Experiment detail for qPCR in this study (doc 78K)
advertisement
")
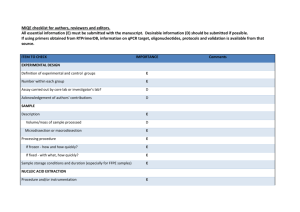
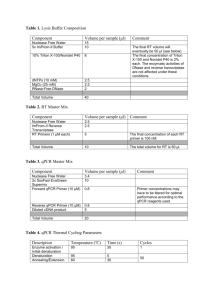
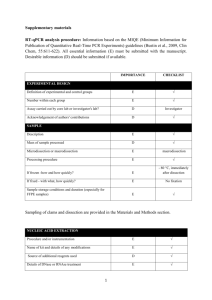
Experiment detail for qPCR in this study Sample acquisition, handling, and preparation All breast and liver tissue samples were collected by the University of Chicago Tissue Core Facility. According to the record of the facility, all samples were frozen at -80℃ immediately after surgery and excluded the possibility of tumor by microscopical examination. Before processing, the sample was embedded in O.C.T. compound (Ted Pella, Redding, CA) and cut into 20μm slice in a cryostat. RNA extraction and quality control RNA was extracted by RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA) according to manufacturer’s recommendation (http://www.qiagen.com/literature/render.aspx?id=170). The quantification and A260/A280 is determined by NanoDrop 1000 (Thermo Scientific, Wilmington, DE). All sample presented an A260/A280 ratio varying from 1.98-2.01, thus indicating that the purity of our RNA sample was rather high. The RNA sample is not subjected to DNase treatment. The integrity of RNA is examined by agrose gel electrophoresis. Reverse transcription cDNA for breast and liver samples was synthesized by using the High Capacity Reverse Transcription Kit (Applied biosystems, Foster City, CA) according to manufacturer’s recommendation. In detail, the reaction mix was setup as following: 10×RT buffer 25×dNTP Mix (100 mM) 10×RT Random Primers MultiScrib Reverse Transcriptase RNase Inhibitor RNA Nuclease-free H2O Total volume Volume (μL) 2.0 0.8 2.0 1.0 1.0 2μg ~ 20 The following thermal condition was used: 25℃ for 10min, 37℃ for 120min, 85℃ for 5sec. The obtained cDNA was stored in -80℃ before using. qPCR assay design The primer for qPCR and related information is listed below. For each gene, the two primers were located in different exons in order to avoid genomic DNA (gDNA) contamination. The secondary structure is determined by mfold (http://mfold.bioinfo.rpi.edu/cgi-bin/dna-form1.cgi) and available on request. The primer specificity was evaluated by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and all primers were unique in human genome. All PCR products were also sequenced to confirm the primer specificity. All qPCR assays presented a single and sharp peak in melt curve. Gene referencea Forward primer (5’-3’) location Reverse primer (5’-3’) location UGT2B4 NM_021139 AACCTGCCAAACCCCTACCG 895-914 TCCACTTGTACAGCCGAGTATTGAGT 1091-1116 UGT2B7 b NM_001074 TGCTTTACTTTGACTTTTGGTTCG 696-719 CCAGGAGTTTCGAATAAGCCATAC 809-832 UGT2B10 NM_001075 ACAGCTTCAGTCCTGGCTACT 537-557 AAGCCATATGTCAGCTTTCCT 773-793 UGT2B15 NM_001076 TGAAGTCAGTCATTAATGACCCTG 1327-1350 CTCGATCCAGGGGCTTCAT 1401-1419 UGT2B17 NM_001077 TATTTTGACTTTTGGTTTCAAGC 701-723 CTCTTCCATTTCCTTAGGCAA 908-928 ß-actin c NG_007992 ACGTGGACATCCGCAAAGAC 2472-2491 CAAGAAAGGGTGTAACGCAACTA 2869-2891 K03432 GTAACCCGTTGAACCCCATT 1718-1737 CCATCCAATCGGTAGTAGCG 1849-1868 18S d aGenbank ID provided. reference 1. cfrom reference 2. d from reference 3. bfrom qPCR setup The qPCR was performed with power SYBR green (AB#4367659, Applied Biosystems) and the exact chemical composition is available in vendor website (https://products.appliedbiosystems.com/ab/en/US/adirect/ab?cmd=catNavigate2&cat ID=602649). The reaction was setup as following: Power SYBR green master mix Forward primer (10μM) Reverse primer (10μM) water cDNA Total volume 10μL 1μL 1μL 3μL 5μL (10 ng) 20μL All qPCR reaction is performed in optical plate (AB#4346906, Applied Biosystems) and sealed with optical adhesive film (AB#4311971, Applied Biosystems). The annealing and extension temperature was fixed at 60℃ according to manufacturer’s recommendation and thermal condition was: 95℃ for 10min; 95℃ for 15sec and 60℃ for 1min, 40cycles. All qPCR were performed in triplicate on a StepOne Plus Real time PCR System (Applied Biosystems). The gDNA contamination was evaluated by qPCR as described in our recent report 4. The result indicated that gDNA contamination is rather low (<5% for most cases). Moreover, since our qPCR primer is in different exons, the gDNA contamination was not likely to influence qPCR result. qPCR assay performance A cDNA mix was 1:5 serial diluted as calibration curve. In each reaction, 100, 20, 4, 0.8, 0.16, 0.032ng cDNA was used and the linear dynamic range covered all sites (all r2>0.99). All unknown samples were within the linear range. The slope and intercept for calibration curve were provided by StepOne software (Applied Biosystems). The PCR efficiency was calculated with the formula 10-1/slope-1 by StepOne software and listed in below table. Gene UGT2B4 UGT2B7 UGT2B10 UGT2B15 UGT2B17 actin 18S aMean Efficiency (mean±standard deviation)a 98.5±4.4% 95.2±3.6% 100.4±2.6% 111.9±3.2% 99.2±6.1% 95.6±1.9% 102.2±2.4% and standard deviation calculated from the replicates. In above table, an efficiency value beyond 100% was observed for multiple genes. Although it is difficult to be interpreted by PCR theory, it is not uncommon in qPCR practice 5-7. In each plate, a non-reverse transcribed RNA sample and H2O sample were included and both of them were not amplified. Data analysis The copy number for each sample was calculated by StepOne software and outputted into spreadsheet. The mean and standard deviation (SD) for each sample was calculated by SPSS 15.0 (SPSS Inc., Chicago, IL). Coefficient of variation (CV) is determined by the formula SD/mean. All samples presented a CV<20%, thus indicating that our result was highly repeatable. In a preliminary experiment, the expression of ß-actin, 18S, GAPDH (primer pair 5’-GAAGGTGAAGGTCGGAGTC-3’ and 5’-GAAGATGGTGATGGGATTTC-3’) 8, and U6 RNA (primer pair 5’-CTCGCTTCGGCAGCACA-3’ and 5’-AACGCTTCACGAATTTGCGT-3’) 9 was evaluated in both breast and liver samples. β-actin and 18S were chosen as reference gene for breast and liver, respectively, due to their conserved expression in these two tissues (result not shown). The final mRNA level is expressed as the ratio between target gene of interest and ß-actin or 18S. After log-transform, all genes were in normal distribution and used for further analysis. Reference 1. Sun C, Di Rienzo A. UGT2B7 is not expressed in normal breast. Breast Cancer Res Treat 2009; 117: 225–226. 2. Liu W, Innocenti F, Wu MH, Desai AA, Dolan ME, Cook EH, Jr., et al. A functional common polymorphism in a Sp1 recognition site of the epidermal growth factor receptor gene promoter. Cancer Res 2005; 65: 46–53. 3. Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 2000; 46: 69–81. 4. Sun C, Southard C, Witonsky DB, Olopade O, Di Rienzo A. Allelic imbalance (AI) identifies novel tissue specific cis-regulatory variation for human UGT2B15. Hum Mutat 2010; 31: 99–107. 5. Steinrigl A, Revilla-Fernández S, Eichinger M, Koefer J, Winter P. Bluetongue virus RNA detection by RT-qPCR in blood samples of sheep vaccinated with a commercially available inactivated BTV-8 vaccine. Vaccine 2010; 28: 5573–5581. 6. Gutha LR, Casassa LF, Harbertson JF, Naidu RA. Modulation of flavonoid biosynthetic pathway genes and anthocyanins due to virus infection in grapevine (Vitis vinifera L.) leaves. BMC Plant Biology 2010; 10: 187. 7. Bernascone I, Janas S, Ikehata M, Trudu M, Corbelli A, Schaeffer C, et al. A transgenic mouse model for uromodulin-associated kidney diseases shows specific tubulo-interstitial damage, urinary concentrating defect and renal failure. Human Molecular Genetics 2010; 19: 2998–3010. 8. Gutala RV, Reddy PH. The use of real-time PCR analysis in a gene expression study of Alzheimer's disease post-mortem brains. J Neurosci Methods 2004; 132: 101–107. 9. Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R, et al. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem 2007; 282: 19575–19588.