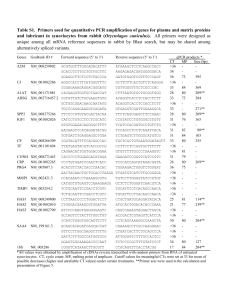
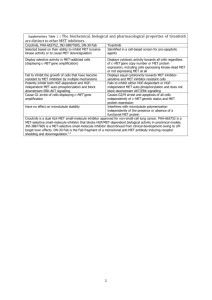
SCALE What is it? Why does it form? Types of scales: 1) Calcium
advertisement
 Calcium")
SCALE What is it? Why does it form? Types of scales: 1) Calcium/Magnesium carbonates - CaCO3 & MgCO3 - formed from the following occurrences: a) Change in formation or system temperatures and/or pressures: As the temperature increases, carbonate scaling tendencies increase. As pressure decreases, carbonate scaling tendencies increase. b) Decrease in solubility due to mixing incompatible waters: If one mixes two or more waters with a Total Dissolved Solids (TDS) difference of more than 20%, carbonate scale can occur. 2) Calcium sulfate - CaSO4 - formed from the following occurrences: a) Change in formation or system temperatures and/or pressures: As the temperature increases, Calcium sulfate scaling tendencies decrease up to —118 degrees F then increase from --118 to —136 degrees F. As pressure decreases, Calcium sulfate scaling tendencies increase. b) Decrease in solubility due to mixing incompatible waters: If one mixes two or more waters with a Total Dissolved Solids (TDS) difference of more than 20%, Calcium sulfate scale can occur. 3) Barium and Strontium sulfates - BaSO4 and SrSO4 respectively are formed from the following occurrences: a) Barium sulfate will generally occur whenever two or more waters are mixed with one of the water(s) containing greater than 2.0 milligrams per liter (mg/1) Barium and the other water(s) containing greater than 150-300 mg/I soluble sulfate. b) Strontium sulfate scales will generally occur based upon either an increase in water temperature and/or a decrease in water pressure as well as oversaturation when two different TDS waters are mixed. SCALE What is it? Why does it form? Types of scales: 1) Calcium/Magnesium carbonates - CaCO3 & MgCO3 - formed from the following occurrences: a) Change in formation or system temperatures and/or pressures: As the temperature increases, carbonate scaling tendencies increase. As pressure decreases, carbonate scaling tendencies increase. b) Decrease in solubility due to mixing incompatible waters: If one mixes two or more waters with a Total Dissolved Solids (TDS) difference of more than 20%, carbonate scale can occur. 2) Calcium sulfate - CaSO4 - formed from the following occurrences: a) Change in formation or system temperatures and/or pressures: As the temperature increases, Calcium sulfate scaling tendencies decrease up to —118 degrees F then increase from --118 to —136 degrees F. As pressure decreases, Calcium sulfate scaling tendencies increase. b) Decrease in solubility due to mixing incompatible waters: If one mixes two or more waters with a Total Dissolved Solids (TDS) difference of more than 20%, Calcium sulfate scale can occur. 3) Barium and Strontium sulfates - BaSO4 and SrSO4 respectively are formed from the following occurrences: a) Barium sulfate will generally occur whenever two or more waters are mixed with one of the water(s) containing greater than 2.0 milligrams per liter (mg/1) Barium and the other water(s) containing greater than 150-300 mg/I soluble sulfate. b) Strontium sulfate scales will generally occur based upon either an increase in water temperature and/or a decrease in water pressure as well as oversaturation when two different TDS waters are mixed. " THE KIT " CONTENTS: 1) XYLENE 2) 15% HYDROCHLORIC ACID 3) BAR MAGNET 4) MAGNIFYING GLASS 5) LEAD ACETATE STRIPS 6) 10% COPPER SULFATE SOLUTION IN 89% DISTILLED WATER + 1% # 2 7) POCKET KNIFE 8) BUTANE LIGHTER 9) POCKET THERMOMETER 10) PLASTIC BAGS 11) DIXIE CUPS 12) SURGICAL SAFETY GLOVES These tests must always be performed at a safe location---completely removed from any oil and gas location/pipeline. Follow the directions and safety precautions for each test. USES: 1) XYLENE- Take a small portion of the solids sample and add to a Dixie cup. Pour a small amount of xylene onto the sample and gently stir. If sample begins to dissolve turning the xylene from clear to brown or black, the solids have Hydrocarbons in them. One can then take a separate sample onto the knife blade and gently heat it with the butane lighter. If the solids sample begins to melt the solids are most probably a form of paraffin. * Safety note- Do not use the open flame butane lighter near the flammable xylene or any oil or gas location. Xylene fumes are hazardous and narcotic and should not be inhaled. 2) 15% HYDROCHLORIC ACID- Take a small portion of the solids sample and add to a Dixie cup. Pour a small amount of 15% Hydrochloric acid onto the sample and gently stir. If the sample begins to bubble gas and dissolve observe the color of the liquid acid in the cup. If the acid remains clear, the sample contains Calcium or Magnesium carbonate. If the liquid acid turns yellow or green, wet with water one end of a lead acetate strip and hold it above but not into the liquid in the Dixie cup. If the lead acetate strip turns brown/black or if a metallic shean is seen on the strip, Hydrogen sulfide is present. This indicates that the sample contains Iron sulfide. If the acid turns yellow and the lead acetate paper remains white take the bar magnet and touch it to the original dry sample, if the sample is magnetic, the solids contain Iron oxide, Mill scale and/or processed metal . If the acid turns yellow, the lead acetate paper remains white and the original dry sample is not magnetic, the solids contain Iron carbonate. *Safety note- 15% HCL acid gives off dangerous fumes and should not be inhaled. Any exposed skin or pipe surface should immediately be washed with lots of water and soap. 3) BAR MAGNET- When applied to a sample of Iron oxide, Mill scale and/or processed metal (steel) will show their magnetic properties. 4) MAGNIFYING GLASS- When used to look closely at a solids sample can easily identify irregular sand grains, pieces of man-made materials such as "0" rings and cotton gloves and/or hexagonal (six sided) Calcium sulfate crystals. 5) LEAD ACETATE STRIPS- When moistened are used to identify the presence of Hydrogen sulfide gas. Can be used for identification of Iron sulfide solids and other safety related issues. *Safety note- Lead acetate strips contain Lead and should not be placed in one's mouth. Lead is a hazardous metal and the strips should be disposed properly. Do not use and throw down on the ground. 6) 10% COPPER SULFATE SOLUTION- When applied to a corrosion coupon or any metal surface will immediately detect visually the absence of a coating and/or corrosion inhibitor. Turns unprotected metal bright copper color on contact. 7) POCKET KNIFE- If left in " The Kit " can be very useful for performing these tests and scrapping out a sample of solids from many locations. * Safety note- The knife provided in " The Kit " is sharp and can cause injury if improperly used. Do not use any metal object including the knife provided in " The Kit " in a manner that can cause a spark in or around any oil and gas location and/or pipeline. 8) BUTANE LIGHTER- Is used in testing solids samples for melting or burning tendencies. The butane lighter produces an open flame. Care should be excercised when using this instrument on any solids sample or around any flammable liquid and/or gas. 9) POCKET THERMOMETER- Can test the relative temperature of practically anything. 10) PLASTIC BAGS- Can contain solids samples for transport. 11) DIXIE CUPS- Nice, disposable test vessels for solids testing. 12) SURGICAL GLOVES- Used to protect users of " The Kit " from the hazardous chemicals contained and used therein. 13) COMMON SENSE- Not supplied in " The Kit " but necessary for use of same. Scale Coupons Most scale coupons sent to this Lab will be of mild steel. The dimensions are x 3", each containing three holes, in addition to the top hole which is used to secure the coupon to the coupon chuck when it is installed in a system. Upon receipt of a scale coupon it will be coated with oil, CaCO3, Iron Oxide, Iron Sulfide and various other materials. In order to determine which materials are present and in what amounts, the following steps should be followed: Scale Coupon Analysis Equipment needed: 1. Analytical balance 2. Beaker (150 ml or 250 ml) 3. Hot plate 4. Drying oven @-160 degrees F. 5. Desiccator 6. Brillo pads Reagents Needed: 1. 2. 3. 4. 5. 6. Toluene Distilled H2O Acetic acid diluted 1:4 with distilled H2O HC1 diluted 1:1 with distilled H2O Acid Inhibitor IPA Procedure: 1. 2. 3. 4. 5. 6. 7. Weigh beaker on balance and record weight. Place coupon in beaker and weigh together. Record Weight. Fill beaker with Toluene until coupon is completely submersed and heat gently on hot plate to a slow boil. Continue heating until all hydrocarbon materials are disolved in the Toluene. Decant liquid and repeat until Toluene remains light in color. Dry beaker and coupon in drying oven. Cool to room temperature in Desiccator and re-weigh and record weight. The weight difference are organic deposits. Fill beaker with 1:4 acetic acid. Heat gently on the hot plate to a slow boil. Continue heating until sample stops effervescencing, decant liquid, repeat if necessary. Dry beaker and coupon in drying oven. Cool to room temperature in Desiccator and re-weigh and record weight. The weight difference are Carbonate deposits. Fill beaker with 1:1 HC1 and acid inhibitor solution: (5-10% inhibitor in 1:1 HC1) to remove the iron compounds present. Heat gently on hot plate to a slow boil, decant liquid, repeat until acid solution remains light in color. Scale Analysis i Continued Page 2. To Determine if iron sulfide (FeS) is present place a damp strip of lead acetate paper over beaker when 1:1 HC1 solution is added. If the paper turns dark FeS is present, report as positive or negative. 8. Dry beaker and coupon in drying oven. Cool to room temperature in desiccator and re-weigh and record weight. The weight differences are iron compound deposits. 9. Remove coupon from beaker and scrub clean using brillo pad and water, rinse coupon in distilled water and damp dry. Place coupon in beaker of Isopropyl alcohol to remove any water remaining. Dry coupon and re-weigh, record weight. Calculations: 1. Weight of coupon with scale equals weight of beaker and coupon minus weight of beaker. 2. Total weight of scale equals weight of coupon with scale minus final weight of cleaned coupon. 3. mg/sq. in = mg scale factor 4. g/sq. in./year = (mg/sq. in) (10-3) (365) days in system 5. mpy = (weight loss of coupon) (factor) days in system 6. Composition of scale: Use the same calculations as are given in the scale analysis procedure. NACE Standard T1A0374-90 Item No. 53023 National Association of Corrosion Engineers Standard Test Method Laboratory Screening Tests to Determine the Ability of Scale Inhibitors to Prevent the Precipitation of Calcium Sulfate and Calcium Carbonate From Solution (For Oil and Gas Production Systems) The National Association of Corrosion Engineers (MACE) issues this standard in conformance with the best current technology regarding the specific subject. This standard represents a consensus of those individual members who have reviewed this document, its scope and provisions. It Is Intended to aid the manufacturer, the consumer, and the general public. Its acceptance does not In any respect preclude anyone, whether he has adopted the standard or not, from manufacturing, marketing, purchasing, or using products, processes or procedures not In conformance with this standard. Nothing contained in this NACE standard is to be construed as granting any right, by implication or otherwise, to manufacture, sell or use In connection with any method, apparatus, or product covered by Letters Patent, or as indemnifying or protecting anyone against liability for infringement of Letters Patent. This standard represents minimum requirements and should in no way be interpreted as a restriction on the use of better procedures or materials. Neither is this standard intended to apply in all cases relating to the subject Unpredictable circumstances may negate the usefulness of this standard in specific instances. NACE assumes no responsibility for the Interpretation or use of this standard by other parties and accepts responsibility for only those official NACE interpretations Issued by NACE In accordance with its governing procedures and policies which preclude the issuance of Interpretations by Individual volunteers. Users of this standard are responsible for reviewing appropriate health, safety, and regulatory documents and for determining their applicability in relation to this standard prior to its use. This NACE standard may not necessarily address aN safety problems and hazards associated with the use of materials, operations, and/or equipment detailed or referred to within this document. CAUTIONARY NOTICE: NACE standards are subject to periodic review, and may be revised or withdrawn at any time without prior notice. NACE requires that action be taken to reaffirm, revise, or withdraw this standard no later than two years from the date of Initial publication. The user is cautioned to obtain the latest edition. Purchasers of MACE standards may receive current Information on all standards and other NACE publications by contacting the NACE Publication Orders Department, P.O. Box 218340, Houston, Texas 77218 (telephone 713/492-0535). Approved November 1974 Revised January 1990 National Association of Corrosion Engineers P.O. Box 218340 Houston, Texas 7 218 713/492-0535 Copyright 1990, National Association of Corrosion Engineers T111011144110 Foreword Scale can be defined as an adherent deptatit of inorganic compounds precipitated from water onto surfaces. Meet oilfield waters are brines containing large amounts of calcium saes. When calcium is deposited as calcium carbonate or calcium sulfate. a lose of production and increased maintenance expenses can result; therefore, scale inhibition is of primary importance to the of producer. Scats inhibitors can be used in many circumstances to control scats formation, thereby reducing production difficulties. Inhibitors are commercially available and are widely used in of and gas production systems. T'he test methods in this standard are designed to provide a relative and quantitative measure of the abilities of inhibitors to prevent the precipitation of solids, a necessary and critical stage in the formation of scale deposits. The laboratory screening tests descridid in this standard cannot and do not allow for the wide variation in water chemistry and system properties seen in field operations. As such they must only be regarded as a starting point in the evaluation of scale inhibition products. The existence and use of these methods allow for a uniform mode of collection of screening test results and facilitates discussion of the results by interested parties. This standard, issued by NACE Group Committee T-1 on Corrosion Control in Petroleum Production. was originally prepared by Task Group T-10-9 and was revised by Task Group T-1D-31, a component of Unit Committee T-1D on Control of Meld Conosion by Chemical Treatment. The members of Task Group T-1D-31 are consumers and producers of scale inhibitors and other interested parties, who use NACE Standard TM0374 (latest revision) or a modification of this standard on a regular basis. The test methods in this standard have been selected by Unit Committee T-10 as a means of comparing, under the specified laboratory conditions, the effectiveness of inhibitors in preverang precipitation of calcium sulfate and calcium carbonate from solution. As the prices of such products change with time and may be unkritem to the tester, no attempt has been made to dilute the inhibitor to a common cost base. Section 1: General 1.1 The test methods described in this standard are static laboratory screening tests designed to give a measure of the ability of inhibitors to prevent the precipitation of calcium carbonate and calcium sulfate from solution at 160°F (71°C). deemed to be outside the scope of this standard. However, field conditions. field brine composition and other variables noted above should be considered at some point in inhibitor evaluation prior to final inhibitor selection for field use. 1.2 These test methods we recommended only for ranking the performance of different chemicals under laboratory conditions set by these methods. They we not intended to provide actual field treating rates. 1.4 Tests should be conducted at various inhibitor concerti*** in order to obtain a better comparison of inhibitors under labor elory conditions set by these methods. The inhibitor concentration mauled for a field applications fa* to be different than That debar-tithed under these laboratory conditions. 1.3 Many factors, such as reaction kinetics, fluid velocity and composition. variable temperatures and pressures, scale adherence and solids dispersion can significantly affect actual scale deposition under field conditions. Detailed consideration of these parameters is 1.5 This standard lists the necessary apparatuses, reagents. and procedures for conducting screening tests. Section 2: Calcium Sulfate Precipitation Test 2.1 This section lists the apparatus and procedure for conducting the calcium sulfate precipitation screening test. 2.2 Apparatuses and solutions 2.2.1 Constant temperature water bath or forced draft oven with the capability of maintaining the specified temperature within `2°F (1°C). 2.2.2 Clean and dust-free glass test cells (4 oz [approximately 125 mL) bottles with a positive sea). 2.2.3 Synthetic brine prepared with distilled or deionized water, as follows: 2.2.3.1 Calcium-containing brine: 7.50 g/L. NaCI (ACS (American Chemical Society, Washington, D.C.] Reagent grade); 11.10 g/L CaCl2 - 2H20 (ACS Reagent grade). 2.2.3.2 Sulfate-containing brine: 7.50 g/L NaCI (ACS Reagent grade): 10.66 g/L Na2S0, (ACS Reagent grade). NACE 2.2.3.3 Note: Insoluble materials in very small quantities will remain after the specified reagents have ccinOstaty dissolved. For corsislency of results, solutions shout/lbw filtered through a 0.45 micron filter. 2.2.4 Apparatus for reproducibly delivering 50 -Lt 0.5 mi., e.g., graduated cylinders or volumetric pipets. 2.2.5 One percent (wt) deionized water solutions of nhltribes ter be tested: 0.1% (wt) inhibitor solutions in deienized water *aid be used for tests where inhibitor loadings are to be betel* 10 mg/l.... 2.2.6 Graduated measuring pipets in the following sizes: 0.1. 0.5 and 1.0 mL. 2.2.7 Standard reagents and apparatus for determination of calcium concentration as per ASTM D 511-88 or D 1126-86, (1) API RP 45,(2) "Standard Methods for the Examination of Water and Wastewater (Part 300)," 3) and other accepted test meth ods. 4 TM0374-90 2.3 Test Procedure 2.3.9 Determine the calcium ion concentration by procedures given in ASTM D 511-88 or D 1126-88, API RP 45, "Standard Methods for the Examination of Water and Wastewater (Part 300)," or another accepted test method. NOTE: Calcium ion concentration values for duplicate test samples often differ by two percent or more. Some analysts consider a five percent difference to be unacceptable and to be cause for rerunning the test 2.3.1 All tests are conducted on the inhibitor on an as-received basis; 1% and 0.1% dilutions are made from the as-received inhibitor. 2.3.2 Using the 1% and 0.1% dilutions, pipet the desired amount of inhibitor into each test cell. Duplicates should be run of each concentration. 2.3.10 Report the average of the duplicate calcium ion concentration values as mg/L calcium sulfate retained in solution for each inhibitor test concentration and both blank concentrations. 2.3.3 Duplicate blanks should be prepared as follows: 2.3.11 Representative data from the evaluation of three inhibitors are given in Table 1. These figures are examples only and do not reflect experimental precision. For a percent inhibition calculation, see Section 4. 2.3.3.1 Two samples of the calcium-containing brine (50 mt. each) are set aside. The blanks before precipitation are determined by measuring the calcium ion concentrations (Paragraph 2.3.9) and dividing each value by 2. "ASTM. 1916 Race St., Philadelphia, PA 19103-1187. American Petroleum Institute (API), 1220 L St., N.W., Washington. DC 20005. (3) American Public Health Association. 1015 15th St.. KW., Washington. DC 20005. ( 2.3.3.2 The blanks after precipitation are prepared and handled as in Paragraphs 2.3.4 through 2.3.9 but do not contain a scale inhibitor. 121 2.3.4 Add 50 mL of sulfate-containing brine to the test cell and mix well. Add 50 mt. of calcium-containing brine to the test cell. TABLE 1 — Calcium Sulfate Retained in Solution (as Calcium Sulfate, mg/L) 2.3.5 Immediately cap the test cell and agitate to mix the brines and the inhibitor thoroughly. Scale 2.3.6 Place all test cells and blanks in a forced draft oven or immerse to 3/4 of their lengths in a water bath at 160°F (71°C) for 24 hours. 2.3.7 Remove the test cells after the 24-hour exposure and avoid agitation. Allow the test cells to cool to 77°F (25°C) s 9°F (5°C) for a time not to exceed two hours. 2.3.8 Pipet 1 ml of the test brine to a suitable vessel, avoiding the transfer of calcium sulfate crystals. and dilute with distilled water, deionized water, or as otherwise specified in the calcium determination method to be used. Inhibitor A B C 1 ppm 3 ppm 5 ppm 10 ppm 20 ppm 5140 4080 4896 5140 4352 5103 5140 4896 5140 5140 5068 5140 5140 5140 5140 Blank (after precipitation) 3808 Blank (before precipitation) 5140 These data indicate that inhibitor A is best. Note: Costs of the inhibitors have not been considered. Section 3: Calcium Carbonate Precipitation Test 3.1 This section lists the apparatus and procedure for conducting the calcium carbonate precipitation screening test. 3.2.4 Fritted-glass gas dispersion tube(s) (medium or coarse porosity rating). 3.2 Apparatus and solutions 3.2.5 Synthetic brines prepared with distilled or deionized water. as follows: 3.2.1 A regulated source of carbon dioxide (CO 2). All recognized grades of CO2 are suitable for this test 3.2.2 Constant temperature water bath or forced draft oven with the capability of maintaining the specified temperature within _t 2"F (1°C). 3.2.3 Clean and dust-free glass test cells (4 oz [approximately 125 mL) bottles with a positive seal). Caution: The amount of vapor space above the test solutions in Paragraph 3.3.6 will affect the test results. To maximize the validity and reproducibility of test results, choose test cells that vary in capacity (volume) when sealed by 5% or less; that is, V, = V i; 0.025 V, where V, equals the desired range of test cell capacities and V equals the mean test capacity. 2 3.2.5.1 Calciuni-containing brine: 12.15 gIL CaCt2 21120 (ACS Reagent grade); 3.68 g/L MgCl 2 - 6H20 (ACS Reagent grade); 33.0 9.11 NaCI (ACS Reagent grade). 3.2.5.2 Bicarbonate-containing brine: 7.36 g/L NaHCO3 (ACS Reagent grade); 33.0 g/L NaCI (ACS Reagent grade). 3.2.5.3 Note: Insoluble materials in very small quantities will remain after the specified reagents have completely dissolved. For consistency of results, the solutions should be filtered through a 0.45 micron filter. NACE • TPA0374-90 3.2.8 Apparatus for reproducibly delivering 50 ± 0.5 mL, e.g., graduated cylinders or volumetric pipets. 3.2.7 One percent (wt) deionized water solutions of inhibitors to be tested: 0.1% (wt) inhibitor solutions in deionized water should be used for tests with loadings below 10 ppm where inhibitor loadings are to be below 10 mg/L. 3.2.8 Graduated measuring pipets in the following sizes: 0.1, 0.5. and 1.0 mL. 3.2.9 Standard reagents and apparatus for determination of calcium concentration as per ASTM D 511-88 or D 1126-86, API RP 45. "Standard Methods for the Examination of Water and Wastewater (Part 300)." and other accepted test methods. 3.3 Test Procedure 3.3.1 All tests are conducted on the inhibitor on an asreceived basis; 1% and 0.1% dilutions are made from the asreceived inhibitor. 3.3.2 Using the 1% and 0.1% dilutions, pipet the desired amount of inhibitor into each test cell. Duplicates should be run of each concentration. 3.3.3 Duplicate blanks should be prepared as follows: 3.3.3.1 Two samples of the calcium-containing brine (50 mL each) are set aside. The blanks before precipitation are determined by measuring the calcium ion concentrations and dividing each value obtained by 2. 3.3.3.2 The blanks after precipitation are prepared and handled as in Paragraphs 3.3.4 through 3.3.10, but do not contain a scale inhibitor. 3.3.4 Both the calcium- and bicarbonate-containing brines should be saturated with CO2 immediately before using. Saturation should be accomplished at room temperature by bubbling CO2 through a bitted-glass gas dispersion tube immersed to the bottom of the container. A rate of 250 rritimin. of CO2 for 30 minutes will be sufficient to saturate up to 1 L of each brine simuttaneously. A tee may be used to spirt the gas flow for this purpose. 3.3.5 Add 50 mL of bicarbonate-containing trine to the test cell and mix well. Add 50 mL of calcium-containing brine to the test cell. 3.3.6 Immediately cap the test cell and agitate to mix brines and inhibitor thoroughly. The cells must be capped tightly to avoid loss of CO2. Note: Pressure will build in the test cells as the CO2saturated test brine approaches and reaches 160°F (71°C). Rupture of the test cells has not been reported, yet it is a potential danger associated with this test procedure. Note also that an improperly sealed test cell may lead to pressure release, a resulting test brine compositional change. and an invalid test result. 3.3.7 Place all test cells and blanks in a forced draft oven or immerse to 3/4 their lengths in a water bath at 160°F (71°C) for 24 hours. 3.3.8 Remove the test cells after the 24-hour exposure and avoid agitation. Allow the test cells to cool to 77°F (25°C) ± 9°F (5°C) for a time not to exceed two hours. 3.3.9 Pipet 1 mt. of the test brine to a suitable vessel, avoiding the transfer of calcium carbonate crystals, and dilute with distilled water, deionized water, or as otherwise specified in calcium determination method to be used. 3.3.10 Determine the calcium ion concentration by procedures given in ASTM D 511-88 or D 1126-86, API RP 45, "Standard Methods for the Examination of Water and Wastewater (Part 300)," or another accepted test method. NOTE: Calcium ion concentration values for duplicate test samples often differ by 2% or more. Some analysts consider a 5% difference to be unacceptable and to be cause for rerunning the test. 3.3.11 Report the average of the duplicate calcium concentration values as mg/L calcium carbonate retained in solution for each inhibitor test concentration and both blank concentrations. 3.3.12 Representative data from the evaluation of three inhibitors are given in Table 2. These figures are examples only and do not reflect experimental precision. For a percent inhibition calculation, see Section 4. TABLE 2 — Calcium Carbonate Retained in Solution (as Calcium Carbonate, mg/L) Scale Inhibitor A B C 1 ppm 3 ppm 5 ppm 10 ppm 20 ppm 3000 3500 3600 3400 4000 4140 3800 4100 4140 4000 4100 4140 4100 4140 4140 Blank (after precipitation) 2600 Blank (before precipitation) 4140 These data indicate that inhibitor C is best. Note: Costs of the inhibitors have not been considered. Section 4: Percent Inhibition Calculation 4.1 Caution: The percent inhibition calculation is for comparative purposes only. It is not intended to reflect the ability of a particular inhibitor to prevent scaling in a field application. 4.2 Percent inhibition values may be calculated as follows: Inhibition (I) = C• Cb x 100 C, - Cb Where: C, = Ca2' concentration in the treated sample after precipitation Cb = Ca2* concentration in the blank after precipitation C, = Ca2 + concentration in the blank before precipitation SPE SPE 15457 SocietkorPetrolesznEncsneere Use of Inhibitors for Scale Control in Brine-Producing Gas and Oil Wells by M.B. Tomson, Rice U.; L.A. Rogers,' Gas Research lnsL; K. Varughese, Aiquatani Pipe Coating Terminal; S.M. Prestwich, U.S. DOE; G.G. Waggett, South Texas College of Law: and M.H. Salimi, Rice U. 'SPE Members This paper was prepared tor presentation at the blot Annual Technical Conference and Exhibition or the Swells, of Petroleum Engineers held in New Orleans. LA October 5-8. 1916. This paper was selected tor presentation by an SPE Program Committee following review of information contained in an abstract submitted by the autrorls). Contents of the paper, as presented. have not been renewed by ihe Society of Petroleum Engineers and an Strisiect io correction by the autlyorni. The material. as presented, ODDS not necessary reflect any position of the Society d PIIIIONWTIEngtners, ns officers. or members Pipers presented at SPE meetings are subject to publication review by Echtonai Committees at the Society of Petroleum Engineers. Permission to Copy n restricted to an abstract of not more than 300 words Illustrabons may not be copied. The abstract should contain =his:victims aCknowndgment of where and by whom the paper is presented. Write Publications Manager. SPE. P O. Boa 933836. Richardson. TX 750113-3836. Telex. 730999 SPEDAL. ABSTRACT IMTRODUCTIOM Field and laboratory work sponsored by the Gas Research Institute (CAI) and the Department of Energy (DOS) have shown that calcium -carbonate scale formation in waters produced with natural gas and oil can be prevented by injection of phosphonate inhibitor into the formation, even if the formation is sandstone without calcite binding material. Inhibitor squeeze jobs have been carried out on DOS's geopressuced -geothermal Gladys McCall brine-gas well and GAI's co-production wells in the Hitchcock field. Following the inhibitor squeeze on Gladys McCall, the well produced over five million barrels of water at • rate of approximately 30,000 BPD without calcium-carbonate scaling. Before the inhibitor squeeze, the well could not be produced above 15,000 BPD without significant scale formation. In the Gil brine-gas co-production field tests, inhibitor squeezes have been used to successfully prevent scaling. Progress has been made toward controlling scale formation from brines often associated with geopressured energy production, co-production wells, and oil wells which make large amounts of water. As brine flows out of the formation and up the well, the pressure drops. This pressure drop causes dissolved carbon dioxide, CO2 to go out of solution, which incr eases the solution pH. The pH rise causes aqueous bicarbonate, HCOm to be converted to carbonate. COm, which tends to initiate calcium carbonate, CaC0s, precipitation Laboratory work has been conducted to determine what types of oil field waters are subject to scaling. This research has led to the development of a saturation index and accompanying nomograph* 3. Core samples from both fields were used in laboratory studies and analytical methods to analyze inhibitors in brine at a low levels were extended. A complete history of field developments and the laboratory backup experiments is inciuded in this paper. The first option, reduced production, generally entails an unacceptable loss in revenue. Injection of inhibitors into the surface equipment, option 2, does not protect the production tubing, and installation of a downhole treat string, option 3, is often prohibitively expensive. The last option, an inhibitor squeeze, can protect the near well bore formation, the production tubing, and the surface equipment. Successful inhibitor squeeze jobs have been which allow prediction of when scale will develop into a problem in brine production. References and illustrations at end of paper. either in the formation pore throats near the well bore, on the production tubing walls or in surface handling equipment. Four scale control options include: 1. 2. 4. Limiting production so that the drop in pressure is not sufficient to induce precipitation (see below for details). Injection of trace concentration of inhibitors in the surface equipment. Injection of trace concentrations of inhibitors downhole via a small diameter treat string or down the annulus; and squeezing inhibitor into the formation in such a manner that the inhibitor will be slowly released when production commences. Use 2 Inhibitors for Scale Control in Brine-Producing Cas and Oil Hells carried out on DOt's geopressured -geothermal Gladys McCall brine-gas well near Grand Chanier, La, and =I's co-production wells in the Hitchcock field near Galveston, Tx, and will be presented. The producing formation of the Gladys McCall well was secondary quartz cemented and contained no calcite, which made a successful inhibitor squeeze design considerably more complicated. Previous laboratory work has led to the development of a method to predict when scale will begin to form and how little inhibitor might be needed to prevent stale. In this paper we will first discuss the field applications and results of phosphonate inhibitor squeezes to prevent formation of CaCOs scale. This will be followed by a description of laboratory experiments and theoretical considerations that led to the development of the inhibitor squeeze techniques employed. FIELD EXPERIMENTS GLADYS MCCALL The first attempts at phosphonate inhibitor squeezes at Gladys McCall were unsuccessful since the inhibitor could not be pumped into the formation. There appeared to be two possible causes for this: (1) Poor surface water quality with high iron and calcium content and (2) Interaction of the phosphonate with formation brine which contained 4000 PPM Ca ion (Table 1). The first of these problems could be handled by using good quality water which had passed through an ion exchange and a good filtering unit to remove any iron hydroxide. The second problem could be handled by using a brine spacer ahead of the inhibitor to force the calcium containing formation water away from the well bore. Based on these conclusions, the inhibitor pill was designed to be injected as follows: 1. 2. 3. 4. 5. 6. 300 B of 15% NaC1 spacer 100 D of 3% inhibitor in 15% ■aC1 (the inhibitor was nitrilotri(methylene phospbonic) acid from Champion Chemical Co., Houston, Tx 100 D of 15% MaCI spacer 100 B of 101 CaCla overflush 500 B of 15% BaCI into the formation as a pusher The well was to be shut in for 24 hrs. to allow reaction In fact, 6% inhibitor was used (step 2). Everything went well with the treatment at first. Pumping rate was held at 2 IPS with only a slight pressure increase over original shut in wellhead pressure. When the 100 D of inhibitor hit the formation, the pressure built up rapidly and the pump rate was reduced to 1 DPW to keep from exceeding fracturing pressure. The 100 D of inhibitor and 100 D of NeC1 spacer was squeezed away at 1 DPW. When the CaCls overflush hit the formation, the pressure built up rapidly to the pre-set limit. Pump rats was reduced to 0.2 BPW and the 100 B of CaCla solution was slowly pumped away. This was followed by approximately 25 II of 15% NaC1 solution to clear the tubing. Then the well was SPE 15457 shut in since all of the inhibitor was in the formation and had been exposed both to calcium from the formation brine and the CaC12 overflush as evidenced by the pressure increases. After 24 hours, the well was allowed to flow back at 100 BPH. Brine samples were taken every 10 barrels during the flow back of the pill and periodically thereafter. These samples were analyzed for numerous elements, in addition to the inhibitor itself (Figure 1). It was found that magnesium was the most distinctive tracer for the formation brine although the early buildup (around 600 barrels of returns) could be due to impurities in the 15% MAC' and some interaction with the phosphonate. By the time 1200 barrels of brine had been produced, Mg, Ca, Na and X had stabilized to their original concentrations in formation brine. About 70% of the inhibitor flowed back with the first few thousand barrels of brine production. The remaining inhibitor was slowly released over the next six months. The concentration of the inhibitor dropped to about 0.1 to 0.2 mg/1 within a few weeks and remained there until the well was shut in for repair and resqueeze in January, 1986. This corresponded to about 701, of the inhibitor remaining in the formation. In order to measure such low inhibitor concentrations in field brine it was necessary to modify standard colormetric phosphonate procedures which will be published elsewhere. Prior to the inhibitor squeeze, production was limited to about 15,000 BPD in order to avoid scale formation. This severely curtailed gas production. After the squeeze it was possible to increase the production rate to about 30,000 BPD, still without scale formation in the production tubing. During this period about 25 SCF of natural gas was separated and sold per barrel of brine produced. At about four months into production, a light scale was observed in the final filters before the disposal well. This was eliminated by addition of 0.25 mg/1 of inhibitor downstream of the choke. After six months of production no indication of scale formation in the production tubing was found, and when the well was shut in, the high pressure side of the choke was observed to be scale free. Thus, the phosphonate inhibitor squeeze had protected the tubing for the production of over five million barrels of brine. Upon analysis of the breakthrough data of the flowback curves from the first squeeze job, it was concluded that the calcium in the formation brine could be used to fors the calcium phosphonato in situ if sufficient mixing could be obtained in a large pill which was pumped a considerable distance from the well bore. This would avoid the pressure increase observed when the calcium of the CaCla overflush hit the formation containing the pill and would greatly simplify the overall operation of the pill application. Based on the observations from the behavior of the first successful phosphonate inhibitor squeeze, a simplified pill procedure was designed as follows: 1. Pump a 100 barrels preflush of 10% illaC1 to push most of the Calcium ion in the reservoir brine away from well bore. SPE 15457 2. 3. 4. 5. M. 8. Towson, ec al. Use 100 barrels of pH neutral 3% (N1(4) phosphonate inhibitor salt in 10% lad solution which will react with calcium from formation water. Overflush with 900 barrels of 10% MaC1 followed by 300 barrels of oil field brine to clean the tubing. Shut in for 36 hours and bring back slowly at 100 barrels per hour. After 2 days resume flow of 30,000 barrels brine per day. Since these fluids had approximately the same density as the formation brine and viscosities were low, the injection rate after preflush was chosen as 6 barrels per minute. An oxygen scavenger (MH4)HS0a and an iron chelating agent (EDTA) was added to all of the brine to prevent formation of iron hydroxide and a 2-1/2 micron filter was used just upstream of the pump suction. In addition. 100 millicuries of I 131 was mixed with the phosphonate pill as • tracer. The pressure curve for the injection is given in Figure 2. No problem was encountered which forced slowing the pumping rate. Maximum pressure rise was approximately 400 to 500 PSI as the pill was pumped into the formation. The well was brought back on stream after 36 hours and produced at the rate of 100 barrels per hour for two days. Flow rate was then increased to approximately 30,000 barrels and has remained there for over six months except for a slow rate decrease that is a function of the reservoir flow properties. Samples were taken on a regular basis during early flowback and once a day thereafter. Figure 3 shows the 1-131 scintillation count data vs. barrels of brine produced. A well defined curve with a maximum at around 1450-1500 barrels of brine flow results. Samples of the brine were also used for determination of the phosphonate concentrations in solution. These are included in Table 2. In this squeeze treatment there was never the tremendous backflow of the calcium phosphonate that had been observed in the earlier squeeze job. This is probably because the phosphonate pill was pushed far enough back into the reservoir to smear it out over a large volume of the sand grains. After 8 days of production at 30,000 BPD the inhibitor concentration dropped to about 0.15 ng/1 and has remained at that level for over six months (Table 2). The successful application of the phosphonate pill has allowed the Gladys McCall well to be produced at the maximum rate available from the reservoir. Over five million barrels of brine have already been produced using this second pill with apparently no reduction in protection from CaCOs deposition. LABORATORY STUDIES AID THEORETICAL CONSIDERATIONS In order to prepare for the field work numerous laboratory experiments were conducted. Core material from the producing formation at the Gladys McCall well was analyzed by x-ray diffraction and by scanning electron microscopy and microprobe analyses. Results are in Table 3. From the information in Table 3 it can be concluded that the producing formation is sandstone cemented by secondary quartz with a small amount of clay. The porosity was found to be about 20%, similar to Brea sandstone. This core material was used in all laboratory column studies of the Gladys McCall well. Column Studies Column studies were conducted using both ground core samples and intact core plugs. Packed columns using ground core material were quick and easy to prepare and allowed numerous variables to be examined. For the experiments using ground core material 14 g portions of core material were gently crushed with a porcelain mortar and pestle and packed into 10 ml plastic pipets fitted for use as chromatography columns. These columns were saturated with synthetic brine (1 M l•Cl and 0.125 CaCla) and then flushed to remove all traces of drilling fluids. These columns had 40% porosity. After preliminary experiments were run to determine appropriate concentration ranges, the following test of the effect of CaCla overflush was performed by injecting the following into a set of columns: 1. 2. 3. 4. 2.0 al 15% lad as a lead spacer 0.5 al 3% active Gyptron T-132 (pH neutral fora of Dequest 2000 from Champion Chas. Houston, Tx.) 0.5 al 15% lad as tail spacer 2.0 al 15% Wel as overflush These columns were shut in for 8 hrs at 85•C (185•F) and than slowly backflushed with 40 al of synthetic brine at 85•C. Columns were then cut into 10 sections and each was analysed for remaining inhibitor. Next, the 2 ml of 15% NaCl overflush (4. above) was replaced by 2 al of 15% CaCla. All experiments were run in duplicate, at least. The overall mass balance of inhibitor in these experiments was generally 901, or better. A fourfold enhanced inhibitor retention is shown in Figure 4 as a consequence of using a CaCla overflush. This corresponded to about 40% of the inhibitor which was loaded. For this reason a CaCla overflush planned to be used is the inhibitor squeeze at the Gladys McCall well. Similar column experiments were conducted using core material from the Delee No. 1 well in the Hitchcock Field. These core materials were calcite cemented. Little difference was observed between the Nadi and CaCla overflush regimes. Columns of intact Gladys McCall core metrial were also prepared. Rectangular pieces of core material 1 cm x 1 cm x S ca were cut and embedded in Epoxy in a 1 in OD steel pipe. The end cap pieces and reducers on the pipe were all filled with Epoxy end later drilled to allow 1/I" 00 high pressure Teflon tubing to contact both ends of the 3 4 Cs. of Inhibitors for Scale Control in Brine-Producing Cas and Oil liens core material (Figure 5). This system was used to evaluate the adsorption and desorption of inhibitors. Columns were first saturated with synthetic brine (1 M Nadi, 0.125 M CaCla, pH . 4.1) and then flushed at 95'C until all traces of drilling fluid were removed. A pill dosage of 0.95 ml of Ti inhibitor in 0.125 M CaCla and 1 M NaC1 was injected into the core. The intact core column was shut in for 24 hrs at 95*C. Desorption of inhibitor into flowing synthetic brine at 95'C was monitored for about 10.000 pore volumes, or sixteen liters. The result is plotted in Figure 6. After 300 pore volumes the inhibitor concentration dropped to about 1 mg/1 (as phosphonic acid) and remained approximately content for another 7000 pore volumes. After about 7000 pore volumes the concentration of inhibitor in the brine rapidly dropped to below detection limit. (31) fora scale (1) -: brie. tubseturated, scale may istssohnt where (Ca14) and (C0:-) represent molar concentrations of calcium and carbonate, respectively, and Kip is the conditional solubility product of CaCO2.1 The solubility product in Equation 1 is a function of temperature T. pressure P, and ionic strength IS, or total dissolved solids TDS. At the pH values normally encountered in sec-pressured brines, the ionic calcium concentration in Equation 1 can be replaced by the total calcium, Tca, which is easily measured. The divalent ionic carbonate concentration, (C01-). in Equation 1 can be expressed in terms of bicarbonate (MC0a) and pH using the second ionization constant of carbonic acid. Below about 8.3 pH, bicarbonate is essentially the same as alkalinity (Alk), which is also readily measured. Finally, by appropriate algebraic substitution the pH can be expressed in terms of Henry's law constant, the alkalinity, and the gas phase partial pressure of carbon dixoide. Oddo and Towson (1962)1 have determined least squared curves for the various equilibrium constants as a function of T, P, and TOE. The SI for most gas and oil well brines has been shown to be:1 SI • lea(Tc. 4)10/71,,,) • SAII • 1.S0410'8 T - 4.21,00* T' - 7.44:10* r - 2.556 Is% • 0.920 IS (2) where Tca(molar) (mg/1 Ca)/40.000; /ilk (solar) . (mg/1 HCO4)/61.000; IS (molar) . conductance (vmho/cm)/66,667 m (mg/1 TD6)/56,500; end ICOa volume or mole fraction of COs in gas phase, with pressure, P, in.psia end temperature, T, in 'F. similar equation for pH was derived.' All of the variables of Equation 2 are readily SI - S I , The driving force for CaCOa precipitation under any solution conditions can be represented by the saturation index, SI, defined by: brim? sup.maturatimil, soy 0: mullibrium measureable. A simple kit to measure Ca, Alk, and TIM on site is available from LaMotte Chemical Co. (Chestertown, Maryland) and the gas phase percent carbon dioxide can be easily measured by e.g. Dreier tubes or gas chromatography. The T and P are generally available at the sample point. To facilitate the use of Equation 2 a monograph was constructed2 and a slide rule is available from Shell Canada (Calgary, Alta, Canada). At initial shut in conditions the downhole SI in Equation 2 is theoretically zero for any formation which is calcite cemented. Therefore, a change in saturation index, ASI, was defined as the sum of the changes resulting from P, T. Ca, Alk, PCO,, and TDS, independently:2 ASI • Saturation Index 'co SPE 15457 sSIP • ASI, aSIA,, • 15110. • &Sins (31)) During production in the absence of scale formation the only variables which change from bottom hole to well head are T and P. Homographs to calculate ASI were also developed.2 A semiquantitative correlation of scale formation process vs SI was developed from field and laboratory studies. For ASI values between zero and 1.1 to 1.4 scale will probably not start to form in equipment free of scale. For AS! values between about 1.4 to 2.3 scale can generally be controlled by trace concentrations of inhibitors. Above ASI of about 2.3 it may not be possible to prevent scale with inhibitors (see below). Below ASI • 0 scale will not form, but corrosion will likely be the prima concern, especially if AU is less than - 1.0. Inhibitors Rosenstein in 1935 found that extremely low concentrations of metaphosphates could be used to prevent scale formation.3 Today several classes of compounds are used for this substoihiometric (threshold) scale inhibition, e.g.: 1. inorganic polyphosphates; 2. polyphosphate esters; 3. phophonates; and 4. low molecular weight polyacrylates; and polymaleates.4 Most of these threshold inhibitors are surface active. They probably prevent nucleation of a new scale phase by interacting with the forming nuclei and preventing formation of a stable solid phase.5 They prevent further growth of an already existing phase by adsorbing onto active growth sites on the surface and preventing incorporation of more lattice ions.' For brines supersaturated with respect to calcite it has been proposed that the lowest concentration of threshold inhibitor needed would be approximately the molar concentration of divalent carbonate. specifically:5 2(CO: -)/Iz(InS -) < 1.0 (4) where parenthesis represent molar concentrations and z is the average charge on the inhibitor or inhibitor unit used to express concentration. The summation sign in Equation 4 suggests strict SPE 15457 5 M. I. Tomos st additivity of mixed inhibitors. In laboratory tests compounds and various combinations of compounds from several chemical classes have been shown to prevent nucleation at concentrations similar to those predicted by Equation 4. Yet, no examples have been found of inhibition of calcite nucleation below the limit suggested by Equation 4. of the iodine has returned by the time 3000 barrel of fluid have been produced. Also, there is an upper limit at which an inhibitor might work. This upper limit is controlled by the solubility of the inhibitor. Inhibitors of precipitation generally form insoluble salts of one of the lattice ions of the phase being inhibited. All of the classes of inhibitors listed above are anions and form insoluble calcium salts. If the concentration of inhibitor is two large a calcium-inhibitor salt (pseudoscale) precipitates. For some of the field systems investigated by the authors this upper limit of inhibitor concentration has been found to be only a few mg/1 or less. concentration found in laboratory tests.1'4 As noted before, with each inhibitor squeeze scale began to form in.the low pressure side of the surface equipment at 4 to 5 - mos. This occurred as the &SI increased above 2.3 to 2.5 (Equation 3),1,2 but an additional 0.25 mg/1 of inhibitor was sufficient to prevent scale in the low pressure region of the surface equipment. When greater than 1 mg/1 of inhibitor was injected in the surface equipment calcium-inhibitor pseudoscale formed in the filter units. DISCUSSION At flowing wellhead conditions the concentration inhibitor, 0.73 sg/1 (1.7 x 10-' N), is only 15% greater than that predicted by Equation 4.1.3 This agreement may be fortuitous, but is similar to the lowest CONCLUSIONS 1. The development of • successful squeeze method to inhibit calcium carbonate depositions from produced brines has been tested. This method has been shown effective in sandstone reservoirs where there is Phosphonate inhibitor squeeze can be used to successfully prevent formation of CaCOs scale in the tubing and surface equipment of brine wells which would normally have scaling problems. Inhibitor concentrations of only 0.15 mg/1 have effectively prevented scale formation in the production tubing. no calcite in the cementing material. The key ingredient in a successful application appears to be that the calcium phosphonate needs to be formed in the reservoir at some distance awe), from 2. the well bore. This can be done by carefully selecting the amounts of preflush to move the brine which contains calcium ion away from the well bore and pumping in excess afterflush to force the Treatments can be successfully carried out in reservoirs where the sand grains are not cemented with calcite. 3. Extreme care should be taken to form the insoluble calcium phosphonste in the reservoir so that it won't block the well bore. 4. The use of a calcium chloride overflush can be phosphonate into the reservoir where it interacts with calcium from the formation water to form the insoluble calcium phosphonste. There is considerable room for improvement in the development of the inhibitor squeeze technique. For example, we are not sure that the 10% RaC1 preflush and afterflush couldn't be done with regularly available oil field brine. Precautions would need to be taken however to see that the Ca and Fe levels in the brine were low. Also, the use of a small pore size filter (preferably 2 microns or less) is a most to prevent blocking of the well bore from any particles in the oil field brine. The mixing data available from the first successful squeeze job at Gladys McCall is quite interesting. The sodium concentration (Figure 1) rises more sharply than expected and than doesn't appear to remain at the high concentration as long as it should. In fact, the sodium concentration has returned almost to its normal formation brine level by the time 900 barrels of brine have been produced, whereas we'd expect high values to cover at least a five hundred barrel spread. Its apparent peak at the same point as the phosphonate peak also is unexpected. avoided by using the natural dispersion properties of the reservoir to induce the mixing of the injected inhibitor with calcium in the formation brine. This greatly simplifies an inhibitor squeeze. Acisgtommigt This work was supported by the Gas Research Institute under contract No. 5084-212-0890, but in on way does this constitute an endorsement by CRI of any products or views contained herein. Manuscript preparation by Leticia Villafranco and artwork by Peggy O'Day are gratefully acknowledged. Egnigang 1. 1583-1590. 2. In the second squeeze inhibitor test the iodine peaks around 1500 barrels (Figure 3) which is about 300 barrels later than would be expected. It is interesting to note however, that if you integrate the area under the iodine curve over 90% Oddo, J. S. and Tomson, M. S.: "Simplified Calculation of CaCOs Saturation at Nigh Temperatures and Pressures in Brine Solutions," J. Pet. Tech. (1982) pp. Tomson, N. B., Natty, J., Durrett, L. R. and Rogers, L.: Saturation Index Predicts Brine's Scale-Forming Tendency," cal salgag L. ga (1985) pp. 97-108. 3. Cowan, J. C. and Weintritt, D. J. Water CONTINUOUS INJECTION OF SCALE INHIBITORS Continuous injection is the preferred method for all scale inhibition since it ensures the presence of proper levels of inhibitor at all times. In this method, the chemical is applied with or without flush by a gasoperated, beam-operated or electric chemical proportioning pump. For proper addition, the scale inhibitors should always be injected into a stream of fluid where adequate turbulence exists to ensure thorough mixing of the inhibitor and to prevent stagnant precipitation of scale inhibitors. Normally continuous feed of scale inhibitors in oil production is required because the systems are once-through. For continuous injection systems, the following are the inhibitor criteria: the inhibitor must be completely soluble in the waters processed at usage concentrations; unlike squeezing, precipitation in continuous injection should be avoided. The solids add to the suspended solids and decrease water quality, as well as promote scale deposition and underdeposit corrosion. The chemical should not be corrosive to pumping equipment or to the system. The scale inhibitor must be thermally and hydrolytically stable. They must be compatible with treating chemicals in the production, processing and water systems; these include: demulsifiers, paraffin compounds, microbiocides, corrosion inhibitors, surfactants and other chemicals. The scale inhibitors do react with many amines, as the scale inhibitors are highly anionic while the other compounds are cationic. Chlorine and reducing microbiocides are also affected by (and affect) scale inhibitors and subsequent performance. Continuous injection scale inhibitors must not be pumped into static brines due to precipitation potential. They should not be pumped directly into water tanks, heater treaters or other vessels. Do not inject ahead of an intermittent operating dump valve. Do not inject into annuli without adequate flush. Do not allow faulty check valves to leak produced water into a scale inhibitor line and do not pump scale inhibitors into lines containing neat corrosion inhibitor. Do not drip feed scale inhibitors. Do maintain chemical pump operation as it is critical to effective scale inhibition. Scale inhibitors generally are injected at 5 to 100 ppm (typically 10 to 25 ppm) in scaling water to effect scale control. They must be injected as far upstream of a scaling point as is feasible. The following are some of the main points where scale inhibitors are applied by continuous injection in the oil field: injection water systems, disposal systems, source wells, producing wells, gas lift wells, flowing wells, hydraulic pumping systems, heater treaters and other vessels. Start treatments high and reduce dosages as possible. The following are some of the generalized recommended continuous injection programs: 1. Source wells-inject chemical down the annulus with slipstream flush at 10-25 ppm. 2. Gas lift wells-inject scale inhibitor highly diluted with fresh water into the lift gas at 10-25 ppm. 3. Producing wells with open annuli-inject the chemical with slipstream flush down the annulus at 15-100 ppm. Problems may occur with high gas production or heading up and unloading out the annulus. A macaroni string should be used for these problems. Continuous Injection of Scale Inhibitors Page 2. 4. Flowlines and laterals-inject the inhibitor at the head of the line at 10 to 100 ppm. 5. Heater treaters-inject the scale inhibitor into the moving fluid stream at the header or other point well upstream of the vessel at 20-100 ppm. 6. Waterfloods and disposal systems-for scale problems downstream of the station, inject the scale inhibitor in the pump suction at 10-25 ppm. Where incompatible waters are involved, add the scale inhibitor to the cation-containing water upstream of the comingling point. 7. Power oil or power water systems-continuously inject the scale inhibitor into the power fluid at 10-25 ppm based upon water production volume rather than power fluid circulation volume. Special note: Do not attempt to add additional scale inhibitor (30-50 ppm) at the injection station or injection wells to protect the producing wells when brakthrough occurs as the scale inhibitor would be adsorbed out within the formation. Unsuccessful attempts have been made to to this. SCALE INHIBITOR SQUEEZES Principles of Squeeze Treatments: 1. Scale inhibition, especially scale inhibition by squeeze treatments, is an art not a science. It involves a certain amount of trial and error and field experience in the development of viable squeeze programs because of the imcompletely understood mechanisms of scale inhibition, the interactions between the chemicals and the water involved and the complexities and unknowns in the formation. 2. For squeeze application of a scale inhibitor to perform for a desired long-term life, the inhibitor must be retained in the formation by one or more retention mechanisms: adsorption, and/or precipitation. Adsorption-a physical interaction between an ionized solution and a solid surface in which molecular forces promote a molecular film by charge attractions between the ions and the solid surfaces. Precipitation-the formation of an insoluble salt from solution by reaction between anion (scale inhibitor) and a cation (divalent metal salt, usually Calcium). 3. The inhibitor retained in the formation must be capable of desorption from the formation surface at a slow enough rate to allow long-term ' inhibition. 4. Adsorption-desorption is the preferred mechanism in scale inhibition in a formation since precipitation involves the development of potentiallyplugging precipitates which could cause long-term or permanent formation damage. 5. In essence, the application of a scale inhibitor squeeze is a batch treatment; however, the performance of the squeeze is a continuous treatment. Requirements of Squeeze Application Inhibitors: 1. The inhibitor utilized must be effective against the scale(d) involved at a low dosage. 2. The inhibitor must have adequate adsorption-desorption characteristics to allow a long-term continuous inhibitor return. 3. The inhibitor must be thermally and hydrolytically stable for long terms under formation conditions. 4. The inhibitor must be compatible with the produced fluids, the formation lithology and other chemicals utilized in squeeze, well treatment and production processing. 5. 6. The inhibitor must not promote or create emulsification. The inhibitor in return fluids must be monitorable at low typical usage concentrations. Scale Inhibitor Squeezes Page 2. 7. Other desirable properties could include solids dispersion capabilities and water-wetting (oil dispersion). Mechanics of Scale Inhibitor Squeezes: 1. A formation squeeze is the placement of a chemical or chemicals into a formation at pressures and rates which are less than fracture pressure for a given formation. 2. Scale inhibitor squeezes are determined arbitrarily, or they have been established by trial and error in field applications. 3. The amount of inhibitor required, the volume of overf lush, and the use of diverting agents or other additives will depend upon: a: Well completion data. b. Amount and flow rates of produced fluids. c. Field experience. 4. Because of the complexity of environments and array of production conditions, there are a number of methods utilized for inhibitor placement in the formation: a. Adsorption squeeze. b. Precipitation squeeze. c. Forced precipitation squeeze. d. Fracturing placement. e. Mixed adsorption-precipitation squeeze (most scale squeezes are probably of this type. Actually, a combination of A & 5. In order for a squeeze treatment to provide maximum benefit, existing scale (and paraffin) deposits must be removed. 6. Potential problems resulting form scale squeeze: a. Formation of a "pseudoscale" due to interaction of improperly applied inhibitor with divalent metal cations in formation brine (Me", Ca++, Ea++, Sr44). Results are scaling, plugging solids and loss of scale inhibitor. b. Emulsions created by surfactancy of some scale inhibitors, especially phosphate esters. Results are decreased productivity following squeeze, presence of chemical emulsion in production and increased demulsifier and heat requirements for oil treating. c. Emulsion blocks are possible anytime foreign fluids are placed into a formation. In addition to, and in relation to, emulsion blocks are the development of zones of water saturation. The result is a loss of production following treatment. Scale Inhibitor Squeezes Page 3. 7. Basic Operation of a scale squeeze: a. Mixing of inhibitor and diluent at a suitable ratio and pumping down tubing or annullus. b. Displacement of the inhibitor to perforations or open hole face. c. Overdisplacement of the inhibitor into the formation several feet from wellbore. d. Shut in period to allow maximum adsorption of the inhibitor and migration of the balance into pores, vugs, fractures and other traps. e. Monitoring of produced brine for inhibitor return. Designing Scale Squeeze Treatments: 1. Scale inhibitor squeeze treatments must be designed for specific conditions on a well-by-well basis. There is no such thing as a standardized squeeze. 2. Only a properly designed scale squeeze will be effective. Application of an optimum inhibitor by improper procedure will result in poor performance or squeeze failure. 3. Scale inhibitor squeezes can be designed for any formation compositionlimestone, dolomite, sandstone, or shale. Returns from sandstone may be 75% or more of the inhibitor during the effective life, 50% or more in limestones and as low as 25% from dolomites. Returns usually improve significantly with future squeezes. 4. The following are the design features of each type of squeeze application. (a) Adsorption Squeeze-preferred squeeze mechanism in which an inhibitor is placed into the formation and the chemical is adsorbed to the formation matrix. Effective in all formation types. (b) Precipitation squeeze-a squeeze in which the acid form of a scale inhibitor is placed in the formation or basic form is mixed with 15% acid and placed in the formation. The acidic scale inhibitor or the HC1 acid reacts with calcium carbonate in the rocks to produce a high level of calcium ion in the solution to promote precipitation of a calcium phosphonate salt which will slowly solubilize as an effective inhibitor. Effective only in limestones, dolomites or calcareous sandstones and calcareous shale. (c) Forced precipitation squeeze-a modified precipitation squeeze which utilizes a calcium chloride solution pumped in the treatment to precipitate the phosphonate salt. Used in non-calcareous sandstones and shales. Scale Inhibitor Squeezes Page 4. (d) Fracturing fluid placement- a scale inhibitor determined compatible with the frac fluids is placed into the formation, pumped at frac pressure and adsorbed or precipitated in the newly developed porosity or fractures. Serves as an initial squeeze. SPECIFIC SCALE INHIBITOR SQUEEZE DESIGNS I. Inhibitor Volume: The first step in developing a scale inhibitor squeeze treatment is to determine how much of the selected scale inhibitor is required. The following calculations allow determination of approximate inhibitor dosages. Normally a minimum of 55 gallons is recommended. A. Radius of Desired Protection (most accurate and preferred procedure)* SI = (Rp 2 - Rw 2 ) xlrx8x TXC or SI = 0.785 (Rp2 -Rw2) x 8 x T where SI = scale inhibitor volume in gallons Rp = radius of desired protection (feet) Rw = radius of wellbore, casing 0.D., or openhole in feet. ?r= 3.14 8 = porosity of formation expressed as a drcimal. T = thickness (length) of pay zone in feet. C = constant; 0.25 based upon experiment results and squeeze case histories. * Use this equation for high volume wells or wells with considerable pay zone. B. Estimated Life Based on Production SI = VwxDxC where SI = scale inhibitor volume in gallons Vw = water production, bwpd D = estimated or desired squeeze life, days C = constant; 0.006 based upon experimental results and squeeze case histories C. Rule of Thumb for Inhibitor Volume 0.62 to 0.7Z of daily water production; 12 in lower volume wells Use no less than 55 gallons per squeeze. BPD x .008 = drums of chemical (12 of H2O prod.) BPD x .006 = drums of chemical (0.72 of H20"prod.) BPD x .005 = drums of chemical (0.62 of H2O Prod.) 42 (based upon 55 = 0.8; 1% = 0.008) II. Displacement Volume: Use the tubing or annular displacement tables in a Halliburton, Dowell or B J-Hughes cementing manual. Use produced water or 2% KC1. Be sure to include 0.1-0.52 suncrwr Specific Scale Inhibitor Squeeze Designs Page 6. • III. Overdisplacement Volume: As a general rule, the amount used is equal to one (1) full days water production, with a 100 barrel minimum (at least 6 foot radial penetration). Do not use more than 250 to 300 barrels except under specific recommendation. Produced water or 2% KC1 brine should always be used. Be sure to include 0.1 to 0.5% SURFAcIRAPT. Radial Penetration (ft.) Over Displacement (BBLS) 1 2 3 23 5 66 6 7 100 130 9 215 IV Inhibitor Dilution Volume Normally, Acci,r...v.recommends 10% in fresh water; some companies recommend 2.5% for phosphate esters and 5% for phosphates in fresh water or produced water. b. If produced water must be used, the proper dilution ratio must be determined with the scale inhibitor added to the water to determine solubility range. Check 1%, 5%, 10% and 20% until a ratio with a clear solution is achieved. Produced water is normally used only when no fresh water is available or due to the presence of watersensitive clays or shale; ACc1;/"41-prefers the use of 2% KC1 brine in these situations rather than produced water. V. Shut-in Time Based upon proven field performance, the shut in time to allow maximum adsorption and migration of the inhibitor should be 24 hours. Some companies may recommend 48 hours, but field experience by several companies have shown excessive time provides no discernible benefit. No less than 12 hours shut-in should be recommended; 24 hours is preferred. VI. Typical Adsorption Squeeze Procedure (no packer) 1. Collect acidized water sample for baseline in residual monitoring. 2. Conduct recommended well clean-out. 3. Close the offside casing and flowline valves and pressure the tubing to 250 psi by pumping 2 to 3 strokes to fill the tubing. 4. Shut the unit in on downstroke. Specific Scale Inhibitor Squeeze Designs Page 7. 5. Pump 10 to 30 barrels of clean produced water or 2% KC1 brine containing 0.5% suRFAc-r4/47- down the annulus. 6. Mix the recommended scale inhibitor volume in fresh water at a 10% ratio of chemical and pump the mixture down the annulus at a rate below frac pressure. 7. Displace the inhibitor-diluent mixture to the perforations or open-hole face with the required volume of produced water (or 2% KC1) containing 0.5% b.v. SURFACT4A)-7-; 8. Overdisplace the inhibitor-diluent mixture into the formation with produced water (or 2% KC1) containing 0.5% b.v. SURFAcro4Afir. Volume should be 100 to 300 barrels depending on the daily water production. 9. Shut the well in for 24 hours and remove treating equipment. 10. Return the well to production and begin monitoring program. Note: For wells producing under packer, the treatment must be down the tubing. As a result, the pump must be unseated in a rod-pumped well to allow passage of treating solution. Displacement volume will change. VII. Staged Inhibitor Squeeze (for wells with large open hole area): 1. Collect acidized water sample for baseline in monitoring. 2. Conduct recommended well cleanout. 3. 4. 5. Premix the recommended scale inhibitor at a 10% ratio in fresh water or 2% KC1 brine and pump 50% of this mixture down the annulus. Pump a 50 barrel pad of water down the annulus. Pump 10 to 20 barrels of diverting agent (typically SO to 100#) down the annulus. The volume necessary will raise the pressure about 150 psi. 6. Pump the remaining scale inhibitor mixture down the annulus. 7. Displace to the perforations with produced water or 2% KC1 containing 0.5% b.v. SURFAc7rAr,11-; 8. Overdisplace the mixture and diverter into the formation with 100 barrels of produced water or 2% KC1 containing 0.5% b.v. SURFA°CrAttn: 9. Shut the well in for 24 hours. 10. Return the well to production and begin monitoring program. Specific Scale Inhibitor Squeeze Designs Page 8. VIII. Typical Precipitation Squeeze: 1. Collect acidized water sample for baseline in monitoring. 2. Conduct the recommended well clean-out. 3. Shut the offside casing and flowline valves. 4. Pressure up on the tubing to seat the pump and prevent leak-by. 5. Mix the recommended dosage of phosphonic acid in fresh water or 2% KC1. Add acid retarder, if needed, at 1% of scale inhibitor volume. 6. Pump the scale inhibitor/diluent mixture down the annulus at adequate pump rate below frac pressure to ensure maximum penetratin of the formation. 7. Displace the inhibitor to the perforations or open hole face with required volume of produced water or 2% KC1 containing 0.5% b.v. P. FACTRArr. 8. Overdisplace the inhibitor into the formation with 100 to 250 barrels of produced water or 2% KC1 containing 0.5% b.v. SURFAC74e1: 9. Shut the well in for 24 hours to allow maximum acid neutralization and precipitation. 10. Return the well to production and initiate the monitoring program. IX. Typical Forced Precipitation Squeeze 1. Collect acidized water sample for baseline in monitoring. 2. Close in offside casing and flowline valves. 3. Pressure up on the tubing. 4. Premix the phosphonic acid with 2% KC1 brine or fresh water at 5% to 10% (optimum) ratio. 5. Pump the mixture down the annulus. 6. Displace the scale inhibitor-diluent mixture with a 1% CaC12 brine (4 lb./barrel). 7. Overflush the scale inhibitor-diluent with 2% KC1 brine or fresh water containing 0.5% b.v. PAR Vitt.c-rott4T to the recommended radial penetration. Specific Scala Inhibitor Squeeze Designs Page 9. Shut the well in for 24 hours (optimum). 9. Put the well back on production and initiate monitoring program. Monitoring Monitoring of the organic phosphate scale inhibitors (phosphate esters, phosphonic acidized phosphonates) can be done by collecting an acidized water sample on at least a monthly basis. Analysis should be ppm of proprietary compound although some chemical companies run total phosphate (no distinction between ortho- and metaphosphate). Polymer residuals are run by specialized procedures (carbonate adsorption bed or dialysis methods), and should not be acidized. Results should be reported on a running report form or graph showing squeeze dates. TYPICAL RETURN CURVE Inhibitor Return X. 8. Time in Days or Months ---> TABLE COMPARISON OF ADVANTAGES AND DISADVANTAGES OF INHIBITOR SQUEEZES AND DOWN HOLE TREAT STRINGS flmarroR sauEgiE Disadvantages Achraniacuti 1. Treat near wel bore formation to prevent plugging during draw down. 1. A squeeze generally must be repeated from every two weeks to two years. 2. All staid surfaces at the well bottom are protected from scale. 2. The rule-of-thumb is that only about one-third of the added inhibitor is actually effective: one-third generally flows back with the first production and about one-third is never returned, although these ratio may improve upon repeated squeezes. 3. A squeeze can be done on old wells without puling tubing. 3. There is virtually no control on the concentration of inhibitor which flows back with the brine. 4. During routine production, tittle maintenance Is required and no on-site power is needed. 4. Performance on a new system is highly unpredictable. 5. Generaiy, a squeeze is a simple procedure for most service comparies. 5. Once a squeeze is started, It is not possible to change the concentration or the chemical, as it is with a treat string. 6. The potential lifetime of a squeeze is virtually unlimited, in theory. 6. There is a real potential for formation damage. 7. Only periodic (about weekly) brine analysis is necessary to detect when to resqueeze. 7. It is difficult and expensive to treat corrosion due the different chemical nature of corrosion inhtitors and the higher concentrations often needed. 8. It is difficult to analyze for most scale inhibitors at the concentrations typically needed. DCMIWQLEIBEALSIERISI Achlaff1808s Disadvantages 1. Chemical type and concentration can be changed as needed. 1. The treat string tubing, generally onefourth in. 00 stainless steel or high alloy steel, etc., is expensive, $0.50 to $5.00 per foot per tube and generally two or more tubes are used per well. 2. Deivery concentration is relabie and can be as the production rate charges. This greatly reduces the need for inhibitor analyst& 2. With old weft installation of a treat string requires a work over. 3. Down Axle pressure can be monitored either directly or by Inference. 3. Operation requires reliable on-site power. Loss of power for a single day may permit serious wale problems to begin. 4. Treat string If °time is potentially unirnited. This yields predictable capital and maintenance costs. 4. It is difficult 13 instal treat strings in slanted brines. 5. Either the same or an additional treat sift can be used to control additional chemical problems, such as corrosion and enaisilicadon. 5. The presence of tubing dips, may complicate some maintenance operations. 6. If any downhole component fails, it is generally necessary to pull the tubing out of the well. IMILL1 The Conditional Sokibilties and Stoichlometdo Coeffidents for Ca - Dlethylenetdaminepenta(Methylene Phosphonate) in 2.0 M Brines at 70° C pH Stoichiornetric Coefficient 4.7 5.0 6.0 7.0 8.0 9.0 0.50 0.48 0.41 0.30 0.31 0.29 Conditional Solublity (K1 4.27 x 104 3.02 x 104 1.92 x 104 1.78 x 104 1.23 x 104 1.20 x 104 SOLUTIONS ARE CHEMICALLY ACTIVE AND EITHER SCALE OR CORROSION DOMINATES BrALLIAMEDIAIEPHOLEM CORROSION: LONGER TO DEVELOP —SOIL G AS 01L X ___________BRINE SCALING TINCENCY NCR. DRAMATICALLY AFTER CHOKE TEMPERAURE ESSENTIALLY CONSTANT PRESSURE DECREASING SCAUNG TENDENCY INCREASNG CORROSION TENDENCY DECREASIVG SC DS CAN PLUG DISPOSAL RESEVOIR i N:stszsA:sr s ;;, CORROSION GENERALLY A PROBLEM DOWNHOLE t t Si t 1 i t ft t it'~ti CONVENTIONAL CURES PARAMETER SCALE CORROSION TaFERATURE DEC. DEC. PRESSURE DEREAS1NG PRESSURE INC. DEC. N RESEVOIR PH DEC. INC. SATURATION INDEX DEC. INC DISSOLVED SALTS INC. DEC. CHEMICAL ADDITIVES MALE Ca180614N PHOSPHONATES AMINES POLYACRYLATES THIO-AMINES POLYMALEATES FATTY ACIDS TO 1 000mg/1 AT 1-10 mg/1 ZINC CHROMATES PROTECTIVE SCALE FIGURE 1. SCHEMATIC DIAGRAM OF A PRODUCTION SYSTEM • THRESHOLD MOTORS MAY NOT WORK ABOVE SI 2.3 RANGE WHERE SCALE CAN PROBABLY BE 2.0 CONTROLLED MTH CHFJAICAL INHIBITORS TRANSMON RANGE FROM NON-SCALING TO SCALING 1.0 RANGE SCALE WHERE FREE INHIBITORS ARE PROBABLY NOT NEEDED IN A SYSTEM EQUILIBRIUM 0.0 SCALE WILL NOT FORM BELOW SI 0.0 SCALE WILL DISSOLVE 1 .0 -1.0 AND BRINE MAY BE VERY CORROSIVE BELOW SI FIGURE 2. DEPICTION OF SI VALUES AND CORRESPONDING SCALE-RELATED PHENOMENA FOR CALCIUM CARBONATE, SCALE FORMING BRINES Polyetnylaneglyco4nnonOnnto Patiel110401011/.00,0W,PHH wo AT ester PE-22 0 Cipost 2000 --- OH 14+0/44HTeM1004nr1.1-410004014:4440/12 HEM Ostn.Ht2ow HO 014 OH I 8 1 HO— P—C — • — OH 0 a tiszed phosphonairearboxylate I VITC-08 CH. 0 44041.04. 044441'1444041.1.40ents\ 04044410n4 200soitax Pe:01 )1 04.04. 24 CHICHI .4,.. I 4404,044 CHI CH.P0s14 0401141120110 PO•14. N 101.10110eCa.°4"W 2144:1444300 Ptanotorals00,044, WTC-11 1 COON CHNC,00.1 I CH, 6,24. ;'0,14 HO —C—COOH CH/000H A 01—,04).-14-404). 1 C. \CH*P03HP4a Cnrle PC/0 VITC-10 -Om AHsPO,Ns, 14000C(CH24— N FIGURE 3. SOME COMMON THRESHOLD SCALE INHIBITORS ais csoPosmil CHEMICAL RESERVOIR CHEMICAL INJECTION POW VALVE 0 FILTER VALVE TREAT STRING RUPTURE DISK CHECK VALVE [ FIGURE 4. SCHEMATIC DIAGRAM OF BASIC INHIBITOR TREAT STRING APPARATUS CALCIUM SULFATE (GYP) SCALE REMOVAL CHEMICALS Scale type deposits have probably been occurring in well bores and surface equipment since the beginning of the industry. The literature shows investigations on paraffinprOblems as early as 1923. (1) And there is a reference for acid being used in wells for, "... dissolving out limestone that has been deposited from the waters...", in Oklahoma in 1928. (2) Generally the inorganic scales were assumed to be calcium carbonate. Where these scales formed they were considered more of a nuisance than a serious problem. The standard solution was acidization of the well and equipment. With the advent of waterflooding and particularly in dolomitic reservoirs, it soon became apparent that, in addition to carbonates, sulfate scales were formed. The principal scale was calcium sulfate with barium and strontium types being occasionally encountered. Since these scales were essentially insoluble in acid the usual treating procedures were no longer effective. It further became apparent in the sixties, that when these scales formed on the reservoir face and in perforations, the productivity of wells would be markedly reduced. As the seriousness of the calcium sulfate problem was recognized there was a concerted effort by both producers and service companies to develop treating chemicals and procedures to solve the problem. EARLY FIELD EXPERIENCE With the damaging effect of scale formation on well productivity fully recognized a major sales effort was directed to this problem by chemical suppliers. In a survey conducted in 1962, twenty-two chemical suppliers were marketing 93 commercial formulations for the prevention or removal of scales. These ranged from well known, low priced materials to complex blends of organics with alleged esoteric actions. Many of the early well treatments failed and by the mid-sixties it was estimated that only 50% of the programs for removal of sulfate scales were even partially effective. A survey conducted in the late sixties listed 35 chemicals being furnished by fifteen suppliers for. the removal of well deposits of sulfate scales. It was obvious from field results that either many of the chemicals were not effective or well treating procedures were inadequate. The extent to which calcium sulfate deposits can impair production of a well is shown by Figure 1, for the period 1961 to 1967. Also it will be noted in 2 Figure 1 there is wide variations in the effectiveness of the clean out procedures, further emphasizing either chemical or treating method inadequacy. In view of frequent treating failures and erratic results,in depth studies of the problem were required. LABORATORY STUDIES ON CALCIUM SULFATE REMOVAL CHEMICALS In the late sixties the laboratory of a major producer began an evaluation of the effectiveness of chemicals being sold for gyp scale removal. Fifteen suppliers furnished 30 chemicals for the tests. The initial program was a screening type to determine which of the chemicals exhibited sufficient activity to warrant additional evaluation. The criteria for passing this test were arbitrarily selected as: A - Effective with a dilution rate of 1:1 with water. B - Compounds formed must be either water or acid soluble. C - Reaction independent of pressure (no CO2 evolution). D.- Effectively convert 75% of scale to a soluble form. The screening tests were on 1/2 inch cubes cut from gypsum rock consisting of fine needle like gyp crystals andarorphous CaSO4 powder. While it was recognized the rock was not comparable to the frequently encountered crystal form, it had many advantages for a screening test. The samples were of a uniform composition and test blocks could be precisely sized. The high porosity and permeability furnished a maximum of surface area for reaction. The bottle results shown in Figure 2 are typical of the performance of many of the chemicals. By visual observation only, the treated sample indicates the reaction has been extensive. But as shown by the test results in the third bottle the rate of conversion has been quite low. As shown in the Table at the bottom of the Figure only four of the chemicals evaluated met the specifications that had been established for acceptability. In view of the inadequacy of most of the chemicals the suppliers were informed of results on their materials and comments requested, permission was also granted to submit additional chemicals for screening. Some suppliers objected to the type of rock used in the testing and it was agreed to conduct future tests on other forms of calcium sulfate. In the second test program 31 chemicals were submitted for evaluation. The testing method was identical to that described for the first program, illustrated in Figure 2. However to assure the form of the calcium sulfate was not influencing the results three variations were used. The rock type used previously, thin clear bladed crystals from a pipe in West Texas carrying Hendricks Reef water, and crystals deposited in tubing in a New Mexico well. The latter were of a variety of shapes and sizes. In the tests they were broken and graded to approximately the size of pea gravel. The test procedure was identical to that presented in Figure 1, except the chemical was used at a 100% concentration. Of the 35 chemicals tested 23 met the requirement for 75% effectiveness. 3 The second screening test was to determine the dilution effect. In most wells the area to be treated will be filled with produced water. Also in many treatments, where a long producing interval is to be covered, it is desirable to dilute the chemical. The criteria for acceptance was that 75% of the scale must be converted to a form soluble in water and/or acid at a 1:1 concentration of the chemical. Figure 3 illustrates typical results on the chemicals meeting this specification and outlines the test procedure followed. This test eliminated 12 of the 23 chemicals. The 11 chemicals meeting the specification all gave results in the 95 to 100 percent efficiency range. The type and speed of reaction indicated all these chemicals to be of the same generic family and of the same approximate concentration. Also the results were so markedly superior to the other 12 chemicals in the program that no further testing was warranted on the other products. The 11 satisfactory products were from nine major chemical supply companies. This assured the availability of a suitable product in all major producing areas. This program established the effectiveness of chemicals and eliminated this as a potential source of treating failures. FACTORS CONTROLLING SCALE DEPOSITS (3 , 4) Once it became recognized that calcium sulfate scales could be plugging the formation there was a tendency to over react. Many operators, without adequate study assumed any abnormal production rate decline was caused by scale deposits. While the ineffectiveness of many of the chemicals resulted in failures, unfortunately many wells were also treated where scale was not the problem. However as the factors governing the precipitation of calcium sulfate scales became more generally known the treating of wells where scale was not the problem markedly decreased. The following discusses the principal items to be reviewed in evaluating the scaling possibilities in a well or field. RESERVOIR ROCK CHARACTERISTICS: It would be assumed that the water, both in the interstices and formation outside the reservoir, would be saturated with the soluble salts contained in the producing interval. The usual mineral sources of the calcium and sulfate ions are anhydrite, gypsum and hemihydrate. If any of these minerals are present it would be assumed the waters contained in the reservoir are saturated with calcium and sulfate ions for the pressure and temperature conditions of the reservoir. It should further be assumed, in flood projects, regardless of the injection water composition, it will become saturated with calcium and sulfate ions, where the above minerals are present, as it moves from injection to producing wells. When calcium sulfate containing minerals, are present in the reservoir, scale deposits should be anticipated during the producing life of the field. However the deposits may be in the producing equipment rather than the well bore. Also the time period over which scales occur will vary with water composition and pressure and temperature conditions in the flow stream,- -- TEMPERATURE EFFECT: The curves on Figure 4 show the solubilities in distilled water of the three mineralized forms of calcium sulfate at various temperatures. While the form and relative position of the curves would be expected to remain the same, the solubility values will change with pressure and other dissolved salts. The relationship between the gypsum and anhydrite illustrates why both types of scales will be encountered in production operations. For the conditions of the curve, at temperatures less than approximately 100°F., gyp crystal 4 scales will develop. Above this temperature the anhydrite should be expected. Since in most wells the temperature drop between reservoir and well bore would be insignificant, this factor is not considered important in most well bore deposits. But as will be noted the solubility of gypsum drops significantly below 100° F. This probably accounts for the heavy crystal deposits in some systems where the surface temperatures in pipelines are below the produced water temperature. PRESSURE EFFECT: Figure 5 illustrates how pressure drop increases the tendency of scale to form from saturated solutions. The pressure drop effect, coupled with the decreasing solubility of gypsum at temperatures below + 100° F. accounts for many of the crystal type scaling problems encountered in shallow, low temperature reservoirs. Also the crystal scale development frequently encountered where pressure drop occurs in surface operations. As shown by Figure 5 the rate of scaling increases with increasing pressure drop. Once scaling has begun the fluid passages will plug rapidly. There have been many instances where calcium sulfate scales have completely plugged fluid passages. As shown in Figure 1, where such deposits occur in perforations or on the formation face the production will be markedly reduced. DISSOLVED SALTS EFFECT: The solubility of calcium sulfate increases as the content of most of the soluble salts normally found in oil field waters increase. Sodium chloride is usually the predominant salt and Figure 6 illustrates how the solubility of calcium sulfate is markedly increased up to a salt concentration of 150 gms. per liter. The variation in calcium sulfate solubility with salt content, when coupled with the pressure effect can be a frequent cause of formation plugging in flood projects. When relatively fresh flood waters contact highly saline formation waters, both of which are saturated with calcium sulfate, the salt concentration in the mixture will be reduced. Under these conditions the solubility of calcium sulfate will be exceeded and scale precipitation occurs. In most of the reservoir this will not significantly reduce the permeability. But when it occurs in the well bore zone or perforations and particularly when coupled with the pressure drop effect, plugging can occur. FACTORS EFFECTING WELL TREATING PROCEDURES (4) Calcium scale removal treatments, being of the chemical conversion type, are time dependent and there are a number of well bore conditions that will markedly influence their effectiveness. The following factors should be evaluated where the conventional "spot and wait" treatments are planned. TREATING TIME: The conversion reaction whereby the calcium sulfate is changed to a water or acid soluble compound is a surface active phenomena. This requires maintaining intimate contact between the scale and the treating compound for a significant period of time. Figure 7 illustrates that for optimum laboratory conditions. With pea sized gravel crystals, a minimum of 6 hours is required for 100% conversion. In this test the total surface areas of the crystals were exposed to 100% concentration of the treating compound. In well treatments the scale would be attached to reservoir face or perforations and the reaction would be occurring only over the exposed surface of the scale. Under these conditions the time for conversion would be greatly increased. Field experience indicates that in developing a procedure for a specific area or field, initial treatments should be at least for a 24-hour period and preferably longer. THIEF ZONES: In low pressure wells, where some strata are essentially depleted, thief zones can markedly reduce treating effectiveness. Should the hydrostatic head used in spotting the treatment be too high, most of the treating chemical may enter the depleted strata. Where thief zones are known to exist a preflush for partially filling the thief zone may be desirable. Also in the spotting of the treatment every effort should be made to minimize the hydrostatic head. WELL BORE WATER: As noted in Figure 3 concentration of the treating chemical is of major importance, with a minimum concentration of 50% being considered necessary. In most wells the producing zone will be filled with water and this may extend up the hole. Also, if the scale is removed first from a high water producing zone, there may be an inflow of water during the treatment. In determining the concentration of the chemical the possibility of these extraneous waters diluting the treatment must be considered. With open annulus spotting procedures dual treatments may be required to obtain good results. LOW PRESSURE ZONES: Where a highly depleted zone is plugged by scaling, once the scale has been removed,there is the possibility of this acting as a thief zone. FIELD TEST DATA: While field experience during the sixties had often been disappointing, with the elimination of inadequate chemicals and the developing of treating procedures the success ratio increased. The experiences in one West Texas field for the period 1970 - 75 are shown in Table I (5). It is obvious from this data, that where calcium sulfate scales are impairing production, successful treatments are possible. AN OPTIMUM WELL TREATING PROCEDURE (5) As the problems associated with scale removal treatments discussed in the previous sections were discovered, procedures were developed to circumvent the difficulties. The method described below, controls the hydrostatic head minimizing the thief zone problem. In addition by isolating the zone to be treated, the possibility of serious dilution of the chemical is prevented. Also by the use of a controlled overflush, that squeezes treatment into the producing interval any skin damage effect, due to scale, will be more directly treated. The technique requires the use of packers and accessories. Careful control is required with regard to-chemical volume and injection procedure. A programmed well clean up is also required to assure maximum treating effectiveness. Obviously the method is far more expensive than the usual spotting method applied in most treatments of this type. Prior to application well performance and reservoir conditions should be reviewed. It is essential that the well have the potential to produce significantly larger volumes of oil, to warrant the additional cost of the treatment. The increases in production that can be obtained with this type of treatment are listed in Table II. These wells in a West Texas field were known to have heavy gyp scale build-ups and with the increases shown, the additional expense and time required for the optimum treatment was certainly justified. TREATING EQUIPMENT AND PROCEDURE: The well equipment is illustrated in Figure 8. It consists of a hook wall packer with a fluid spotting control valve, coupled to the packer by a tail pipe, to position the valve close to the bottom of the zone to be treated. The spring in the control valve is adjusted to the wells hydrostatic head, under closed in conditions. When the assembly has been run and the slips set the treatment is pumped into the well. The recommended volume is 1-1/2 to 2 times that required to cover the zone to be treated. The chemical mixture is displaced into the well with the volume of oil required to spot the treatment over the zone to be treated. The packer is then set and the remaining volume of chemical in the tubing is displaced into the formation. The well is closed in for 24 hours, but preferably longer on initial treatments. The well is returned to production, by swabbing the tubing dry, and dropping a bar to shear the pin in the valve and open the block out sleeve. The swabbing should be continued for two to four hours. This is to recover the residue of the reaction and prevent any unconverted small crystals from being forced back into the formation and acting as a nuclei for scale growth. The well should be acidized for a final clean-up and stimulation. CONCLUSIONS 1. Laboratory and field test programs have proved the calcium sulfate scales can be successfully removed from the well bore face and perforations. 2. Marked increases in oil production can be obtained from reservoirs having the potential for increased production. 3. Where large increases in production are possible the additional expenses of optimum treatments are warranted. 4. Successful scale removal treatment should be followed by scale inhibitor squeeze type treatments to minimize further scaling. REFERENCES (1) Mills R. : Van A. : The Paraffin Problems in Oil Wells, U.S. Bur. Mines R. I. 2550 (1923) (2) History of Petroleum Engineering, Pg. 598, American Petroleum Institute. (3) NGSMA Handbook (4) Richard S. Fulford, Effects of Brine Concentration and Pressure Drop on Gypsum Scaling in Oil Wells, AIME SPE-1803, 1967 (5) Jerry N. Crane, An Engineered Approach For Successful Removal of CaSO4 Scale Deposition; Petroleum Engineer, July 1972 FIGURE CAPTIONS Fig. 1. - Calcium Sulfate Scale Plugging And Clean Out Results In A West Texas Well Fig. 2. - Results With Gyp Scale Removal Chemicals Fig. 3. - Average Effect Of Dilution On Gyp Scale Removal Chemicals Fig. 4. - Solubility Of Calcium Sulfate Scales In Water Fig. 5. - Pressure Effect On Scaling Tendency Of Calcium Sulfate Fig. 6. - Effect Of Salt Concentration On Solubility Of Calcium Sulfate Fig. 7. - Rate Of Gyp Crystals Conversion Versus Time Fig. 8. - Optimum Treating Procedure Table I. - Gyp Scale Removal Treatdents In a West Texas Field Table II.- Results With Optimum Well Treating Procedure inhibitor senleeze 4000 -- 0 ZL - TIAPPY.L.C/1101MI 1 3000 2000 _ I 1000 -1 1- CLEAN OUT PROGRAMS -i 62 I 63 I 64 1 65 _ r ___ 1 66 1 67 . "•"."."'"."••••,-4,.•• 0 Li «C id, Vb 4 a E ". U a 0 " C•4 0 a . . 0 a t V •I•.• •• tu -.1 alat; "o W S i n 8.tua IL Val •••• t" * la •IP 0 -1.1 .0 • 0 14 : ...In 31 2 c t:t %.... V 0 E 7: WEIGHT OF SAMPLE — 16.16 0. SAMPLE LOSS-4.71 gr E :E o to 0 *0 TREATING EFFICIENCY —29% 0 g GYP SCALE REMOVAL CHEMICALS C FiVIURP1 - 2 p ab i .r■ . WEIGHT OF SAMPLE-11.45 grs. TESTRESULTS TREATED SAMPLE 11.":4■••■'" '•••"7"." .., .0 4.0 k....2 — 3 0_ a c ;-, r... E "v t I a a -- 0 a ... .1 AVERAGE EFFECT OF DILUTION ON GYP SCALE REMOVAL CHEMICALS GYP SCALE CRYSTALS CONVERTED -% Hot Water 100 80 iii 15°/0HCI 11 11 — Nr- 60 40 20 0 10 100 50 25 CHEMICAL CONCENTRATION —% FInURT: - 4 S OLUB I LI TY OF SCALES IN WATER ZOW I AtMI 1401YDRATE (C460-.11/420) 2400 2200 2000 ,GYPSUM (CaS0.2H20) 1100 , 1600 i 1400 1200 100 1111 Ilk ANwroarr a (Ca604 33 IS 104 140 171 212 240 214 120 36$ 0 am TEMPERATURE-'F SCALE PRECIPITATED 700 (6000 ppm in solution @ 100'F. & 100 gm/1 P4sCI) z 600 2 see 2 0 7 3116 lo o / /tut io c a 0 2 10 12 14 PRESSURE DROP in nein 100 111 SOLUBILITY OF CALCIUM VS SALT CONCENTRATION 4000 z 0 n 5000 3 a ♦000 a 3000 2000 0 50 100 150 Gill Conaminition—ipme.11 IOW R PERCENT CRYSTALS CONVERTED GYP CRYSTALS CONVERSION PERCENT VS TIME (optimum for laboratory tests) PACKER AND SPOTTING CONTROL VALVE (unknown fluid level) SLIPS SET PACKER OPEN PACKER CLOSED OPEN BY-PASS CLOSED SPOTTING CHEMICAL CHEMICAL ISOLATED TAILPIPE SPOTTING VALVE OPEN CLOSED TABLE I GYP SCALE REMOVAL TREATMENTS IN A WEST TEXAS FIELD YEAR 10 91 V • 73TAL WELLS 17 42 31 50 33 17 rya BEFORE OIL WATER 17 10 29 20 36 29 45 81 46 100 23 110 36 59 AFTER OIL 60 50 64 61 62 35 56 WATER 69 81 113 142 146 175 121 INCREASE OIL 43 21 28 18 16 12 20 WATER 59 61 84 61 46 65 62 - 1 1 TREATING RESULTS Well No. 1 2 3 4 5 6 7 8 9 10 Total Prior Production Water 12 0 7 3 21 8 8 21 17 13 44 0 35 9 61 5 8 13 37 16 88 250 Oil Oil Production After Water 197 46 37 37 120 102 31 393 61 56 89 97 81 68 96 31 72 104 35 96 880 969