell division, how big would your clones be? If you get clones you can examine the phenotype in a tissue. To make this more directed, you could express FLP specifically in the eye, so that clones would be produced in this location. How would you do this? Can you explain how you might use the system you worked out to do a screen for new mutations that are required for proper development of the eye? Note this is what an eye looks like if the fly carries a eyelessGAL4 (eye specific), a UAS-FLP, heterozygous for w+ insert on II R, and has an FRT at base of II R. In this case there is no mutation on II R that is required for development of the eye. A Little about the fly eye The following is a cool site with a beautiful animation illustrating the cellular anatomy of the fly eye, or more precisely the anatomy of an ommatidium – one of the 800 units that comprise the compound eye of a fly. http://www.sdbonline.org/fly/vdevlhom/movie.htm Because each ommatidia is a sophisticated multicellular structure and 800 of these units are precisely arrayed in the normal eye, it constitutes a remarkably sensitive reporter for accurate developmental and cell biological performance. The level of integrity of eye structure allows grading scoring of defects from extremely subtle to incredibly severe. LEFT: Example of analysis of essential genes in eye clones. Using eyeless FLP, clones are produced specifically in the eye. P, Q and R show pairs of images – the left is a light micrograph in which the sister clones of mitotic recombination (induced by FLP) are marked red (w+) or white (w-). The image on the left is a scanning electron micrograph that shows the lenses of the individual ommatidia and the inter-ommadidial hair. In P and P` the white tissue is wild type, in Q and Q` it is mutant for mitochondrial enzyme (cytochrome oxidase). Clearly the mutation messes up development of ommatidia, but the mutant cells largely survive. Not shown here are data suggesting that the mitochondrial defect induces a "checkpoint" that arrests the cell cycle by promoting p53 dependent destruction of cyclin E (key cell cycle protein). R and R` show that when the fly has only one copy of one of the key subunits of the proteosome (protein degradation system hypothesized to degrad cyclin E) the phenotype of the cytochrome oxidase loss is partially suppressed. . Making the eye homozygous for one chromosome arm e.g. 2L Wt FRT GRM-hid FRT GMR-hid/FRT + EGUF/+ FRT GMR-hid CL/FRT +EGUF GMR = a promoter that drives expression late in eye development hid – a gene whose expression induces an apoptotic program Here GMR is directly driving hid expression and the construct is on 2L, and an FRT is on the base of the same chromosome arm. B) When hid expression is driven by GMR almost all eye cells are produced but then die EGUF = shorthand for eyeless-GAL4, UAS-FLP eyeless is a promoter that drives early and continuous expression in the eye precursor cells. The EGUF constructs are not on chromosome 2 C) In this eye there is an FRT on both maternal and paternal 2Ls and recombination promoted by EGUF produces two populations of cells – ones with and ones without GMR-hid. When development reaches the late stages of eye development – half of the cells commit apoptosis when they express hid. Thus the eye is reduced. CL = a cell lethal mutation (also on 2L along with GMR-hid). Note that this is recessive lethal, and the cells will only die when they become homozygous. D) Early expression of FLP begins to produce homozygous cells early during development. The cells homozygous for CL die and surviving cells compensate by increased growth and proliferation. The population of heterozygous cells declines as the cells proliferate, because with each cycle, FLP has a chance (~50%) of converting the cells to homozygosity. The CL/CL cells will die and be replaced. The "w.t." cells will accumulate. Finally, late in development GMR-hid turns on and kills the few remaining heterozygous cells, and the cells that are homozygous for the chromosome arm lacking GMR-hid, and CL go on to produce a normal eye. This eye is now homozygous for one arm of one chromosome. So what? Well if the chromosome had been mutagenized and carried a mutant, it would be homozygous just in the eye. Phenotypes could be scored, and because you don't need two flies to bread a homozygote, the screen can be done with single flies (mass breading, no vials and one less generation – work it out – it makes things easier and allows screening for a new category of mutants). SEE - Stowers RS, et al. (2002) Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 36(6):1063-77 Flip out cassette – Clonal expression FLP is also useful in different strategy in which one induces clonal expression and/or loss of expression of transgene. FLP mediated recombination between tandem FRT elements excises the intervening DNA and leaves single FRT element. In the design illustrated yellow, y, includes transcriptional termination signals and is bracketed by FRTs. FLP expression induces excision of y, simultaneously allowing expression of X. If X expression is compatible with cell survival, you will see y marked clones in the adult that also are expressing X. Positively Marked Clones Genetic markers work well but cannot be followed immediately after making a clone, preventing analysis of the early consequence of change in gene function. For this molecular markers. such as GFP, are useful. However, while recessive genetic markers are only detected in the homozygous cells of the clone, GFP is detected in the heterozygous cells. Consequently, the usual way of setting up clones with molecular markers is to follow the loss of the marker in the clone. This makes it very difficult to follow small clones in a sea of positive signal. The GAL80 repressor provides a solution. A cell that loses GAL80 can now express GAL4 dependent genes and thus can be marked by expression of a UAS-GFP (see diagram). This is called the MARCM technique for “Mosaic Analysis with a Repressible Cell Marker” It is powerful enough to mark even single cells. It is flexible because the marked cell will also express any other UAS gene that is present. RNAi in flies Long dsRNA Flies works Worms works Cell autonomy autonomous Transitive/* intransitive Amplification Intransitive * Spreads throughout (except nervous system) Transitive Apparently not Amplifies signal Mammals Doesn’t work: induces interferon response that nonspecifically shuts down translation (viral defense strategy) ? ? ? * Transitive: If you make dsRNA complementary to the right half of transcript, the biological response will produce RNAi against both the left and right half of the transcript. This is apparently due to RNA "replicase" (RNA dependent RNA polymerase) that can extend RNA primers to produce new regions of dsRNA and hence program RISC complexes with RNA from all contiguous sequences. Intransitive: You only get RNAi to dsRNA sequences added. Importance: If you use dsRNA representing the nonconserved 3` untranslated region of the actin 5C gene, RNAi against the conserved part of the gene will not be produced and the treatment will not target the other 4 highly similar Actin genes. RNAi in cell culture Cell culture provides advantages for RNAi screening and RNAi in Drosophila cell culture is uniquely simple. Advantages of Drosophila S2 cells: 1. Take up long dsRNA directly from media 2. Because long dsRNA works, library construction is simple – robotic PCR from genomic DNA, followed by symmetric PCR to dsRNAs of about 500bp to all of the genes of the genome. 3. The whole genome can be screened in a time frame of 2 weeks to 2 months. 4. RNAi in S2 cells also provides a terrific system for experimental dissection. One, two, three or more dsRNAs can be used to knockout combinations of genes, and film cellular phenotypes (e.g. cytokinesis defect) can be filmed in real time using GFP tagged markers. Echard, A., Hickson, GRX, Foley, E., and O’Farrell, PH. (2004) Terminal Cytokinesis Events Uncovered after an RNAi Screen. Curr. Biol. 14(18):1685-93 Foldback transgenes http://www.vdrc.at/rnai-library/ Two libraries –1. The GD Library: 2. The second library has inserts (10,714) in a common site. Similar except a new transgenic technology that uses a phage site specific recombinase (phiC31) to insert RNAi constructs at specific docking sites. The benefit is that the context effect on gene expression is removed. Disadvantage – can’t be combined in large numbers because they are allelic. In addition to VDRC, there are two other large libraries of RNAi constructs – one in Boston and one in Kyoto. Boston also provides reagents and technical help for cell based RNAi screens RNAi knockdowns – in space and time Since RNAi transgenes are under the control of UAS, they can be expressed ubiquitously or in any tissue with the appropriate driver and they can be regulated by GAL80ts for temporal control, or by GAL80 in the MARCM system to specifically inactivate the gene in individual marked cells. Using gene dose, and temperature the severity of knockdown can be adjusted. RNAi by injection dsRNA can be injected into the early embryo and maternal RNAs can be eliminated or accumulation of zygotic product blocked – maternally provided proteins are of course immune to this. Triple RNAi of the three mitotic cyclins of Drosophila by dsRNA injection into the anterior (left) pole blocks subsequent mitoses and the domain of action expands with time to halt successive waves of mitosis. McCleland ML and O’Farrell, PH (2008) RNAi of Mitotic Cyclins in Drosophila Uncouples the Nuclear and Centrosome Cycle. Curr Biol. 18(4):245-54 There are three large banks of RNAi stocks – Vienna (VDRC), Boston (DRSC) and Japan (NIG-FLY).