Supplementary materials RT-qPCR analysis procedure: Information
advertisement

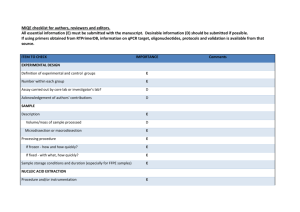
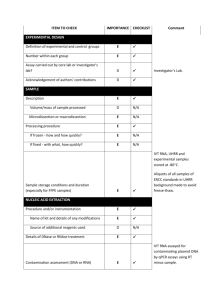
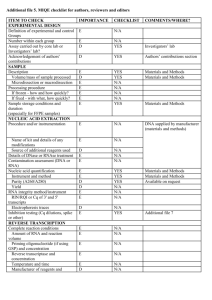
Supplementary materials RT-qPCR analysis procedure: Information based on the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines (Bustin et al., 2009, Clin Chem, 55:611-622). All essential information (E) must be submitted with the manuscript. Desirable information (D) should be submitted if available. IMPORTANCE CHECKLIST Definition of experimental and control groups E √ Number within each group E √ Assay carried out by core lab or investigator's lab? D Investigator Acknowledgement of authors' contributions D √ Description E √ Mass of sample processed D √ Microdissection or macrodissection E macrodissection Processing procedure E √ If frozen -how and how quickly? E - 80 °C, immediately after dissection If fixed - with what, how quickly? E No fixation Sample storage conditions and duration (especially for FFPE samples) E √ EXPERIMENTAL DESIGN SAMPLE Sampling of clams and dissection are provided in the Materials and Methods section. NUCLEIC NUCLEIC ACID EXTRACTION Procedure and/or instrumentation E √ Name of kit and details of any modifications E √ Source of additional reagents used D √ Details of DNase or RNAse treatment E √ 1 Contamination assessment (DNA or RNA) E √ Nucleic acid quantification E √ Instrument and method E √ Purity (A260/A280) D √ Yield D √ RNA integrity method/instrument E √ RIN/RQI or Cq of 3' and 5' transcripts E √ Electrophoresis traces D √ Inhibition testing (Cq dilutions, spike or other) E √ Total RNA was extracted using the Absolutely RNA Miniprep Kit (Agilent technologies) according to the manufacturer's instructions. A step of phenol:chloroform:isoamylic alcohol (25:24:1, Sigma) extraction was added. For removing traces of genomic DNA, RNA was treated with RNAse-free DNase (Agilent technologies) according to the manufacturer's instructions. RNA qualities and quantities were determined using a microplate spectrophotometer (Epoch, Biotek). The quality of RNA with a 260/280 ratio between of 1.92.1 and a 260/230 ratio of 1.8-2.2 was considered satisfactory for use in our study. Also, integrity of RNA from each extract was confirmed by inspecting the bands after electrophoresis of 1 µL RNA on an agarose gel. The extracted RNA was stored in Eppendorf tubes at -80°C until further use. Possible contaminations of the RNA from all samples were assessed with “no reverse transcription” by qPCR. Furthermore, a melting curve analysis was performed as standard in order to detect DNA contamination of the RNA which would be visible as further unspecific peak. REVERSE TRANSCRIPTION Complete reaction conditions E √ Amount of RNA and reaction volume E √ Priming oligonucleotide (if using GSP) and concentration E √ Reverse transcriptase and concentration E √ Temperature and time E √ Manufacturer of reagents and catalogue numbers D √ 2 Cqs with and without RT D √ Storage conditions of cDNA D √ Reverse transcription was performed with AffinityScript cDNA Synthesis Kit (Agilent technologies). cDNA was synthesized from DNase-treated total RNA according to the manufacturer's instructions. First-strand cDNA was synthesized from 5 µg total using 1 µL of random primers (0.1 µg/μL), 1 µL Oligo(dT) primer (0.5 μg/μL), 0,8 µL dNTPs (25 mM each), 2 µL of 10× AffinityScript RT buffer, 1 µL of AffinityScript Multiple Temperature RT, 0.5 µL RNase Block Ribonuclease Inhibitor (40 U/μL) and RNase free water in a final volume of 20 µL. The retro-transcription was performed by incubating the reactions during 60 min at 42°C. The cDNA was stored in Eppendorf tubes at -20°C until further analysis. For the performed qPCR studies, in “no reverse transcription control samples” (RNA not treated with reverse transcription enzyme) no amplification was detected. qPCR TARGET INFORMATION If multiplex, efficiency and LOD of each assay. E only singleplex Sequence accession number E √ Location of amplicon D √ Amplicon length E √ In silico specificity screen (BLAST, etc) E √ Pseudogenes, retropseudogenes or other homologs? D Sequence alignment D √ Secondary structure analysis of amplicon D √ Location of each primer by exon or intron (if applicable) E NA What splice variants are targeted? E NA Sequence accession numbers and amplicon lengths are listed in Table S2. The primer sets of each gene were analyzed with NCBI Blast to ensure specificity. 3 qPCR OLIGONUCLEOTIDES √ Primer sequences E RT Primer DB Identification Number D Probe sequences D Location and identity of any modifications E NA Manufacturer of oligonucleotides D √ Purification method D √ Primers purified by desalting (DLS) were purchased from Sigma-Aldrich. qPCR PROTOCOL Complete reaction conditions E √ Reaction volume and amount of cDNA/DNA E √ Primer, (probe), Mg++ and dNTP concentrations E √ Polymerase identity and concentration E √ Buffer/kit identity and manufacturer E √ Exact chemical constitution of the buffer D Additives (SYBR Green I, DMSO, etc.) E SYBR Green I Manufacturer of plates/tubes and catalog number D √ Complete thermocycling parameters E √ Reaction setup (manual/robotic) D √ Manufacturer of qPCR instrument E √ qPCR analyses were carried out in optical 96-well plates (ABgene, Thermo Fisher Scientific, USA) in a Mx3000P QPCR System (Stratagene, Agilent technologies). Each 20 µL reaction contained 1 µL of reverse-transcribed product template, 10 µL of 2x SYBR Green QPCR Master mix (Agilent technologies), 2 µL of the gene-specific primer pairs (at a final concentration of 300 nM for each primer) and 7 µL of H2O. Cycling parameters are specified in the Materials and Methods section. 4 qPCR VALIDATION Evidence of optimisation (from gradients) D no gradient Specificity (gel, sequence, melt, or digest) E √ For SYBR Green I, Cq of the NTC E √ Standard curves with slope and y-intercept E √ PCR efficiency calculated from slope E √ Confidence interval for PCR efficiency or standard error D r2 of standard curve E √ Linear dynamic range E √ Cq variation at lower limit E √ Confidence intervals throughout range D Evidence for limit of detection E √ If multiplex, efficiency and LOD of each assay. E only singleplex The specificity of the amplification products has been confirmed by size estimations on an agarose gel and by analyzing their melting curves. Serial 10-fold dilutions of cDNAs were used to calculate the standard curve and measure the amplification efficiency for each target and housekeeping genes (Table S2). DATA ANALYSIS qPCR analysis program (source, version) E √ Cq method determination E √ Outlier identification and disposition E √ Results of NTCs E √ Justification of number and choice of reference gene E only one, justified Description of normalisation method E √ Number and concordance of biological replicates D √ Number and stage (RT or qPCR) of technical replicates E √ Repeatability (intra-assay variation) E √ Reproducibility (inter-assay variation, %CV) D √ 5 Power analysis D Statistical methods for result significance E √ Software (source, version) E √ Cq or raw data submission using RDML D - qPCR analysis program: Stratagene software (Agilent technologies) Obtained data were analyzed using the comparative Ct (threshold cycle) method. Cq’s were determined by setting the threshold automatically Outliers for which Ct was equal or above that of the negative control (without DNA) were excluded Results of NTCs: no amplification products present thus no Ct Justification of choice of reference gene: displayed in Materials and Methods section Description of normalization method: endogenous reference gene: see Materials and Methods section Number and concordance of biological replicates: Five independent biological replicates were analyzed. Number and stage of technical replicates: two technical replicate reactions for each biological replicate Table S1 Physico-chemical parameters measured at investigated sites Control Z1 Z2 Z3 Temperature (°C) 16.0 17.9 17.2 18.1 Salinity (psu) 33.1 36.0 35.9 35.5 pH 8.82 8.57 8.49 8.46 6 Table S2 Specific primer pairs used in the quantitative PCR analysis of studied Ruditapes decussatus genes Gene name and function Mitochondrial metabolism cox1 16S rRNA Accession number Forward primer Reverse primer Amplicon length (pb) Efficiency of primer pairs DQ184830 AJ417846 ATATGGCATTCCCTCGT TGCAACGAGAGTTGTACTAAG CGTTACAGCGATGCAC ACATCGAGGTCGCAAA 302 357 1.90 1.86 Oxidative stress response sod AY377969 GTGGTTTGAAGCCAGG CAGCGTGAACGACAAG 258 1.92 Protein reparation and protection hsp70 EU380904 CTTCGGTGGTGGTACT CTTCGGCACTGCTTGA 245 1.92 Detoxification system mt AJ249687 CGTGTAATTGTATTGAGACTGG ACTTTGCAGCCTGAAC 124 1.90 Reference 18S rRNA EF105249 GAGCAATAACAGGTCTGTG GGCAGGGACGTAATCAA 210 1.94 cox1: cytochrome C oxidase subunit I; 16S rRNA: ribosomal RNA 16S; mt: metallothionein; sod: superoxide dismutase; hsp70: heat shock protein 70; 18S rRNA: ribosomal RNA 18S 7 Table S3 Bioaccumulation factor of trace metals in R. decussatus sampled from the Tunis lagoon and controls through surface sediments Sampling site Cd Pb Hg Cu Zn Control - 0.28 1.47 2.45 2.21 Z1 1.28 0.09 0.44 0.54 0.95 Z2 0.22 0.02 0.92 0.46 0.29 Z3 0.58 0.04 1.31 0.35 0.98 Table S4 Sorted rotated factor loading (pattern) of 19 variables on the principal factors Axes Factor 1 Factor 2 % Variance 72 % 13 % GST 0,927 -0,323 CAT 0,878 -0,353 MDA 0,826 0,141 AChE -0,940 -0,067 cox1 0,845 0,254 16S 0,841 0,188 sod 0,891 -0,272 hsp70 0,847 -0,420 mt 0,892 -0,225 [Cd] clam 0,821 0,548 [Pb] clam 0,666 0,615 [Hg] clam 0,907 0,338 [Cu] clam 0,815 0,177 [Zn] clam 0,920 0,255 [Cd] sed 0,870 -0,462 [Pb] sed 0,912 -0,362 [Hg] sed 0,480 0,698 [Cu] sed 0,958 0,109 [Zn] sed 0,844 -0,468 8