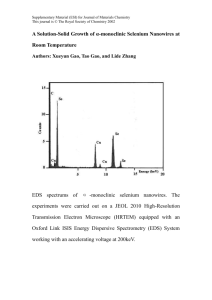
First principle study of excitons in dopped zinc oxide nanowire

First principle study of excitons in dopped zinc oxide nanowire
Quantum confinement and orientation
Kavous Mirabbaszadeh, Sara Yazdani
Department of Physics, Amir Kabir University of Technology, Tehran, Iran
First principle studies have been carried out on long dopped ZnO nanowires. The density functional theory-local density approximation (DFT+LDA+U) electronic structure has been obtained via a plane-wave code. In addition, Response-function of homogeneous electric field and the dielectric constant of dopped nanowires have been calculated. The electron-hole binding energy is calculated using an excitonic code. Quantum confinement effects and the orientation of the freestanding nanowires have been considered in the calculations. Finally, the results have been illustrated and compared with experimental values which are in agreement.
Reference:
Mauro Bruno, Maurizia Palummo, Andrea Marini, Rodolfo Del Sole, Valeria Olevano,
Alexandre N. Kholod and Stefano Ossicini, PHYSICAL REVIEW B 72, 153310 (2005)
F